
Abstract
Micronuclei are extra-nuclear bodies mainly derived from ana-telophase lagging chromosomes/chromatins (LCs) that are not incorporated into primary nuclei at mitotic exit. Unlike primary nuclei, most micronuclei are enclosed by nuclear envelope (NE) that is highly susceptible to spontaneous and irreparable rupture. Ruptured micronuclei act as triggers of chromothripsis-like chaotic chromosomal rearrangements and cGAS-mediated innate immunity and inflammation, raising the view that micronuclei play active roles in human aging and tumorigenesis. Thus, understanding the ways in which micronuclear envelope (mNE) goes awry acquires increased importance. Here, we review the data to present a general framework for this question. We firstly describe NE reassembly after mitosis and NE repair during interphase. Simultaneously, we briefly discuss how mNE is organized and how mNE rupture controls the fate of micronuclei and micronucleated cells. As a focus of this review, we highlight current knowledge about why mNE is rupture-prone and irreparable. For this, we survey observations from a series of elegant studies to provide a systematic overview. We conclude that the birth of rupture-prone and irreparable micronuclei may be the cumulative effects of their intracellular geographic origins, biophysical properties, and specific mNE features. We propose that DNA damage and immunogenicity in micronuclei increase stepwise from altered mNE components, mNE rupture, and refractory to repair. Throughout our discussion, we note interesting issues in mNE fragility that have yet to be resolved.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
A conserved feature of most eukaryotic organisms is their genomes are packed into linear chromosomes. All these chromosomes are organized into nucleus that is enclosed by nuclear envelope (NE). Faithful inheritance of every chromosome from parent cells to daughter cells is essential for proper development and thus is tightly controlled during mitosis. Higher eukaryotes usually undergo “open” mitosis, which involves an early event of NE breakdown (NEBD) to facilitate the movement and separation of the duplicated chromosomes. Once sister chromatids are separated, NE components bind to the surface of daughter chromatin discs and rapidly engulf each chromatin disc. This process, termed as NE reassembly (NER), is critical for generating intact and functional NE to enclose all daughter chromosomes within a single nucleus (Ungricht and Kutay 2017).
To ensure mitotic fidelity, several sophisticated cell cycle checkpoints have evolved in eukaryotic cells to inhibit the progression into or through mitosis if errors are detected (Maiato et al. 2015; Sacristan and Kops 2015; Nähse et al. 2017). Therefore, segregation errors occur in a relative low frequency (Thompson and Compton 2011; Wilhelm et al. 2019). During the ana-telophases of mitosis, missegregated chromosomes typically lag behind from the chromatin discs. If these lagging chromosomes/chromatins (LCs) are far enough from the rest of chromatin disc, they recruit their own NE and form into isolated micronuclei (Guo et al. 2019). While micronuclei are rare in healthy cells, they are frequent in aging, cancer, and many other diseased cells (Guo et al. 2019).
Although micronuclei and primary nuclei locate in same cells, they are enclosed by remarkably different envelopes. Unlike NE, micronuclear envelope (mNE) possesses altered protein components. Moreover, mNE exhibits a high frequency of spontaneous rupture and such rupture is always irreparable (Hatch et al. 2013). While the frequency of spontaneous mNE rupture is relative low in healthy and young cells, it is considerably high in aging (nearly 40%) and tumor cells (up to 60%) (Hatch et al. 2013; Barroso-Vilares et al. 2020). Ruptured micronuclei have several downstream consequences including chromothripsis and innate immunity that are critical for cancer, aging, and inflammation, raising the view that micronuclei may play active roles in aging, tumorigenesis, and many other diseases (Guo et al. 2019). Thus, definitively establishing the origin of rupture-prone and irreparable micronuclei has been acquiring increased importance in the past few years. Recent work has uncovered multiple mechanisms by which defects in mNE reassembly (mNER) cause irreparable ruptures of mNE in systems ranging from flies to humans.
In this review, we highlight recent works to illuminate how mNE is organized, why mNER is deficient and why mNE is rupture-prone, why ruptured mNE is irreparable, and how irreparable mNE influences the fates of micronuclei and micronucleated cells. We also outline important gaps in the causes and consequences of mNE fragility that remain to be clarified. To avoid confusion, “NE” and “NER” will be used in context of primary nuclei and “mNE” and “mNER” are used in context of micronuclei in the following sections.
NE is a spatiotemporally controlled structure
Prior to discuss why mNE are rupture-prone and irreparable, it may be informative to first understand the complexity of eukaryotic NE, especially how NEBD and NER are spatiotemporally controlled in a highly coordinated manner throughout the mitosis. Eukaryotic NE is physically connected with various cytoskeletal proteins and is functionally organized by nuclear membrane, nuclear lamina, and nuclear pore complexes (NPCs) (Fig. 1). Nuclear membrane is a phospholipid bilayer membrane that can be separated into outer and inner nuclear membranes (ONM and INM, respectively). ONM faces the cytoplasm and connects with endoplasmic reticulum (ER), while INM enclosing nucleoplasm (Ungricht and Kutay 2017). ONM and INM are merged at numerous sites of NPCs, a type of huge structures that are composed of multiple copies of different nucleoporins (Nups) and are used for transport and communication between nucleus and cytoplasm (Beck and Hurt 2017). Nuclear lamina is a relatively thin protein meshwork located under INM. The major structural components of nuclear lamina are the A-type (lamin-A and lamin-C) and B-type lamins (lamin-B1 and B2). B-type lamins are crucial to NE integrity while A-type lamins provide mechanical rigidity to NE (Cho et al. 2019; Nmezi et al. 2019). Through such a coordinated way, nuclear lamins interact with each other and hundreds of binding partners (such as LBR and emerin) to provide structural support to nucleus and help to maintain NE stability overtime (de Leeuw et al. 2018).

An architectural overview of nuclear envelope (NE) and micronuclear envelope (mNE). The nucleus of eukaryotic cells contains the nuclear genome that is organized into several discrete chromosomes. These chromosomes occupy distinct territories within the nucleus in a non-random manner. The nucleoplasm is separated from the cytoplasm by NE, which is comprised by nuclear membrane, nuclear pore complexes (NPCs), and nuclear lamina. The nuclear membrane is a specialized membrane structure of the endoplasmic reticulum (ER), and it consists of the inner nuclear membrane (INM) and the outer nuclear membrane (ONM). Interactions between nuclear lamina and INM proteins provide mechanical supports to the NE and facilitate chromatin organization. Some selected INM proteins including SUN1/2, LEM, emerin, and LBR are shown. These proteins interact with HP1 and BAF on chromatin to link NE and chromatin. INM and ONM are continuous and are joined at the sites of NPCs, which mediate the transport and communication between the nucleus and cytoplasm in a rapid while highly controlled way. The architecture of NPCs is highly complex and includes three features: concentric inner, outer, and membrane ring assemblies; cytosolic filaments; and nuclear basket. The nucleus is connected to the cytoskeleton network (such as actin filament, microtubule, and intermediate filament) mediated by nesprin and its various isoforms. The connection between nucleus and cytoskeleton network is essential for nuclear shape, position, and many other cellular processes. Compared to NE, mNE has altered protein components. The distribution, function, and structure of NPCs in mNE are heterogeneous. The integrity of nuclear lamin-B1 is lost in most micronuclei, and lamin-B1 is totally absent in some micronuclei. In contrast, some proteins such as LBR and emerin are relatively enriched in mNE. Deficient mNE protein components impair DNA repair, replication, and transcription in micronuclei and, more importantly, lead the mNE prone to rupture
Intracellular separation of nucleus and cytoplasm by NE holds advantages for eukaryotic cells to survive in the complicated environments. However, it presents a big challenge for dividing cells to precisely segregate the replicated chromosomes into daughter cells (Fig. 2). At the onset of open mitosis, NEBD is triggered by the phosphorylation of NPCs, lamins, LBR, and other INM proteins by several mitotic kinases including CDK1, PLK1, and Aurora-B (Linder et al. 2017). Upon phosphorylation, the affinity of these proteins to chromatin and to each other is significantly decreased. In prophase, cytoplasmic dynein is recruited to the outside of NE, where dynein generates pulling forces to trigger the tearing of nuclear membrane (Salina et al. 2002). Meanwhile, the growing microtubules facilitate the disruption of nuclear lamina (Beaudouin et al. 2002). The complete disassociation of chromatins from nuclear membranes is critical for the organization and segregation of mitotic chromosomes (Champion et al. 2019). After NEBD, the collapsed NE components are completely retracted by interconnected ER (Ungricht and Kutay 2017). During late prophase, nuclear protein Ki-67 coats each mitotic chromosomes to ensure their individuality and enables a complete set of chromosomes to move independently to each of the emerging daughter nuclei during ana-telophase (Cuylen et al. 2016).

The processes of nuclear envelope (NE) breakdown and reassembly during open mitosis. After replication, all chromatins are enclosed by NE in an interphasic nucleus. During early prophase, the chromatins begin to condense and cells separate the duplicated centrosomes, from which the mitotic spindle starts to assemble. NE breakdown is initiated in late prophase (prometaphase), and the progressive phosphorylation of NE components by mitotic kinases is required to dissociate them from chromatin. Upon breakdown, NE membrane proteins are retracted into the interconnected endoplasmic reticulum (ER). At metaphase, the chromosomes are captured by microtubules and aligned in the equator of the spindle. After all chromosomes have been organized at the metaphase plate, the sister chromatids are synchronously separated to form two daughter chromatin discs, and the anaphase is initiated. During anaphase, the central spindle microtubules are formed between the segregating chromatin discs and serve as a hub for signaling molecules required for the karyokinesis and cytokinesis. NE reassembly (NER) starts by the re-binding of ER membranes to chromatin discs at late anaphase and continues during chromatin decondensation at telophase. The process of NER is associated with a global activation of phosphatases such as PP1 and PP2A, which are required to counteract kinase activity on chromosomes, to dephosphorylate the components of reforming NE, and to promote chromosome decondensation. The organization of NE proteins during NER is spatially regulated: the parts of each chromatin disc that face the spindle pole and midzone are termed as the “core” region, whereas the other regions that perpendicular to midzone axis are termed as “non-core” region. Thus, the NE proteins that are initially reassembled into the core or non-core regions are called core or non-core proteins, respectively. A closed NE that contains nuclear pore complexes (NPCs) is formed around each chromatin disc at the end of telophase, which marks the end of mitosis. Cytokinesis completes cell division and involves splitting the cytoplasm by the ingression of a cleavage furrow followed by abscission mediated by midzone ring. Aurora-B is a mitotic kinase that localizes to the kinetochores and centromeres from prophase to metaphase and then locates to the cleavage furrow and midbody during cytokinesis. Such alteration of Aurora-B position is critical for Aurora-B in ensuring the fidelity of chromosome segregation
Upon entering anaphase, the phosphorylation of NE protein components is counteracted by concomitantly inactivating mitotic kinases and activating phosphatases such as PP1/PP2A (Mehsen et al. 2018). The chromokinesin Kid localizes to the boundaries of chromatin discs and helps to maintain their compressed state, which is prerequisite for ensuring proper NER and generating a single nucleus in each emerging daughter cell (Ohsugi et al. 2008). When chromosome separation is finished, the BAF cross-bridges anaphase chromatids and forms a meshwork that forces the reforming NE only load to the surface of each chromatin disc, thereby guiding the formation of a single nuclei (Samwer et al. 2017).
At the end of anaphase, NER begins when ER is targeted to the rims of chromatin discs (Schellhaus et al. 2016). Most of NE membrane proteins are inserted into ER immediately after they are translated from ER-bound ribosomes (Schellhaus et al. 2016). Thus, the diffusion within ER leads them to reach ONM and INM easily and quickly. Besides, many NE proteins possess a general affinity to chromatin (Schellhaus et al. 2016). Nuclear delivery of ER membrane is associated with the transition of ER shapes from tubules to sheets (Anderson and Hetzer 2007, 2008). Once the reassembling nuclear membrane engulfs chromatin discs, Nups start to assemble into NPCs to prevent the formation of NPC-free NE (De Magistris and Antonin 2018). The heteromultimeric protein machinery ESCRT-III functions to monitor and clear defective NPC intermediates (Webster et al. 2014). After NPCs reassembly, the recruitments of nuclear transport receptors like importins and exportins are needed to fully create permeability of NPCs (Lowe et al. 2015). Studies of nuclear lamins reassembly after mitosis are extremely rare and whether lamins reassembly occurs before or after the insertion of NPCs remains poorly understood.
The final steps of NER include the severing of spindle microtubules that remain connected to the core regions and the sealing of NE gaps where microtubules intersected. Membrane fission machinery ESCRT-III, ESCRT-III polymerization regulator CC2D1B, ESCRT-III disassemblase VPS4, microtubule-severing enzyme spastin, and INM protein LEM2 cooperate to spatiotemporally regulate microtubule disassembly and NE sealing at NE-microtubule intersection sites (McCullough et al. 2018). In addition, ER sheet insertion regulated by local lipid synthesis helps to narrow large NE holes intersected by microtubules, and this process is independent on ESCRT-III system (Penfield et al. 2020).
During NER, the organization of NE proteins on chromatin disc is spatially regulated (Afonso et al. 2014; LaJoie and Ullman 2017). To specify such spatial organization, the regions of each chromatin disc that face the spindle pole and midzone are termed as “core” region, whereas other regions that are perpendicular to midzone axis are termed as “non-core” region (Fig. 2). NE proteins initially reassembled into core and non-core regions are therefore called core and non-core proteins, respectively. Core protein BAF is required to establish such a transient spatial organization of chromatin disc (Haraguchi et al. 2008) and are recruited to the core regions by the central spindle microtubules (CSMs) (LaJoie and Ullman 2017). However, CSMs and kinetochore microtubules impede the access of ER membranes to core regions, leading these regions possess a relatively lower density of non-core proteins initially (De Magistris and Antonin 2018).
Uncontrolled NE rupture during interphase can be repaired quickly
Although NE is extremely dynamic during mitosis, it is also unstable during interphase. Multiple studies have demonstrated that interphase NE is prone to uncontrolled ruptures (Shah et al. 2017). Interphase NE rupture occurs most frequently when cells migrate through confined constrictions, where mechanical stress causes chromatin or nucleoplasm to herniate through the weak sites of nuclear lamina (Denais et al. 2016; Raab et al. 2016). Although cells benefit from the associated cytoskeletal proteins in regulating nuclear structure, position and function, NE stability, and NEBD (Kirby and Lammerding 2018; Patteson et al. 2019), NE rupture can be induced by actin- and dynein-derived compression forces imposed on nucleus, even in non-migrating cells (Hatch and Hetzer 2016; Takaki et al. 2017; Penfield et al. 2018). In addition, uncontrolled NE ruptures are particularly prominent in cells of laminopathies, a number of diseases that are associated with defects in lamin meshwork (de Leeuw et al. 2018). NE ruptures in these cells are frequently observed in static conditions and can be significantly increased by cytoskeletal mechanical stresses (Chen et al. 2018; Chen et al. 2019). Moreover, spontaneous NE rupture also occurs in cancer cells without lamin deficiency and extraneous mechanical stress (Vargas et al. 2012), probably due to the loss of either p53 or Rb pathway (Yang et al. 2017).
Depending on biological contexts, NE rupture confers both physiological roles and pathogenic consequences. NE ruptures in migrating cells could release intra-nuclear pressure and facilitate nuclear deformation (Shah et al. 2017), which is critical for leukocytes that need to rapidly migrate to the wound sites when injury occurs (Raab et al. 2016). NE rupture in migrating leukocytes therefore has significant roles in normal immune responses. However, NE rupture is also accompanied by uncontrolled exchange between nucleoplasm and cytoplasm, giving rise to DNA damage and nucleus fragmentation (Denais et al. 2016; Raab et al. 2016). To minimize the detrimental consequences, neutrophils reduce the rigidity of nucleus by expressing a relative low level of lamin-A, which enables low frequencies of NE rupture and DNA damage during long-distance migration (Rowat et al. 2013). Another reason for why normal cells are insusceptible to NE rupture-induced DNA damage is they cannot survive to genomic variation due to robust checkpoints, apoptosis pathways, and DNA repair machinery (Chen et al. 2019). Although cancer cells also alter the expression of NE proteins to modify nuclear shape and rigidity, NE rupture is frequent and has been evaluated as a major pathway for generating random genomic variation in metastatic cancer cells (Denais et al. 2016; Raab et al. 2016; Irianto et al. 2017).
Since persistent interspersion of contents in cytoplasm and nucleoplasm is detrimental for cell viability (Chen et al. 2018), ruptured NE can be efficiently repaired within minutes to several hours (Hatch et al. 2013; Irianto et al. 2017). As what occurs during NER, rapid repair of NE depends on ESCRT-III system (Denais et al. 2016; Raab et al. 2016). CHMP7, a core component of ESCRT-III system, is recruited to NE by the INM protein LEM2 to guide the assembly of ESCRT-III to ruptured NE (Gu et al. 2017). Besides, studies in mice fibroblasts revealed that BAF, together with transmembrane LEM-domain proteins, is required to repair NE rupture (Halfmann et al. 2019). Smaller ruptures and larger ones have different requirements for BAF: BAF is specifically recruited to larger ruptures to increase the recruitment of transmembrane NE proteins to the exposed chromatin, whereas smaller ruptures can be rapidly repaired without BAF (Young et al. 2020). In contrast, ESCRT-III system is thought to be highly efficient in repairing small NE ruptures (< 100 nm) (Lusk and Ader 2020). These data suggest two non-exclusive pathways for NE repair, one is ESCRT-III-dependent pathway that is used to repair smaller NE ruptures and another is BAF-dependent that is used to repair larger NE ruptures. In addition, lamins are required to counteracts forces generated from dynein during rupture repair, thereby ensuring stable NE repair and prevent catastrophic NE collapse (Penfield et al. 2018).
How mNE is organized
Micronuclei are spatially isolated nucleus-like bodies (Fig. 3a) that can be induced by diverse pathways (Guo et al. 2019). In most cases, micronuclei are caused by chromosome missegregation in mitosis. Micronuclei can also arise from resolving chromatin bridges and the fragmentation of nuclear blebs. In this sense, micronuclei can be used as index of chromosomal instability and intermediates of chromosome loss (Guo et al. 2020). Micronuclei analysis such as cytokinesis-block micronucleus (CBMN) assay (Fig. 3b) is widely used for measuring environmental genotoxic factors and for monitoring an individual’s genome health (Fenech et al. 2020).

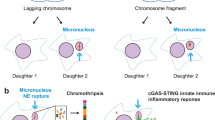
Images of micronuclei in human cancer cells. a Micronuclei are small nucleus-like bodies that spatially localized to the surround of primary nucleus. b An example of the binucleated cell harbors a micronucleus, as scored in the cytokinesis-block micronucleus (CBMN) assay. Missegregated chromosomes, such as lagging chromosomes, can be identified as micronuclei in binucleated daughter cells after cytochalasin-B induced cytokinesis block. Thus, the CBMN assay is widely used as a measure of chromosome instability. c and d Images of HeLa cells after exit the mitosis in a nocodazole challenged condition showing multiple micronuclei that are homogeneous in size (c) or heterogeneous in size (d). (a and b) Human HCC1806 breast cancer cells were stained for DNA (blue) and β-actin (red). Micronuclei are indicated by arrowheads. (c and d) Giemsa-stained DNA is in red, and the cytoplasm is in blue. Scale bars, 10 μm
A major feature of micronuclei is that their mNE exhibits high degree of structural and conformational alterations. Compared to primary nucleus within the same cell, most micronucleus recruits an altered set of NE components during mNER. It has shown that NPCs are reassembled in an extremely low density in mNE, resulting deficient import and export in micronuclei (Crasta et al. 2012; Hatch et al. 2013; Maass et al. 2018). In addition, most mNE loss the integrity of lamin-B1, and 30–50% of these are absent of lamin-B1 (Okamoto et al. 2012). In some cases, mNE also exhibit a high heterogeneity in lamin-A/C (de Castro et al. 2018). Since the presence of NPCs and lamins are morphological and biochemical features to distinguish NE from ER, loss of NPCs and lamins makes mNE to be biochemically more similar to ER. Moreover, most mNE have significantly increased the amounts of LBR and emerin, but the underlying mechanisms remain unclear (Maass et al. 2018). While LBR and emerin are normally localized at the periphery of NE, they diffuse in the nucleoplasm of most micronuclei (Maass et al. 2018). These results suggest that mNE organization is highly heterogenic (Fig. 1). Indeed, a recent study has shown that micronuclei are covered by three types of mNE: intact mNE assembly (in which both core and non-core NE proteins were evenly reassembled), type I mNE (in which only core NE proteins were reassembled), and type II mNE (some regions were deficient for both core and non-core NE proteins) (Miyazaki et al. 2020).
Why mNER is deficient and why mNE is prone to rupture
Compromised mNE, such as loss in the integrity of lamins, makes mNE to be rupture-prone (Hatch et al. 2013; Chen et al. 2018). The frequent observation of mNE rupture in cancers and the biological relevance of ruptured micronuclei underscore the need to dissecting the ways in which mNE goes awry. In principle, exploring the process of mNER would aid in better understanding the root causes of mNE fragility. In this section, we summarize our current knowledge about how mNER goes awry and why mNE is rupture-prone.
CSMs impede the recruitment of non-core NE proteins to mNE
Experimentally inducing microtubules’ mislocalization from centrosomes results in defective NER and abnormal nuclear shapes in HeLa cells (Kawaguchi et al. 2015). Moreover, aberrantly stable microtubules interfere the assembly of a continuous lamin-B1 network at mitosis exit (Naso et al. 2020). These findings underscore that NER requires a temporally and spatially restricted microtubule assembly. Since micronuclei are largely originated from LCs that typically embedded in CSMs during ana-telophase (Fig. 4a), it is interesting to ask whether CSMs induce mNER defects.

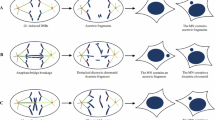
A summary of current knowledge about why micronuclear envelope reassembly (mNER) is deficient and why micronuclear envelope (mNE) is rupture-prone. a When the spindle assembly checkpoint is inactivated or dysfunctional, metaphases with aberrantly attached chromosomes can progress into anaphase, leading to a high frequency of chromosome missegregation. There are two types of missegregated chromosomes: peripherally localized chromosomes/chromatins and, more frequently, lagging chromosomes/chromatins (LCs). If these missegregated chromosomes are sufficiently far from the rest of the chromatin disc, they form into micronuclei and recruits mNE at mitotic exit. The LCs are embedded in the dense central spindle microtubules (CSMs), which impair the mNER around LCs. Recruitment of the mNE components around peripheral chromosomes is efficient, and the resulting peripheral micronuclei are therefore relatively stable. However, some of the peripheral micronuclei may undergo actin-dependent rupture, although the underlying molecular mechanisms remain incompletely understand. b At the metaphase-to-anaphase transition, Aurora-B is highly concentrated in the spindle midzone to form an Aurora-B gradient. Unlike chromatin discs, LCs are surrounded by Aurora-B gradient, which impairs mNER around LCs through several non-exclusive pathways. Firstly, Aurora-B gradient induces a midzone PLK-1 activity by directly phosphorylating PLK-1, and the phosphorylated PLK-1 impedes the recruitment of importin β and NPCs around LCs. Secondly, Aurora-B preferentially phosphorylates histone H3(Ser10) on LCs. The phosphorylated H3 prevents chromatin from associating with HP1α, resulting to localized inhibition of NER. Finally, Aurora-B gradient may generate a compact meshwork of central spindle microtubules (CSMs), which in turn causes most of LCs to undergo deficient mNER during the ana-telophase. c Aurora-B-mediated mNER delay ensures LCs to reintegrate the daughter nuclei. During anaphase, acentric LCs are connected to centric sister chromatids inside chromatin discs by Aurora-B-coated DNA tethers. In addition, acentric LCs are mechanically linked to peripheral microtubules, which work together with chromokinesin Klp3a to drive poleward movement of acentric LCs. Meanwhile, Aurora-B gradient phosphorylates H3(Ser10) on acentric LCs, which in turn prevents HP1α from associating with acentrics and inhibits mNER around acentrics. As a result, acentric LCs are encompassed by mNE that contain nuclear membranes but devoid of lamin and NPCs. The ectopic Aurora-B on DNA tethers induces localized nuclear envelope (NE) channels by delaying NE reassembly (NER) in inner core of daughter nuclei. To rejoin the acentric LCs, lamins around NE channels extend outward and retract back as acentric LCs passed through channels. After acentric LCs entering, membrane fusion protein NSF and ESCRT-III system drive membrane fusion between mNE and NE. Interestingly, the refused daughter nuclei undergo global morphological changes, probably reflecting a NE remodeling process to cover the channels with functional NE. In construct, NE channel cannot be induced by centric LCs, which makes them unable to reintegrate chromatin discs. As a result, they recruit a deficient mNE and form into rupture-prone micronuclei. d Since lamin-B1 is too stiff to bend, the localization of lamin-B1 at nuclear membrane is curvature dependent. Lamin-B1 is less likely to be located within smaller structures like mNE that have a high curvature but can stably interact with NE that have a low curvature
A recent study tried to answer this question through a series of elegant experiments. Liu et al. (2018) found that micronuclei formed from LCs that are located inside CSMs (termed here as central micronuclei) fail to recruit non-core proteins (e.g., Nup133, Nup107, and LBR) in HeLa cells. As a consequence, over 50% of central micronuclei undergo NE rupture. This effect can be counteracted by depolymerizing or relaxing the CSMs. More precisely, non-core proteins are absent from domains of LCs that are associated with CSMs but can be efficiently recruited to domains that are devoid of CMSs. These findings provide convinced evidence to support that excess contacts of LCs with CSMs would impede mNER around LCs. Besides, once LCs are released from CSMs when cells exit mitosis, LCs still fail to establish functional mNE, highlighting the importance of the fine order of mNER to establish a functional mNE (Liu et al. 2018).
How CSMs might limit the access of non-core NE proteins to LCs was not formally established. A likely possibility is that core and non-core NE proteins are distributed in different regions of ER. Since the nuclear delivery of ER membrane needs the transition of ER tubules to ER sheets (Anderson and Hetzer 2007, 2008), it is likely non-core proteins are enriched in ER sheets that are too large to penetrate CSMs, whereas the core proteins are enriched in ER tubules that are able to enter CSMs (Samwer and Gerlich 2018). In supporting this view, non-core NE proteins were reported to assemble within fenestrated ER sheets (Otsuka et al. 2018). Besides, limiting the delivery of ER sheets to LCs by CSMs may also impede the closure of nuclear membrane around LCs because ER sheet insertion is required to narrow NE holes (Penfield et al. 2020). These data indicate that CSMs not only disturb the recruitment of core proteins but also block the closure of mNE.
Midzone Aurora-B gradient delays mNER
Aurora-B is a protein kinase with a major role in monitoring mitotic fidelity (Afonso et al. 2017). By acting as a key regulator of the spindle assembly checkpoint (Sacristan and Kops 2015), chromosome separation checkpoint (CSC) (Maiato et al. 2015), and abscission checkpoint (Nähse et al. 2017), Aurora-B coordinates chromosome alignment and segregation, anaphase chromosome compaction, and cytokinesis to occur in a correct order. To fulfill these roles, Aurora-B position is changeable throughout mitosis (Fig. 2). Aurora-B first localizes along the chromosome during prophase and becomes concentrated at the inner centromere and kinetochores region during prometaphase and metaphase (Joukov and De Nicolo 2018). At the metaphase–anaphase transition, Aurora-B translocates to CSMs and later on concentrates at the midzone to form a spatial Aurora-B gradient (Fuller et al. 2008).
Aurora-B was found to inhibit NER around decondensed sperm chromatin in Xenopus interphase egg extract (Ramadan et al. 2007). More importantly, it has shown that the concentrated Aurora-B gradient prevents efficient non-core mNER around LCs in Drosophila and human U2OS osteosarcoma cells (Afonso et al. 2014). When Aurora-B is mislocalized away from the spindle, Nup107 binds to LCs with the same kinetics as to the chromatin discs (Afonso et al. 2014). A similar observation was reported in HeLa cells, where inhibition of Aurora-B allows the NPCs component mAb414 loading onto LCs normally (de Castro et al. 2018). These evidences indicate that Aurora-B may disturb mNER.
While this is a compelling hypothesis, some data argue against this possibility. Because there is a positive feedback loop in which Aurora-B activity organizes CSMs and CSMs is required to continuously maintain Aurora-B activity (Fuller et al. 2008), the negative effect of Aurora-B on mNER could be an indirect consequence of increased mass and bundling of CSMs result from Aurora-B. Indeed, inhibition of Aurora-B and microtubule polymerization displays a remarkably similar effect on the kinetics of non-core mNER (Liu et al. 2018). To separate the interdependent effects between Aurora-B and microtubules, Liu et al. (2018) used paclitaxel to prevent CSMs disassembly after inhibiting Aurora-B by ZM447439, and they found that the recruitment of Nup133 to LCs is deficient. Subsequently, they loosened the bundling of CSMs by siRNA-mediated knockdown of kinesin KIF4A, and no ZM447439 was added, and they found that KIF4A depletion increased the recruitment of Nup107, Nup133, and LBR to LCs. Based on these observations, Liu et al. concluded that the CSMs function independent on Aurora-B to inhibit non-core mNER (Liu et al. 2018).
Therefore, whether Aurora-B is involved in delaying mNER around LCs is still controversial, and further studies are warranted to the systematic identification of downstream targets of Aurora-B that participate in NER and how they are spatiotemporally regulated by Aurora-B. In our opinion, several potential pathways should be considered in future studies (Fig. 4b). Firstly, Aurora-B gradient induces a midzone PLK-1 activity by directly phosphorylating PLK-1, and the phosphorylated PLK-1 negatively regulates the recruitment of importin-β and NPCs around LCs (de Castro et al. 2018), indicating that the loading of NPCs to LCs is spatially monitored by Aurora-B-dependent PLK-1 activity. Importantly, PLK-1 induced mNER delay is not likely depending on CSMs, because PLK-1 could enhance the activity of depolymerase Kif2a to increase microtubule disassembly (Jang et al. 2009). Secondly, Aurora-B preferentially phosphorylates histone H3 Ser10 on acentric LCs to prevent it from associating with HP1α in Drosophila (Warecki and Sullivan 2018). Loss of HP1α would result local inhibition of mNER, as HP1α anchors the peripheral heterochromatin to INM and mediates NER in a dynamic manner (Kourmouli et al. 2000). Since HP1α is also required for the generation of a layer of heterochromatin at the nuclear periphery (MacPherson and Spakowitz 2019), loss of HP1α binding may explain the lack of peripheral heterochromatin at the periphery of most micronuclei (Sutyagina et al. 2019). Whether Aurora-B phosphorylates the H3 in centric LCs is still unclear. Finally, if LCs are trapped in midzone, Aurora-B mediates the abscission checkpoint to delay abscission by phosphorylating ESCRT-III component CHMP4C and retaining VPS4 within midzone (Thoresen et al. 2014). Since VPS4 is a disassemblase of ESCRT-III (Lata et al. 2008), retention of VPS4 inhibit ESCRT-III assembly and may impede mNE sealing. Interestingly, CSMs can disassemble normally in cells which the abscission checkpoint is activated (Steigemann et al. 2009), again suggesting Aurora-B may delay mNER around LCs independent on CSMs.
mNER delay is required for acentric LCs to reintegrate the daughter nuclei
Higher eukaryotic cells have sophisticated mechanisms to maintain their genome in a single nucleus. Considering this, one might have expected that eukaryotic cells would have mechanisms to ensure LCs to be enclosed into daughter nuclei. Several studies in Drosophila models have shed light on this question. Early evidence from fission yeasts has shown that spindle elongation rate is slowed in cells with LCs and these LCs appear to interact with microtubules for poleward movement (Pidoux et al. 2000). In agreement with this find, acentric LCs in Drosophila neuroblasts lag during anaphase due to the lack of centromeres, but over 95% of them ultimately segregate poleward (Royou et al. 2010). Acentric LCs are connected to centric sister chromatids located in chromatin discs by DNA tethers, which are coated with BubR1, Polo, INCENP, and Aurora-B and are essential for the reintegration of acentric LCs (Royou et al. 2010). Moreover, acentric LCs are mechanically linked to peripheral microtubules, and the chromokinesin Klp3a plays key roles in driving poleward movement of acentric LCs in Drosophila neuroblasts (Karg et al. 2017). Further studies found that mNER around acentric LCs is delayed and localized NE channels are induced by acentric LCs in nascent daughter nuclei, which provide means for acentric LCs to reintegrate the daughter nuclei (Karg et al. 2015).
Notably, a series of studies from Sullivan and colleagues provided insights into how NE channels are formed and how acentric LCs are reintegrated in Drosophila (Fig. 4c). Aurora-B gradient preferentially phosphorylates H3(S10) on acentric LCs (Warecki and Sullivan 2018). The phosphorylated H3 prevents HP1α from associating with LCs and results in localized inhibition of mNER around acentric LCs (Warecki and Sullivan 2018). As a result, acentric LCs are encompassed by mNE that contain nuclear membranes but devoid of lamins and NPCs (Warecki et al. 2020). On the other hand, ectopic Aurora-B on DNA tethers induces localized NE channels by delaying NER around the NE-tether intersection sites (Karg et al. 2015). Thus, lamins, NPCs, and nuclear membranes are all lacked in NE channels (Warecki et al. 2020). To rejoin the acentric LCs, lamins around NE channels extend outward and retract back as acentric LCs passed through channels (Warecki et al. 2020). After acentric LCs entering, membrane fusion protein NSF and ESCRT-III system drive membrane fusion between mNE on acentric LCs and NE on daughter nuclei (Warecki et al. 2020). Interestingly, the refused daughter nuclei undergo global morphological changes, probably reflecting a NE remodeling process to restructure NE architecture (Warecki et al. 2020). Overall, Aurora-B mediates CSC that provides a spatial cue to delay NER and mNER until acentric LCs can be incorporated within chromatin disc to form a single nucleus in Drosophila (Karg et al. 2015; Warecki and Sullivan 2018). Meanwhile, abscission checkpoint is coordinately mediated by Aurora-B gradient to provide sufficient time for acentric LCs reintegration before the completion of cytokinesis (Nähse et al. 2017).
These findings raise an interesting question of whether Aurora-B-mediated CSC is evolutionarily conserved. Nevertheless, this question has never been systemically studied in higher eukaryotes. It has been found that the most majority of LCs would ultimately end up in micronuclei in human cell lines. For example, ~ 50% of LCs induced by spindle assembly checkpoint inhibitors ultimately end up in micronuclei in RPE-1 cells (Soto et al. 2017). 61.5–86.7% of LCs induced by nocodazole are entrapped in micronuclei in HCT116 cells (Huang et al. 2012). In U2OS cells, 78% of anaphases with spontaneous LCs result in at least one daughter cell with micronuclei (Thompson and Compton 2011), despite inconsistent result is reported (Girão et al. 2019). In RPE-1 cells, 100% of spontaneous LCs and 81.4–94.4% of LCs induced by replication stress end up in micronuclei (Wilhelm et al. 2019). Therefore, it has been proposed that human cells appear to lack a strict Aurora-B-based CSC checkpoint to reintegrate LCs (Lusk and King 2018). However, it is premature to conclude it because most of these LCs are centrics, which have been reported to be unable to induce NE channels in Drosophila (Karg et al. 2015). How does CSC function differently between acentric and centric LCs remain to be clarified. Interestingly, recent evidence suggests that acentrics can be tethered to centric sister chromatids by MDC1-TOPBP1 pathway in human mitotic cells, and deficiency in this pathway results in the formation micronuclei (Leimbacher et al. 2019). Thus, whether Aurora-B participates in ensuring acentric LCs to reintegrate in human cells will be a topic of study in the coming years.
It is interesting to note that over 80% of centric LCs resulting from induced chromosome alignment defects are able to reintegrate the nascent daughter nucleus in RPE-1 cells (Fonseca et al. 2019). Centric LCs derived from unaligned chromosomes display defects in interchromosomal compaction and abnormal lamin-A/C distribution and rejoin the chromatin disc to form multilobed daughter nuclei (Fonseca et al. 2019). How reintegration of unaligned LCs is molecularly regulated in these cells is not known. Coupled these results with previous data that nearly 80% of LCs derived from aligned but misattached chromosomes end up into micronuclei in U2OS cells (Thompson and Compton 2011), it is indicative that the types of molecular defects giving rise to centric LCs may determine whether centric LCs reintegrate the chromatin disc or not (Orr and Maiato 2019). Although the reintegration of unaligned LCs in RPE-1 cells supports the existence of CSC in human cells (Orr and Maiato 2019), whether such reintegration is mediated by Aurora-B remains unclear. It is interesting to see whether mNER around unaligned LCs is fundamentally different from this of misattached ones.
Collectively, delaying mNER around LCs by Aurora-B gradient is an adaptive response to promote the reintegration of acentric LCs into chromatin discs and ultimately contributes to preserving genome integrity. However, such response may cause deficient mNER around centric LCs that cannot reintegrate chromatin discs (Fig. 4c). A comprehensive picture on this model will require data from human systems.
Peripheral micronuclei undergo actin-dependent mNE rupture
As mentioned above, delaying NER around LCs by CSMs, Aurora-B gradient or some other unappreciated factors are an adaptive response to the presence of LCs at the midzone. This indicates that rupture-prone is an intrinsic property of LCs-derived micronuclei and raises the interesting question of whether micronuclei not derived from LCs have a stable mNE.
To this end, Liu et al. developed an elegant protocol to move the unaligned chromosomes outside the regions of spindle microtubules (Fig. 4a). Micronuclei formed from these peripheral chromosomes (termed as peripheral micronuclei) are able to recruit both core and none-core NE proteins and form functional NPCs into NE. In line with these results, ~ 10% of the peripheral micronuclei in HeLa cells underwent spontaneous NE rupture though 50% of central micronuclei showed mNE ruptured (Liu et al. 2018). However, peripheral micronuclei in RPE-1 cells showed a high rate of NE disruption (~ 32%). Moreover, they found that treating RPE-1 cells with actin assembly inhibitor latrunculin A results a statistically significant reduction of NE rupture in peripheral micronuclei (reduced to ~ 10%) but not in central micronuclei. However, actin-dependent micronuclei rupture was not observed in HeLa cells whose peripheral micronuclei possessed a lower frequency of rupture (Liu et al. 2018). Interestingly, the proportion of ruptured to intact micronculei in U2OS cells was not altered after treating with actin assembly inhibitor cytochalasin D (Hatch and Hetzer 2016). One limitation of this study was that various groups of micronculei (such as peripheral and central micronuclei) have not been distinguished. Overall, these finding suggests that the induction of peripheral micronuclei rupture by actin filaments (F-actin) is context dependent (Fig. 4a).
The finding that peripheral micronuclei undergo actin-dependent rupture is not unexpected. It has shown that NE rupture in U2OS cells is inhibited by the loss of contractile actin bundles whereas disrupting F-actin or contractile actin fiber significantly prevents NE rupture (Hatch and Hetzer 2016). Similar observation has been noted in HeLa cells, whereby contractile actin fibers locally compress the nucleus, leading to NE rupture (Takaki et al. 2017). In addition, disrupting F-actin by cytochalasin-D reduces NE ruptures in mouse embryonic fibroblasts lacking nuclear lamins (Chen et al. 2018). Recently, F-actin is found to grow within nuclear lamina and sprout spikes to pry nuclear membranes away from nuclear lamina and segregate NPCs into conglomerates, which leave stretches of bare membranes that are prone to rupture in starfish oocytes (Wesolowska et al. 2020).
Note that F-actins induce peripheral micronuclei rupture should be borne in mind when using CBMN assay for evaluating micronuclei rupture. Micronuclei scoring in CBMN assay is specially restricted to once-divided cells, which are recognized by their binucleated appearance (Fig. 3b) after blocking cytokinesis with cytochalasin-B (Fenech 2007). Given micronuclei rupture in some cases can be induced by F-actin, cytochalasin-B might reduce the frequency of NE rupture in micronuclei in CBMN assay. In addition, the cleavage furrow ingression is needed for CSMs to be bundled and compacted into an intercellular bridge (Fig. 2). Nevertheless, the inhibition of cleavage furrow by cytochalasin-B may further reduce the frequency of CSM-induced micronuclei rupture. Thus, there are probably caveats to the applicability of CBMN assay to measure the frequency of inherited and induced micronuclei rupture.
Overall, microtubules and F-actins participate in inducing unstable mNE. This viewpoint is further supported by the observation that micronuclei induced by colchicine or colchicine and cytochalasin-B in HCT116 cells are largely intact (Kneissig et al. 2019). These findings lay the foundation for future studies in dissecting how cytoskeleton networks regulate the membrane dynamics of mNE.
Lamin-B1 is devoid from highly curved regions of mNE
Among all lamin subtypes, lamin-B1 is essential for mNE integrity because loss of lamin-B, but not lamin-B2 or lamin-A/C, is specifically linked to mNE rupture (Hatch et al. 2013). It has found that both central and peripheral micronuclei develop discontinuity in lamin-B1 nuclear rim, and this discontinuity cannot be restored by actin or microtubule assembly inhibitors (Liu et al. 2018). Similarly, up to 42% of micronuclei induced by colchicine alone or colchicine and cytochalasin-B in HCT116 cells is devoid of lamin-B1 (Kneissig et al. 2019). These findings suggest that cytoskeleton can partly explain deficient recruitment of lamin-B1 in micronuclei. Instead, lamin-B1 discontinuity may be induced by factors intrinsic to mNE, such as high curvature (Fig. 4d).
It has found that the localization of lamin-B1, but not lamin-A/C, at NE is curvature-dependent: in circular nuclei, lamin-B1 is evenly distributed around NE, while in more ellipsoidal nuclei, lamin-B1 is rarely distributed at the pole regions (Nmezi et al. 2019). Similarly, highly curving regions of nuclei, such as nuclear blebs imposed by cell migration through small pores, consistently associates with lamin-B1 dilution (Xia et al. 2019). These data suggest that high-curvature regions in elongated nuclei can result in dilution or loss of the lamin-B1 meshwork, although it remains incompletely understand to what extent the curving NE directly disrupts lamin-B1 assembly. In light of this finding, lamin-B1 is less likely to be located within smaller structures like micronuclei where the membrane curvature is high. Consistent with this point, LCs always fail to recruit lamin-B1 (Afonso et al. 2014) but can efficiently assemble lamin-A/C (de Castro et al. 2018), and micronuclei with high curvature display low lamin-B intensity (Xia et al. 2019).
Typically, lamin-B filaments have a high affinity for NE because this process is mediated by LBR (LaJoie and Ullman 2017). Given that most mNE possess a relative high level of LBR (Maass et al. 2018), it is interesting to ask why lamin-B1 is excluded from high-curvature regions. Lamin-B filaments are very stiff with the persistence length (a stiffness parameter) is ~ 0.5 μm in mouse cells (Turgay et al. 2017). In light of this observation, Xia et al. (2019) proposed that lamin-B filaments are too stiff to bend along high-curvature membrane areas that have small diameters. This model could generally apply to highly curved membranes like mNE. Indeed, lamin-B1-negative micronuclei have a significant small diameter (1.9 μm) than this of lamin-B1-positive ones (2.7 μm) regardless the number of encapsulated chromosomes (Kneissig et al. 2019). Peripheral micronuclei are typically larger and assemble more lamin-B1 as compared with central micronuclei (Liu et al. 2018). These results indicate that the proper assembly of lamin-B1 depends on the curvature of mNE (Kneissig et al. 2019).
In addition, membrane curvature seems not to be the only factor to regulate recruitment of lmain-B1 to mNE. Double minutes are a specific type of micronucleus that usually derived from extrachromosomal DNA and commonly seen in cancer genome (Guo et al. 2019). It has shown that lamin-B recruitment is much more efficient in micronuclei bearing double minutes than those bearing chromosomes or chromatin fragments in human Colo320 cells (Utani et al. 2007). Thus, it is important to define whether lamin-B1 can be recruited to high-curvature mNE by other pathways. Although loss of lamin-B2 does not induce mNE rupture, its overexpression suppresses mNE rupture (Hatch et al. 2013). One open question arising from this finding is whether recruitment of lamin-B1 to mNE is facilitated by lamin-B2.
The dilution or loss of lamin-B may promote NE rupture at high-curvature sites. High nuclear curvatures imposed by external probes can rapidly rupture NE without disrupting plasma membrane (Xia et al. 2018; Xia et al. 2019). In line with this, several recent studies have identified that NE rupture occurs at the leading tip of the nuclei migrating through a constriction (Denais et al. 2016; Raab et al. 2016; Bakhoum et al. 2018). Besides, lamin-B1 dilution clearly correlates spatiotemporally with loss of NE integrity (Xia et al. 2019). Thus, high curvature favors NE rupture after lamin-B1 disruption. This model could generally apply to high-curved mNE. Indeed, small micronuclei have a high trend than large micronuclei to rupture (Xia et al. 2019), although it has also reported that mNE rupture is not related to micronuclei size (Hatch et al. 2013).
High curvature not only restricts the distribution of lamin-B1, but it also compromises the retention of DNA repair factors after NE rupture (Crasta et al. 2012). High nuclear curvatures imposed by an external probe or by cell attachment to either aligned collagen fibers or stiff matrix promote the mislocalization of multiple DNA repair factors (including KU-70 and KU-80) to the cytoplasm (Xia et al. 2018). As a result, ruptured nuclei with higher levels of DNA repair factor mislocalization have higher levels of DNA damages, as indicated by γH2AX (Xia et al. 2018). This model could generally apply to micronuclei, as small micronuclei harbor high cGAS and γH2AX but low lamin-B and KU70/KU80 (Xia et al. 2019).
Over the last several years, the curvature-dependent effects of NE have received increasing attention. To induce different levels of NE curvature, the conventional approaches employ the external probes, pores, or channels. Micronuclei generated during cell division vary in size and therefore in curvature. For example, high dose of colchicine could induce up to 22 micronuclei with varied size (from 1 to ~ 8 μm) in PtK1 (Potorous tridacylis kidney) cells (Géraud et al. 1989). Figure 3 c and d shows nocodazole-treated HeLa cells containing multiple micronuclei that are homogeneous or heterogeneous in size, respectively. In addition, BAF depletion in HeLa cells causes a remarkable micronucleation phenotype, and most micronuclei are formed around individual chromosomes, giving these micronuclei a highly varied curvature (Samwer et al. 2017). Such a great variation in size of induced micronuclei raises the possibility to use the micronucleation as a more rapid and direct approach to study the curvature-dependent effects of NE (Xia et al. 2019). However, we should note that the commonly-used micronuclei-inducing agents also disturb mNER, which is a potential obstacle in using micronuclei to study curvature-related properties of NE.
Lamin-B1 in mNE is specifically degraded by autophagy
Autophagy is a process in which intracellular components and dysfunctional organelles are delivered to the lysosome for degradation and recycling. Nucleophagy is a selective autophagy that can function physiologically to clean up nuclear wastes produced by nuclear damage (Park et al. 2009). During oncogene-induced senescence, several NE proteins (such as lamins, LBR, and emerin) can be degraded by nucleophagy (Dou et al. 2015; Lenain et al. 2015). Interestingly, micronuclei have been reported to be degraded by autophagy in frog (Chmielewska et al. 2018) and human cells (Rello-Varona et al. 2012; Bartsch et al. 2017). Specifically, lamin-B1 is a primary substrate for nucleophagy in mNE (Dou et al. 2015). Unlike what occurs in NE (Lenain et al. 2015), lamin-A/C and lamin-B2 in mNE are not degraded by nucleophagy (Dou et al. 2015). The liability of lamin-B1 to be degraded underlies the rupture-prone nature of mNE because the expression of nondegradable lamin-B1 significantly reduces mNE rupture (Hatch et al. 2013).
Why ruptured mNE is irreparable
As we mentioned above, ESCRT-III system is responsible for NE sealing during later mitosis and NE repairing during interphase. To specifically target ESCRT-III system to nuclear membrane, the INM protein LEM2 condenses into a liquid-like phase and associates with CHMP7, which in turn directly recruits core ESCRT-III subunits including CHMP4B and other downstream ESCRT-III proteins to NE (Gu et al. 2017; von Appen et al. 2020). In cells with ruptured NE, CHMP7 is diffused into nucleus through NPCs or ruptured sites and become activated after binding with LEM2. The activated CHMP7 promotes the polymerization of ESCRT-III system at the ruptured sites, which in turn induces nuclear membrane expansion and remodeling to accelerate NE repair (Thaller et al. 2019). LEM2 can also interact with another INM protein Nur1 to recruit ESCRT-III to the sites of NE seal and repair (Pieper et al. 2020). Precise control of CHMP7 level at INM is critical for NE repair because the mutant CHMP7 that persistently locates in INM induces unlimited polymerization of ESCRT-III and dramatic NE deformation at NE. Thus, CHMP7 is pulled apart from LEM2 by Vps4 (Pieper et al. 2020) and is actively excluded from nucleus by export transport receptor Xpo1 upon the completion of NE repair (Vietri et al. 2019).
However, this model of membrane seal and repair was developed through analysis of NE ruptures. Ruptures of mNE differ from these of NE in that they are usually irreparable (Hatch et al. 2013). Given the importance of mNE rupture, several attempts were made to define why ruptured mNE are refractory to repair. To answer this question, it should firstly understand whether and how ruptured mNE can be detected by membrane repair system. As what occurs in NE, CHMP7 is needed to ensure mNE integrity. After binding with LEM2, CHMP7 accumulates selectively on micronuclei that have discontinuous or no lamina and have reduced or no NPCs (Willan et al. 2019). Meanwhile, CHMP7 accumulation correlates well with ER membrane invasion (Willan et al. 2019). However, despite ESCRT-III system and ER can be timely recruited to the ruptured sites of mNE by CHMP7 and LEM2, it is not associated with the initiation of NE repair. Instead, micronuclei inherently lack the capacity to restrict the accumulation of CHMP7, which drives unrestrained ESCRT-III recruitment and induces membrane deformation and secondary ruptures (Vietri et al. 2019). Similarly, mNE ruptures are increased in cells depleted with VPS4, an AAA ATPase that promotes ESCRT-III turnover (Willan et al. 2019). How ESCRT-III accumulation induces mNE deformation is unclear. Nevertheless, it seems that ESCRT-III is not the primary cause of mNE rupture, since the fractions of micronucleated cells and those with ruptured mNE are significantly increased in cells depleted with CHMP7 (Willan et al. 2019). These results demonstrate that while baseline level of ESCRT-III is required for ensuring mNE integrity, persistent ESCRT-III accumulation upon excessive nuclear influx of CHMP7 not only fails to repair the ruptured mNE but also deforms the mNE and further promotes NE rupture. Interestingly, ESCRT-III accumulation may have more adverse consequences to mNE. For example, ESCRT-III may destabilize and clear defective NPCs in mNE (Webster et al. 2014).
Overall, ESCRT-III system has opposite roles in maintaining the integrity of NE and mNE (Fig. 5). ESCRT-III exhibits exquisite sensitivity to rapidly response and repair NE ruptures, whereas it fails to repair, and even further deforms, the ruptured mNE because it is unlimitedly polymerized and hyperactivated at ruptured sites of mNE. Why micronuclei inherently fail to restrict the accumulation of CHMP7 remains an important question. A recent study establish that CHMP7 (~ 70 kD) is small enough to passively leak through the NPCs but is actively exported from nucleus by Xpo1 (Thaller et al. 2019). These data indicate that the establishing of functional ESCRT-III systems to ruptured NE depends on the robustness of NPCs. Since NPCs in mNE is severely compromised, it explains why micronuclei are unable to limit the recruitment of CHMP7. In addition, CHMP7 is preferentially accumulated in acentric micronuclei (Willan et al. 2019). It is therefore interesting to determine whether centromere is involved in restraining the access of CHMP7 to mNE. Besides, impaired ubiquitin proteasome system in micronuclei (Maass et al. 2018) may prevent the turnover of CHMP7 and other ESCRT-III subunits.

A summary of current knowledge about why ruptured micronuclei are irreparable. When nuclear envelope (NE) rupture occurs in primary nucleus, CHMP7 rapidly diffuses into nucleus through the ruptured site. Then, CHMP7 forms complexes with LEMD2 surrounding the ruptured site to activate ESCRT-III system. After all ruptures are completely repaired, CHMP7 dissociates with LEMD2 and is exported to the cytoplasm in an Xpo1-dependent way by nuclear pore complexes (NPCs). In contrast, when micronuclear NE (mNE) rupture, CHMP7 diffuses into the micronuclei and quickly spreads along the whole micronuclei surface. Because Xpo1-dependent nuclear-to-cytoplasmic export pathway is unavailable in mNE, CHMP7 accumulates rapidly within micronuclei and combines with all available LEM2 protein in inner nuclear membrane. Excess CHMP7-LEM2 complexes cause unrestrained recruitment of ESCRT-III to the ruptured sites, which not only fails to repair the mNE but also drives extreme membrane deformation and catastrophic DNA damages (e.g., generating DNA double-strand breaks and single-strand DNA) in micronuclei
Together, altered NE protein components in mNE not only leads micronuclei susceptible to future rupture but also makes ruptured mNE refractory to repair. Obviously, mechanisms implicated to intrinsic irreparability of ruptured mNE have only started to be recognized. Interestingly, nuclear lamina counteracts dynein-generated forces on damaged nuclei and ensures stable NE repair (Penfield et al. 2018). Times for repairing ruptured NE are more prolonged in cells that knockout all lamins than this of cells that only knockout lamin-B1, and irreparable NE ruptures is frequently observed in cells knockout all lamins (Chen et al. 2018). In this context, an interesting question remains to be proved is whether lamin-B1 discontinuity or absence contributes to the irreparability of mNE ruptures.
The problems with ruptured and irreparable mNE
Compromised mNE has significant downstream consequences to the fate of micronuclei and micronucleated cells (Fig. 1). Low density of NPCs in mNE impairs the recruitment of DNA replication and repair factors, resulting in delayed replication and deficient DNA repair capacity in micronuclei. Premature condensation then causes the micronucleated chromosomes to shatter into multiple fragments, which would be rejoined by non-homologous end joining (NHEJ) and results in chromothripsis-like complex rearrangements (Zhang et al. 2015; Ly et al. 2017). In addition, a direct outcome after mNE rupture is the extrusion of double-strand DNA (dsDNA) fragments into cytoplasm, which act as a trigger of innate immunity mediated by cGAS-STING pathway (Ablasser and Chen 2019). Activation of cGAS-STING pathway has multiple consequences to micronucleated cells, such as inflammation, metastasis, senescence, or apoptosis (Guo et al. 2019). In addition, nuclear DNA derived from collapsing micronuclei can be loaded to exosomes, a type of extracellular vehicles (Yokoi et al. 2019). Meanwhile, nucleosomes released from ruptured micronuclei can be secreted outside of the cell by amphisomes, a new model for active secretion of extracellular DNA (Yokoi et al. 2019). These data indicate that ruptured micronuclei serve as important mediators for intercellular genetic communication and sources of cell-free DNA in cancers.
Refractory to mNE repair is expected to increase DNA damage and immunogenicity in micronuclei. Unrestrained ESCRT-III recruitment drives a high degree of catastrophic DNA damage to the micronucleated chromosomes. Micronuclei enriched for CHMP4B preferentially accumulate a high level of single-stranded DNA (ssDNA) (Vietri et al. 2019; Willan et al. 2019). Accumulation of CHMP7 is important for generating ssDNA in micronuclei because the proportion of micronuclei that are enriched for ssDNA is significantly reduced upon CHMP7 depletion (Willan et al. 2019). Moreover, unregulated accumulation of ESCRT-III could drive the fragmentation of micronucleated chromosomes, although the underlying mechanism remains unclear (Vietri et al. 2019). The authors reasoned that it could be the result of DNA replication stress and premature chromosome condensation in micronuclei (Vietri et al. 2019). Nevertheless, this cannot explain why these catastrophic DNA damages are tightly associated with ESCRT-III accumulation. Interestingly, ESCRT-III accumulation also recruits exonucleases, such as TREX1, to ruptured micronuclei (Vietri et al. 2019). Accumulation of TREX1 may explain such association because TREX1 is found to drive ssDNA and chromothriptic fragmentation (Maciejowski et al. 2019). This also raises the interesting question of whether TREX1 represents a NHEJ-independent pathway to drive chromothripsis in micronuclei. In addition, binding of cGAS to micronuclei is also enhanced by CHMP7 accumulation (Willan et al. 2019). This cannot be the result of accumulated ssDNA in CHMP7-enriched micronuclei because cGAS has a dramatically reduced affinity for ssDNA (Ablasser and Chen 2019). Thus, it is intriguing to speculate that CHMP7 alters the structure of micronucleated chromatin to facilitate cGAS-mediated cytosolic DNA recognition.
Based on these evidences, DNA damage and immunogenicity in micronuclei are increased stepwise from mNE structure alteration, rupture, and refractory to repair (Fig. 6). Micronuclei with altered NPCs are unable to recruit sufficient DNA replication and repair factors to maintain replication fidelity (Crasta et al. 2012; Hatch et al. 2013; Xia et al. 2019). As a consequence, replication stress induces a relative low level of DNA damage within intact micronuclei that have no immunogenicity. Upon mNE rupture, either spontaneous or induced from premature chromosome condensation, damaged dsDNA and DNA repair factors are extruded into cytoplasm, which activate the immunogenicity of micronuclei and further increase DNA damage within ruptured micronuclei (Zhang et al. 2015; Harding et al. 2017; Mackenzie et al. 2017; Xia et al. 2019). Finally, persistent recruitment of ESCRT-III to ruptured micronuclei not only fails to repair mNE but further drives chaotic chromosome damage and alters chromatin structure to enhance cGAS binding (Vietri et al. 2019; Willan et al. 2019). Although mNE rupture induces DNA damage in micronuclei, DNA damage in micronuclei per se cannot induce mNE rupture (Hatch et al. 2013).

Framework outlining the steps of acquiring DNA damages and immunogenicity in micronuclei. Current evidence presented in this Review supports that DNA damage and immunogenicity in micronuclei are increased stepwise from altered micronuclear envelope (mNE) components, mNE rupture, and refractory to repair. See further description in text
Overall, substantial advances in our understanding about the biological relevance of micronuclei moved our view of micronuclei from a passive index of chromosomal instability to a possible active player in human aging, cancer and other diseases. This also emphasizes that mNE rupture analysis should be included in micronuclei-based genotoxic studies and human bio-monitoring studies. Doing so could allow ruptured micronuclei, rather than total micronuclei, to be used as a more specific and precise biomarker in genotoxicology and human aging and diseases.
Conclusions and outlooks
Driven by technological advances, the last two decades have seen substantial progresses toward the understanding of the biological functions of micronuclei. Ruptured micronuclei play active roles in chromothripsis, senescence, inflammation, and even tumor initiation and evolution. This review has summarized the current knowledge on the birth of rupture-prone and irreparable micronuclei, an emerging theme of micronuclei biology. Based on these results, a complicated picture of the causes of mNE fragility is beginning to emerge. We conclude that irreparable micronuclei rupture may be the cumulative effects of their intracellular geographic origins, biophysical properties, and specific mNE features, although these are more complex than the currently conceived models. Considering this complexity, full understanding how and why mNE undergo irreparable rupture remains ongoing challenges ahead.
Moving forward, it will be interesting to explore whether the kinesin Eg5 disturbs mNER around LCs, because ~ 80% of micronuclei derived from LCs induced by Eg5 inhibition do not experience mNE rupture in PtK1 cells (He et al. 2019). Since a balance of cytoskeletal forces is needed to maintain NE architecture and integrity, whether mNE is also associated with cytoskeleton and, if so, whether and how this process is imbalanced are challenging questions to test. In addition, spatiotemporal regulation of membrane lipid synthesis is required for NE integrity (Barbosa et al. 2019; Penfield et al. 2020). Further study is crucial to define whether spatiotemporal regulation of lipid synthesis is disturbed in mNE. Considering the rupture size is a critical factor in determining the efficiency of NE repair, it is tempting to consider whether mNE ruptures are typically large that beyond the repair capacity of ESCRT-III system and whether BAF-mediated membrane repair pathway is relatively efficient in repairing mNE ruptures. Besides, amounting evidence has shown that ruptured micronuclei are associated with the invasion of ER tubules inside micronucleated chromatin (Hatch et al. 2013; Maass et al. 2018; Willan et al. 2019). Nevertheless, how ER is invaded and what is the biological significance of this phenotype are poorly understood. A likely possibility for the former scenario is that ER invasion is mediated by microtubules which stick into micronuclei before or after mNER, because ER tubules usually slide on microtubules for its extension (Chen et al. 2013). Whether ER invasion is a reflection of ruptured micronuclei or whether invaded ER prevents mNE ruptures from being sealed need to be tested.
Elucidating the origins and fates of rupture-prone micronuclei not only provides a glimpse into the molecular regulation of normal NER but also helps to interprete the pathology of genetic diseases associated with NE malfunction. Given the diverse biological consequences of ruptured micronuclei, a more mechanistic understanding of why mNE is prone to irreparable rupture may also have a broad relevance in identifying mechanisms contributing to multiple cellular processes, such as chromothripsis, senescence, inflammation, autoimmune, and even to tumorigenesis and cancers immunotherapy. Considering this, studying the structure, organization, and function of mNE is likely to be new frontiers in cell biology.
Data availability
Not applicable.
References
Ablasser A, Chen ZJ (2019) cGAS in action: expanding roles in immunity and inflammation. Science 363:eaat8657. https://doi.org/10.1126/science.aat8657
Afonso O, Matos I, Pereira AJ, Aguiar P, Lampson MA, Maiato H (2014) Feedback control of chromosome separation by a midzone Aurora B gradient. Science 345:332–336. https://doi.org/10.1126/science.1251121
Afonso O, Figueiredo AC, Maiato H (2017) Late mitotic functions of Aurora kinases. Chromosoma 126:93–103. https://doi.org/10.1007/s00412-016-0594-5
Anderson DJ, Hetzer MW (2007) Nuclear envelope formation by chromatin-mediated reorganization of the endoplasmic reticulum. Nat Cell Biol 9:1160–1166. https://doi.org/10.1038/ncb1636
Anderson DJ, Hetzer MW (2008) Reshaping of the endoplasmic reticulum limits the rate for nuclear envelope formation. J Cell Biol 182:911–924. https://doi.org/10.1083/jcb.200805140
Bakhoum SF, Ngo B, Laughney AM, Cavallo J-A, Murphy CJ, Ly P, Shah P, Sriram RK, Watkins TBK, Taunk NK, Duran M, Pauli C, Shaw C, Chadalavada K, Rajasekhar VK, Genovese G, Venkatesan S, Birkbak NJ, McGranahan N, Lundquist M, LaPlant Q, Healey JH, Elemento O, Chung CH, Lee NY, Imielenski M, Nanjangud G, Pe’er D, Cleveland DW, Powell SN, Lammerding J, Swanton C, Cantley LC (2018) Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 553:467–472. https://doi.org/10.1038/nature25432
Barbosa AD, Lim K, Mari M, Edgar JR, Gal L, Sterk P, Jenkins BJ, Koulman A, Savage DB, Schuldiner M, Reggiori F, Wigge PA, Siniossoglou S (2019) Compartmentalized synthesis of triacylglycerol at the inner nuclear membrane regulates nuclear organization. Dev Cell 50:755–766. https://doi.org/10.1016/j.devcel.2019.07.009
Barroso-Vilares M, Macedo JC, Reis M, Warren JD, Compton D, Logarinho E (2020) Small-molecule inhibition of aging-associated chromosomal instability delays cellular senescence. EMBO Rep:e49248. https://doi.org/10.15252/embr.201949248
Bartsch K, Knittler K, Borowski C, Rudnik S, Damme M, Aden K, Spehlmann ME, Frey N, Saftig P, Chalaris A, Rabe B (2017) Absence of RNase H2 triggers generation of immunogenic micronuclei removed by autophagy. Hum Mol Genet 26:3960–3972. https://doi.org/10.1093/hmg/ddx283
Beaudouin J, Gerlich D, Daigle N, Eils R, Ellenberg J (2002) Nuclear envelope breakdown proceeds by microtubule-induced tearing of the lamina. Cell 108:83–96. https://doi.org/10.1016/S0092-8674(01)00627-4
Beck M, Hurt E (2017) The nuclear pore complex: understanding its function through structural insight. Nat Rev Mol Cell Biol 18:73–89. https://doi.org/10.1038/nrm.2016.147
Champion L, Pawar S, Luithle N, Ungricht R, Kutay U (2019) Dissociation of membrane–chromatin contacts is required for proper chromosome segregation in mitosis. Mol Biol Cell 30:427–440
Chen S, Novick P, Ferro-Novick S (2013) ER structure and function. Curr Opin Cell Biol 25:428–433. https://doi.org/10.1016/j.ceb.2013.02.006
Chen NY, Kim P, Weston TA, Edillo L, Tu Y, Fong LG, Young SG (2018) Fibroblasts lacking nuclear lamins do not have nuclear blebs or protrusions but nevertheless have frequent nuclear membrane ruptures. Proc Natl Acad Sci U S A 115:10100–10105. https://doi.org/10.1073/pnas.1812622115
Chen NY, Yang Y, Weston TA, Belling JN, Heizer P, Tu Y, Kim P, Edillo L, Jonas SJ, Weiss PS, Fong LG, Young SG (2019) An absence of Lamin B1 in migrating neurons causes nuclear membrane ruptures and cell death. Proc Natl Acad Sci U S A 116:25870–25879. https://doi.org/10.1073/pnas.1917225116
Chmielewska M, Dedukh D, Haczkiewicz K, Rozenblut-Kościsty B, Kaźmierczak M, Kolenda K, Serwa E, Pietras-Lebioda A, Krasikova A, Ogielska M (2018) The programmed DNA elimination and formation of micronuclei in germ line cells of the natural hybridogenetic water frog Pelophylax esculentus. Sci Rep 8:7870. https://doi.org/10.1038/s41598-018-26168-z
Cho S, Vashisth M, Abbas A, Majkut S, Vogel K, Xia Y, Ivanovska IL, Irianto J, Tewari M, Zhu K, Tichy ED, Mourkioti F, Tang H-Y, Greenberg RA, Prosser BL, Discher DE (2019) Mechanosensing by the lamina protects against nuclear rupture, DNA damage, and cell-cycle arrest. Dev Cell 49:920–935. https://doi.org/10.1016/j.devcel.2019.04.020
Crasta K, Ganem NJ, Dagher R, Lantermann AB, Ivanova EV, Pan Y, Nezi L, Protopopov A, Chowdhury D, Pellman D (2012) DNA breaks and chromosome pulverization from errors in mitosis. Nature 482:53–58. https://doi.org/10.1038/nature10802
Cuylen S, Blaukopf C, Politi AZ, Müller-Reichert T, Neumann B, Poser I, Ellenberg J, Hyman AA, Gerlich DW (2016) Ki-67 acts as a biological surfactant to disperse mitotic chromosomes. Nature 535:308–312. https://doi.org/10.1038/nature18610
de Castro IJ, Gil RS, Ligammari L, Di Giacinto ML, Paola V (2018) CDK1 and PLK1 coordinate the disassembly and reassembly of the nuclear envelope in vertebrate mitosis. Oncotarget 9:7763–7773. https://doi.org/10.18632/oncotarget.23666
de Leeuw R, Gruenbaum Y, Medalia O (2018) Nuclear lamins: thin filaments with major functions. Trends Cell Biol 28:34–45. https://doi.org/10.1016/j.tcb.2017.08.004
De Magistris P, Antonin W (2018) The dynamic nature of the nuclear envelope. Curr Biol 28:R847–R897
Denais CM, Gilbert RM, Isermann P, McGregor AL, te Lindert M, Weigelin B, Davidson PM, Friedl P, Wolf K, Lammerding J (2016) Nuclear envelope rupture and repair during cancer cell migration. Science 352:353–358. https://doi.org/10.1126/science.aad7297
Dou Z, Xu C, Donahue G, Shimi T, Pan J-A, Zhu J, Ivanov A, Capell BC, Drake AM, Shah PP, Catanzaro JM, Daniel Ricketts M, Lamark T, Adam SA, Marmorstein R, Zong WX, Johansen T, Goldman RD, Adams PD, Berger SL (2015) Autophagy mediates degradation of nuclear lamina. Nature 527:105–109. https://doi.org/10.1038/nature15548
Fenech M (2007) Cytokinesis-block micronucleus cytome assay. Nat Protoc 2:1084–1104. https://doi.org/10.1038/nprot.2007.77
Fenech M, Holland N, Kirsch-Volders M, Knudsen LE, Wagner K-H, Stopper H, Knasmueller S, Bolognesi C, El-Zein R, Bonassi S (2020) Micronuclei and disease – report of HUMN project workshop at Rennes 2019 EEMGS conference. Mutat Res Genet Toxicol Environ Mutagen 850-851:503133. https://doi.org/10.1016/j.mrgentox.2020.503133
Fonseca CL, Malaby HL, Sepaniac LA, Martin W, Byers C, Czechanski A, Messinger D, Tang M, Ohi R, Reinholdt LG (2019) Mitotic chromosome alignment ensures mitotic fidelity by promoting interchromosomal compaction during anaphase. J Cell Biol 218:1148–1163. https://doi.org/10.1083/jcb.201807228
Fuller BG, Lampson MA, Foley EA, Rosasco-Nitcher S, Le KV, Tobelmann P, Brautigan DL, Stukenberg PT, Kapoor TM (2008) Midzone activation of aurora B in anaphase produces an intracellular phosphorylation gradient. Nature 453:1132–1136. https://doi.org/10.1038/nature06923
Géraud G, Laquerrière F, Masson C, Arnoult J, Labidi B, Hernandez-Verdun D (1989) Three-dimensional organization of micronuclei induced by colchicine in PtK1 cells. Exp Cell Res 181:27–39. https://doi.org/10.1016/0014-4827(89)90179-1
Girão H, Okada N, Rodrigues TA, Silva AO, Figueiredo AC, Garcia Z, Moutinho-Santos T, Hayashi I, Azevedo JE, Macedo-Ribeiro S, Maiato H (2019) CLASP2 binding to curved microtubule tips promotes flux and stabilizes kinetochore attachments. J Cell Biol 219:e201905080. https://doi.org/10.1083/jcb.201905080
Gu M, LaJoie D, Chen OS, von Appen A, Ladinsky MS, Redd MJ, Nikolova L, Bjorkman PJ, Sundquist WI, Ullman KS, Frost A (2017) LEM2 recruits CHMP7 for ESCRT-mediated nuclear envelope closure in fission yeast and human cells. Proc Natl Acad Sci U S A 114:E2166–E2175. https://doi.org/10.1073/pnas.1613916114
Guo X, Ni J, Liang Z, Xue J, Fenech MF, Wang X (2019) The molecular origins and pathophysiological consequences of micronuclei: new insights into an age-old problem. Mutat Res Rev Mutat Res 779:1–35. https://doi.org/10.1016/j.mrrev.2018.11.001
Guo X, Dai X, Zhou T, Wang H, Ni J, Xue J, Wang X (2020) Mosaic loss of human Y chromosome: what, how and why. Hum Genet 139:421–446. https://doi.org/10.1007/s00439-020-02114-w
Halfmann CT, Sears RM, Katiyar A, Busselman BW, Aman LK, Zhang Q, O’Bryan CS, Angelini TE, Lele TP, Roux KJ (2019) Repair of nuclear ruptures requires barrier-to-autointegration factor. J Cell Biol 218:2136–2149. https://doi.org/10.1083/jcb.201901116
Haraguchi T, Kojidani T, Koujin T, Shimi T, Osakada H, Mori C, Yamamoto A, Hiraoka Y (2008) Live cell imaging and electron microscopy reveal dynamic processes of BAF-directed nuclear envelope assembly. J Cell Sci 121:2540–2554. https://doi.org/10.1242/jcs.033597
Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA (2017) Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 548:466–470. https://doi.org/10.1038/nature23470
Hatch EM, Hetzer MW (2016) Nuclear envelope rupture is induced by actin-based nucleus confinement. J Cell Biol 215:27–36. https://doi.org/10.1083/jcb.201603053
Hatch EM, Fischer AH, Deerinck TJ, Hetzer MW (2013) Catastrophic nuclear envelope collapse in cancer cell micronuclei. Cell 154:47–60. https://doi.org/10.1016/j.cell.2013.06.007
He B, Gnawali N, Hinman AW, Mattingly AJ, Osimani A, Cimini D (2019) Chromosomes missegregated into micronuclei contribute to chromosomal instability by missegregating at the next division. Oncotarget 10:2660–2674. https://doi.org/10.18632/oncotarget.26853
Huang Y, Jiang L, Yi Q, Lv L, Wang Z, Zhao X, Zhong L, Jiang H, Rasool S, Hao Q, Guo Z, Cooke HJ, Fenech M, Shi Q (2012) Lagging chromosomes entrapped in micronuclei are not 'lost' by cells. Cell Res 22:932–935
Irianto J, Xia Y, Pfeifer CR, Athirasala A, Ji J, Alvey C, Tewari M, Bennett RR, Harding SM, Liu AJ, Greenberg RA, Discher DE (2017) DNA damage follows repair factor depletion and portends genome variation in cancer cells after pore migration. Curr Biol 27:210–223. https://doi.org/10.1016/j.cub.2016.11.049
Jang C-Y, Coppinger JA, Seki A, Yates JR, Fang G (2009) Plk1 and Aurora A regulate the depolymerase activity and the cellular localization of Kif2a. J Cell Sci 122:1334–1341. https://doi.org/10.1242/jcs.044321
Joukov V, De Nicolo A (2018) Aurora-PLK1 cascades as key signaling modules in the regulation of mitosis. Sci Signal 11:eaar4195. https://doi.org/10.1126/scisignal.aar4195
Karg T, Warecki B, Sullivan W (2015) Aurora B–mediated localized delays in nuclear envelope formation facilitate inclusion of late-segregating chromosome fragments. Mol Biol Cell 26:2227–2241. https://doi.org/10.1091/mbc.E15-01-0026
Karg T, Elting MW, Vicars H, Dumont S, Sullivan W (2017) The chromokinesin Klp3a and microtubules facilitate acentric chromosome segregation. J Cell Biol 216:1597–1608
Kawaguchi A, Asaka MN, Matsumoto K, Nagata K (2015) Centrosome maturation requires YB-1 to regulate dynamic instability of microtubules for nucleus reassembly. Sci Rep 5:8768. https://doi.org/10.1038/srep08768
Kirby TJ, Lammerding J (2018) Emerging views of the nucleus as a cellular mechanosensor. Nat Cell Biol 20:373–381
Kneissig M, Keuper K, de Pagter MS, van Roosmalen MJ, Martin J, Otto H, Passerini V, Campos Sparr A, Renkens I, Kropveld F, Vasudevan A, Sheltzer JM, Kloosterman WP, Storchova Z (2019) Micronuclei-based model system reveals functional consequences of chromothripsis in human cells. eLife 8:e50292. https://doi.org/10.7554/eLife.50292
Kourmouli N, Theodoropoulos PA, Dialynas G, Bakou A, Politou AS, Cowell IG, Singh PB, Georgatos SD (2000) Dynamic associations of heterochromatin protein 1 with the nuclear envelope. EMBO J 19:6558–6568. https://doi.org/10.1093/emboj/19.23.6558
LaJoie D, Ullman KS (2017) Coordinated events of nuclear assembly. Curr Opin Cell Biol 46:39–45. https://doi.org/10.1016/j.ceb.2016.12.008
Lata S, Schoehn G, Jain A, Pires R, Piehler J, Gőttlinger HG, Weissenhorn W (2008) Helical structures of ESCRT-III are disassembled by VPS4. Science 321:1354–1357. https://doi.org/10.1126/science.1161070
Leimbacher P-A, Jones SE, Shorrocks A-MK, de Marco Zompit M, Day M, Blaauwendraad J, Bundschuh D, Bonham S, Fischer R, Fink D, Kessler BM, Oliver AW, Pearl LH, Blackford AN, Stucki M (2019) MDC1 interacts with TOPBP1 to maintain chromosomal stability during mitosis. Mol Cell 74:571–583. https://doi.org/10.1016/j.molcel.2019.02.014
Lenain C, Gusyatiner O, Douma S, van den Broek B, Peeper DS (2015) Autophagy-mediated degradation of nuclear envelope proteins during oncogene-induced senescence. Carcinogenesis 36:1263–1274. https://doi.org/10.1093/carcin/bgv124
Linder MI, Köhler M, Boersema P, Weberruss M, Wandke C, Marino J, Ashiono C, Picotti P, Antonin W, Kutay U (2017) Mitotic disassembly of nuclear pore complexes involves CDK1- and PLK1-mediated phosphorylation of key interconnecting nucleoporins. Dev Cell 43:141–156. https://doi.org/10.1016/j.devcel.2017.08.020
Liu S, Kwon M, Mannino M, Yang N, Renda F, Khodjakov A, Pellman D (2018) Nuclear envelope assembly defects link mitotic errors to chromothripsis. Nature 561:551–555. https://doi.org/10.1101/263392
Lowe AR, Tang JH, Yassif J, Graf M, Huang WYC, Groves JT, Weis K, Liphardt JT (2015) Importin-β modulates the permeability of the nuclear pore complex in a ran-dependent manner. eLife 4:e04052. https://doi.org/10.7554/eLife.04052
Lusk CP, Ader NR (2020) CHMPions of repair: emerging perspectives on sensing and repairing the nuclear envelope barrier. Curr Opin Cell Biol 64:25–33. https://doi.org/10.1016/j.ceb.2020.01.011
Lusk CP, King MC (2018) Rotten to the core: why micronuclei rupture. Dev Cell 47:265–266. https://doi.org/10.1016/j.devcel.2018.10.023
Ly P, Teitz LS, Kim DH, Shoshani O, Skaletsky H, Fachinetti D, Page DC, Cleveland DW (2017) Selective Y centromere inactivation triggers chromosome shattering in micronuclei and repair by non-homologous end joining. Nat Cell Biol 19:68–75. https://doi.org/10.1038/ncb3450
Maass KK, Rosing F, Ronchi P, Willmund KV, Devens F, Hergt M, Herrmann H, Lichter P, Ernst A (2018) Altered nuclear envelope structure and proteasome function of micronuclei. Exp Cell Res 371:353–363. https://doi.org/10.1016/j.yexcr.2018.08.029
Maciejowski J, Chatzipli A, Dananberg A, de Lange T, Campbell PJ (2019) APOBEC3B-dependent kataegis and TREX1-driven chromothripsis in telomere crisis. bioRxiv:725366. https://doi.org/10.1101/725366
Mackenzie KJ, Carroll P, Martin C-A, Murina O, Fluteau A, Simpson DJ, Olova N, Sutcliffe H, Rainger JK, Leitch A, Osborn RT, Wheeler AP, Nowotny M, Gilbert N, Chandra T, Reijns MAM, Jackson AP (2017) cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 548:461–465. https://doi.org/10.1038/nature23449
MacPherson Q, Spakowitz AJ (2019) Chromosomal condensation leads to a preference for peripheral heterochromatin. bioRxiv:714360. https://doi.org/10.1101/714360
Maiato H, Afonso O, Matos I (2015) A chromosome separation checkpoint. BioEssays 37:257–266. https://doi.org/10.1002/bies.201400140
McCullough J, Frost A, Sundquist WI (2018) Structures, functions, and dynamics of ESCRT-III/Vps4 membrane remodeling and fission complexes. Annu Rev Cell Dev Biol 34:85–109. https://doi.org/10.1146/annurev-cellbio-100616-060600
Mehsen H, Boudreau V, Garrido D, Bourouh M, Larouche M, Maddox PS, Swan A, Archambault V (2018) PP2A-B55 promotes nuclear envelope reformation after mitosis in Drosophila. J Cell Biol 217:4106–4123. https://doi.org/10.1083/jcb.201804018
Miyazaki K, Ichikawa Y, Saitoh N, Saitoh H (2020) Three types of nuclear envelope assemblies associated with micronuclei. CellBio 9:14–28
Nähse V, Christ L, Stenmark H, Campsteijn C (2017) The abscission checkpoint: making it to the final cut. Trends Cell Biol 27:1–11. https://doi.org/10.1016/j.tcb.2016.10.001
Naso FD, Sterbini V, Crecca E, Asteriti IA, Russo AD, Giubettini M, Cundari E, Lindon C, Rosa A, Guarguaglini G (2020) Excess TPX2 interferes with microtubule disassembly and nuclei reformation at mitotic exit. Cells 9:374. https://doi.org/10.3390/cells9020374
Nmezi B, Xu J, Fu R, Armiger TJ, Rodriguez-Bey G, Powell JS, Ma H, Sullivan M, Tu Y, Chen NY, Young SG, Stolz DB, Dahl KN, Liu Y, Padiath QS (2019) Concentric organization of A- and B-type lamins predicts their distinct roles in the spatial organization and stability of the nuclear lamina. Proc Natl Acad Sci U S A 116:4307–4315. https://doi.org/10.1073/pnas.1810070116
Ohsugi M, Adachi K, Horai R, Kakuta S, Sudo K, Kotaki H, Tokai-Nishizumi N, Sagara H, Iwakura Y, Yamamoto T (2008) Kid-mediated chromosome compaction ensures proper nuclear envelope formation. Cell 132:771–782. https://doi.org/10.1016/j.cell.2008.01.029
Okamoto A, Utani K-I, Shimizu N (2012) DNA replication occurs in all lamina positive micronuclei, but never in lamina negative micronuclei. Mutagenesis 27:323–327. https://doi.org/10.1093/mutage/ger082
Orr B, Maiato H (2019) No chromosome left behind: the importance of metaphase alignment for mitotic fidelity. J Cell Biol 218:1086–1088. https://doi.org/10.1083/jcb.201902041
Otsuka S, Steyer AM, Schorb M, Hériché J-K, Hossain MJ, Sethi S, Kueblbeck M, Schwab Y, Beck M, Ellenberg J (2018) Postmitotic nuclear pore assembly proceeds by radial dilation of small membrane openings. Nat Struct Mol Biol 25:21–28. https://doi.org/10.1038/s41594-017-0001-9
Park Y-E, Hayashi YK, Bonne G, Arimura T, Noguchi S, Nonaka I, Nishino I (2009) Autophagic degradation of nuclear components in mammalian cells. Autophagy 5:795–804. https://doi.org/10.4161/auto.8901
Patteson AE, Vahabikashi A, Pogoda K, Adam SA, Mandal K, Kittisopikul M, Sivagurunathan S, Goldman A, Goldman RD, Janmey PA (2019) Vimentin protects cells against nuclear rupture and DNA damage during migration. J Cell Biol 218:4079–4092. https://doi.org/10.1083/jcb.201902046
Penfield L, Wysolmerski B, Mauro M, Farhadifar R, Martinez MA, Biggs R, Wu H-Y, Broberg C, Needleman D, Bahmanyar S, Spang A (2018) Dynein pulling forces counteract Lamin-mediated nuclear stability during nuclear envelope repair. Mol Biol Cell 29:852–868. https://doi.org/10.1091/mbc.E17-06-0374
Penfield L, Shankar R, Szentgyörgyi E, Laffitte A, Mauro M, Audhya A, Müller-Reichert T, Bahmanyar S (2020) Regulated lipid synthesis and LEM2/CHMP7 jointly control nuclear envelope closure. J Cell Biol 219:e201908179. https://doi.org/10.1083/jcb.201908179
Pidoux AL, Uzawa S, Perry PE, Cande WZ, Allshire RC (2000) Live analysis of lagging chromosomes during anaphase and their effect on spindle elongation rate in fission yeast. J Cell Sci 113:4177–4191
Pieper GH, Sprenger S, Teis D, Oliferenko S (2020) ESCRT-III/Vps4 controls heterochromatin-nuclear envelope attachments. Dev Cell 53:27–41. https://doi.org/10.1016/j.devcel.2020.01.028
Raab M, Gentili M, de Belly H, Thiam H-R, Vargas P, Jimenez AJ, Lautenschlaeger F, Voituriez R, Lennon-Duménil A-M, Manel N, Piel M (2016) ESCRT III repairs nuclear envelope ruptures during cell migration to limit DNA damage and cell death. Science 352:359–362. https://doi.org/10.1126/science.aad7611
Ramadan K, Bruderer R, Spiga FM, Popp O, Baur T, Gotta M, Meyer HH (2007) Cdc48/p97 promotes reformation of the nucleus by extracting the kinase Aurora B from chromatin. Nature 450:1258–1262. https://doi.org/10.1038/nature06388
Rello-Varona S, Lissa D, Shen S, Niso-Santano M, Senovilla L, Marino G, Vitale I, Jemaa M, Harper F, Pierron G (2012) Autophagic removal of micronuclei. Cell Cycle 11:170–176
Rowat AC, Jaalouk DE, Zwerger M, Ung WL, Eydelnant IA, Olins DE, Olins AL, Herrmann H, Weitz DA, Lammerding J (2013) Nuclear envelope composition determines the ability of neutrophil-type cells to passage through micron-scale constrictions. J Biol Chem 288:8610–8618. https://doi.org/10.1074/jbc.M112.441535
Royou A, Gagou ME, Karess R, Sullivan W (2010) BubR1- and polo-coated DNA tethers facilitate poleward segregation of acentric chromatids. Cell 140:235–245. https://doi.org/10.1016/j.cell.2009.12.043
Sacristan C, Kops GJPL (2015) Joined at the hip: kinetochores, microtubules, and spindle assembly checkpoint signaling. Trends Cell Biol 25:21–28. https://doi.org/10.1016/j.tcb.2014.08.006
Salina D, Bodoor K, Eckley DM, Schroer TA, Rattner JB, Burke B (2002) Cytoplasmic dynein as a facilitator of nuclear envelope breakdown. Cell 108:97–107. https://doi.org/10.1016/S0092-8674(01)00628-6
Samwer M, Gerlich DW (2018) A core problem in nuclear assembly. Nature 561:467–468. https://doi.org/10.1038/d41586-018-06668-8
Samwer M, Schneider MWG, Hoefler R, Schmalhorst PS, Jude JG, Zuber J, Gerlich DW (2017) DNA cross-bridging shapes a single nucleus from a set of mitotic chromosomes. Cell 170:956–972. https://doi.org/10.1016/j.cell.2017.07.038
Schellhaus AK, De Magistris P, Antonin W (2016) Nuclear reformation at the end of mitosis. J Mol Biol 428:1962–1985. https://doi.org/10.1016/j.jmb.2015.09.016
Shah P, Wolf K, Lammerding J (2017) Bursting the bubble – nuclear envelope rupture as a path to genomic instability? Trends Cell Biol 27:546–555. https://doi.org/10.1016/j.tcb.2017.02.008
Soto M, Raaijmakers JA, Bakker B, Spierings DCJ, Lansdorp PM, Foijer F, Medema RH (2017) p53 prohibits propagation of chromosome segregation errors that produce structural aneuploidies. Cell Rep 19:2423–2431. https://doi.org/10.1016/j.celrep.2017.05.055
Steigemann P, Wurzenberger C, Schmitz MH, Held M, Guizetti J, Maar S, Gerlich DW (2009) Aurora B-mediated abscission checkpoint protects against tetraploidization. Cell 136:473–484
Sutyagina OI, Kisurina-Evgenieva OP, Onishchenko GE (2019) Micronuclei elimination in MCF-7 human breast adenocarcinoma cells. Cell Tissue Biol 13:207–218. https://doi.org/10.1134/s1990519x19030106
Takaki T, Montagner M, Serres MP, Le Berre M, Russell M, Collinson L, Szuhai K, Howell M, Boulton SJ, Sahai E, Petronczki M (2017) Actomyosin drives cancer cell nuclear dysmorphia and threatens genome stability. Nat Commun 8:16013. https://doi.org/10.1038/ncomms16013
Thaller DJ, Allegretti M, Borah S, Ronchi P, Beck M, Lusk CP (2019) An ESCRT-LEM protein surveillance system is poised to directly monitor the nuclear envelope and nuclear transport system. eLife 8: e45284. doi: https://doi.org/10.7554/eLife.45284
Thompson SL, Compton DA (2011) Chromosome missegregation in human cells arises through specific types of kinetochore–microtubule attachment errors. Proc Natl Acad Sci U S A 108:17974–17978. https://doi.org/10.1073/pnas.1109720108
Thoresen SB, Campsteijn C, Vietri M, Schink KO, Liestøl K, Andersen JS, Raiborg C, Stenmark H (2014) ANCHR mediates Aurora-B-dependent abscission checkpoint control through retention of VPS4. Nat Cell Biol 16:547–557. https://doi.org/10.1038/ncb2959
Turgay Y, Eibauer M, Goldman AE, Shimi T, Khayat M, Ben-Harush K, Dubrovsky-Gaupp A, Sapra KT, Goldman RD, Medalia O (2017) The molecular architecture of lamins in somatic cells. Nature 543:261–264. https://doi.org/10.1038/nature21382
Ungricht R, Kutay U (2017) Mechanisms and functions of nuclear envelope remodelling. Nat Rev Mol Cell Biol 18:229–245. https://doi.org/10.1038/nrm.2016.153
Utani K-I, Kawamoto J-K, Shimizu N (2007) Micronuclei bearing acentric extrachromosomal chromatin are transcriptionally competent and may perturb the cancer cell phenotype. Mol Cancer Res 5:695–704. https://doi.org/10.1158/1541-7786.mcr-07-0031
Vargas JD, Hatch EM, Anderson DJ, Hetzer MW (2012) Transient nuclear envelope rupturing during interphase in human cancer cells. Nucleus 3:88–100. https://doi.org/10.4161/nucl.18954
Vietri M, Schultz S, Bellanger A, Jones C, Raiborg C, Skarpen E, Pedurupillay CR, Kip E, Timmer R, Jain A, Collas P, Knorr R, Grellscheid S, Kusumaatmaja H, Brech A et al (2019) Unrestrained ESCRT-III drives chromosome fragmentation and micronuclear catastrophe. bioRxiv:517011. https://doi.org/10.1101/517011
von Appen A, LaJoie D, Johnson IE, Trnka MJ, Pick SM, Burlingame AL, Ullman KS, Frost A (2020) LEM2 phase separation promotes ESCRT-mediated nuclear envelope reformation. Nature 582:115–118. https://doi.org/10.1038/s41586-020-2232-x
Warecki B, Sullivan W (2018) Micronuclei formation is prevented by Aurora B-mediated exclusion of HP1a from late-segregating chromatin in Drosophila. Genetics 210:171–187. https://doi.org/10.1101/268912
Warecki B, Ling X, Bast I, Sullivan W (2020) ESCRT-III–mediated membrane fusion drives chromosome fragments through nuclear envelope channels. J Cell Biol 219:e201905091. https://doi.org/10.1083/jcb.201905091
Webster BM, Colombi P, Jäger J, Lusk CP (2014) Surveillance of nuclear pore complex assembly by ESCRT-III/Vps4. Cell 159:388–401. https://doi.org/10.1016/j.cell.2014.09.012
Wesolowska N, Avilov I, Machado P, Geiss C, Kondo H, Mori M, Lenart P (2020) Actin assembly ruptures the nuclear envelope by prying the lamina away from nuclear pores and nuclear membranes in starfish oocytes. eLife 9:e49774. https://doi.org/10.7554/eLife.49774
Wilhelm T, Olziersky A-M, Harry D, De Sousa F, Vassal H, Eskat A, Meraldi P (2019) Mild replication stress causes chromosome mis-segregation via premature centriole disengagement. Nat Commun 10:3585. https://doi.org/10.1038/s41467-019-11584-0
Willan J, Cleasby AJ, Flores-Rodriguez N, Stefani F, Rinaldo C, Pisciottani A, Grant E, Woodman P, Bryant HE, Ciani B (2019) ESCRT-III is necessary for the integrity of the nuclear envelope in micronuclei but is aberrant at ruptured micronuclear envelopes generating damage. Oncogenesis 8:29. https://doi.org/10.1038/s41389-019-0136-0
Xia Y, Ivanovska IL, Zhu K, Smith L, Irianto J, Pfeifer CR, Alvey CM, Ji J, Liu D, Cho S, Bennett RR, Liu AJ, Greenberg RA, Discher DE (2018) Nuclear rupture at sites of high curvature compromises retention of DNA repair factors. J Cell Biol 217:3796–3808. https://doi.org/10.1083/jcb.201711161
Xia Y, Pfeifer CR, Zhu K, Irianto J, Liu D, Pannell K, Chen EJ, Dooling LJ, Tobin MP, Wang M, Ivanovska IL, Smith LR, Greenberg RA, Discher DE (2019) Rescue of DNA damage after constricted migration reveals a mechano-regulated threshold for cell cycle. J Cell Biol 218:2545–2563. https://doi.org/10.1083/jcb.201811100
Yang Z, Maciejowski J, de Lange T (2017) Nuclear envelope rupture is enhanced by loss of p53 or Rb. Mol Cancer Res 15:1579–1586. https://doi.org/10.1158/1541-7786.mcr-17-0084
Yokoi A, Villar-Prados A, Oliphint PA, Zhang J, Song X, De Hoff P, Morey R, Liu J, Roszik J, Clise-Dwyer K, Burks JK, O’Halloran TJ, Laurent LC, Sood AK (2019) Mechanisms of nuclear content loading to exosomes. Sci Adv 5:eaax8849. https://doi.org/10.1126/sciadv.aax8849
Young AM, Gunn AL, Hatch EM (2020) BAF facilitates interphase nuclear envelope repair through recruitment of nuclear transmembrane proteins. Mol Biol Cell:mbc.E20-01-0009. https://doi.org/10.1091/mbc.E20-01-0009
Zhang C-Z, Spektor A, Cornils H, Francis JM, Jackson EK, Liu S, Meyerson M, Pellman D (2015) Chromothripsis from DNA damage in micronuclei. Nature 522:179–184. https://doi.org/10.1038/nature14493
Acknowledgments
We thank the anonymous reviewers for helpful comments and suggestions.
Code availability
Not applicable.
Funding
This work was funded by the National Natural Science Foundation of China (Project No. 31860301 to X.W. and No. 31900410 to X.G.).
Author information
Authors and Affiliations
Contributions
X.G. contributed towards the design and structure of the manuscript. X.G. and X.D. reviewed the literature, designed the figures, and drafted the manuscript. X.W., T.Z., and J.N. contributed to the discussion. J.X. and X.W. contributed to editing the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Guo, X., Dai, X., Wu, X. et al. Understanding the birth of rupture-prone and irreparable micronuclei. Chromosoma 129, 181–200 (2020). https://doi.org/10.1007/s00412-020-00741-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00412-020-00741-w