
Abstract
Formation of stable kinetochore-microtubule attachments is essential for accurate chromosome segregation in human cells and depends on the NDC80 complex. We recently showed that Chmp4c, an endosomal sorting complex required for transport protein involved in membrane remodelling, localises to prometaphase kinetochores and promotes cold-stable kinetochore microtubules, faithful chromosome alignment and segregation. In the present study, we show that Chmp4c associates with the NDC80 components Hec1 and Nuf2 and is required for optimal NDC80 stability and Hec1-Nuf2 localisation to kinetochores in prometaphase. However, Chmp4c-depletion does not cause a gross disassembly of outer or inner kinetochore complexes. Conversely, Nuf2 is required for Chmp4c kinetochore targeting. Constitutive Chmp4c kinetochore tethering partially rescues cold-stable microtubule polymers in cells depleted of the endogenous Nuf2, showing that Chmp4c also contributes to kinetochore-microtubule stability independently of regulating Hec1 and Nuf2 localisation. Chmp4c interacts with tubulin in cell extracts, and binds and bundles microtubules in vitro through its highly basic N-terminal region (amino acids 1–77). Furthermore, the N-terminal region of Chmp4c is required for cold-stable kinetochore microtubules and efficient chromosome alignment. We propose that Chmp4c promotes stable kinetochore-microtubule attachments by regulating Hec1–Nuf2 localisation to kinetochores in prometaphase and by binding to spindle microtubules. These results identify Chmp4c as a novel protein that regulates kinetochore-microtubule interactions to promote accurate chromosome segregation in human cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Successful chromosome segregation during cell division requires formation of stable attachments between kinetochores and spindle microtubules in human cells. Kinetochores are formed at the periphery of sister centromeres and the site of kinetochore assembly is specified by CENP-A, a modified histone H3 (Perpelescu and Fukagawa 2011). The inner kinetochore domain contains the constitutive centromere-associated network, a complex of proteins that links CENP-A-containing chromatin to the KMN protein network (named for the KNL1, MIS12 and NDC80 complexes) within the outer kinetochore (Maiato et al. 2004; Varma and Salmon 2012). In turn, the outer kinetochore interacts with spindle microtubules and also mediates recruitment of spindle checkpoint proteins to delay anaphase onset in the presence of unattached kinetochores (Foley and Kapoor 2013).
Initial interactions between chromosomes and spindle microtubules occur predominantly along the microtubule lattice and are mediated by motor proteins. These interactions mature into “end-on” attachments consisting of spindle microtubules embedded in the outer kinetochore, which depend on the NDC80 complex (Foley and Kapoor 2013). The NDC80 is a heterotetramer comprising the Spc24–Spc25 proteins which anchor the complex into the kinetochore and the Hec1–Nuf2 proteins, which interact with the spindle microtubules (Alushin et al. 2010; Cheeseman et al. 2006; Ciferri et al. 2008; DeLuca et al. 2006). Hec1 binds to microtubules through its calponin-homology domain and also through a positively charged N-terminus tail of approximately 80 amino acids in length (Alushin et al. 2010; Ciferri et al. 2008; Sundin et al. 2011). The Nuf2 protein does not directly bind to kinetochore microtubules (Alushin et al. 2010; Sundin et al. 2011); however, depletion of either Hec1 or Nuf2 by siRNA causes reduction of both proteins at kinetochores, unstable kinetochore-microtubule attachments and chromosome misalignment (DeLuca et al. 2003; DeLuca et al. 2002; Sundin et al. 2011). The Astrin-SKAP complex stabilises mature “end-on” attachments and Ska proteins facilitate kinetochore binding to depolymerising microtubules during chromosome movement; however, the list of proteins that regulate kinetochore-microtubule attachments is likely incomplete (Friese et al. 2016; Hanisch et al. 2006; Kern et al. 2017; Schmidt et al. 2012).
We recently showed that Chmp4c, a component of the endosomal sorting complex required for transport machinery (ESCRT; Campsteijn et al. 2016; Hurley 2015), localises to kinetochores in prometaphase but is reduced from kinetochores aligned at the metaphase plate (Petsalaki et al. 2018). Chmp4c binds to the spindle checkpoint protein ZW10 and promotes localisation of the Rod-ZW10-Zwilch (RZZ) and Mad1–Mad2 complexes to unattached kinetochores. Furthermore, Chmp4c is required for timely mitotic progression, faithful chromosome alignment and segregation (Petsalaki et al. 2018). In addition, Chmp4c promotes cold-stable kinetochore microtubules, optimal spindle and centromere length through unidentified mechanisms (Petsalaki et al. 2018). In the present study, we find that Chmp4c associates with Hec1 and Nuf2 and promotes Hec1–Nuf2 kinetochore localisation in prometaphase. We also find that the N-terminal region of Chmp4c binds to microtubules and is required for proper kinetochore–microtubule interactions and chromosome alignment. These results identify Chmp4c as a novel protein that promotes stable kinetochore–microtubule attachments.
Results
Chmp4c promotes Hec1 and Nuf2 kinetochore localisation
Stable kinetochore–microtubule attachments depend on the NDC80 complex. Depletion of the endogenous Chmp4c by siRNA located in the 3′-untranslated region (siChmp4c) reduced localisation of Nuf2 or Hec1 to prometaphase kinetochores by approximately 50% compared with controls and this was not due to reduced total levels of Nuf2 or Hec1 proteins (Fig. 1a–c; Online Resource Fig. S1a,b). In cells expressing Nuf2 fused to GFP (Nuf2:GFP), Chmp4c-depletion also reduced Nuf2:GFP kinetochore staining compared with controls (Fig. 1d). However, Chmp4c-depleted and control cells exhibited similar total and kinetochore levels of Spc24 and Spc25 proteins (Fig. 1e–g; Online Resource Fig. S1c). Furthermore, Chmp4c-depletion did not impair localisation of Spc24 fused to GFP (Spc24:GFP) to kinetochores (Fig. 1h). Expression of Chmp4c:GFP which is resistant to siChmp4c-mediated degradation, but not GFP alone, rescued Hec1 kinetochore localisation after depletion of the endogenous Chmp4c compared with control cells (Fig. 2a, b; Online Resource Fig. S1a). Depletion of Chmp4c reduced the Spc24-Nuf2 interaction by co-immunoprecipitation (as judged by the Nuf2/Spc24 signal ratio) in cells enriched in mitosis by nocodazole treatment and shake-off compared with controls, and this interaction was rescued after expression of Chmp4c:GFP (Fig. 2c). These results show that Chmp4c is required for optimal integrity of the NDC80 complex. However, localisation of Knl1, Mis12 and CENP-H (a component of the constitutive centromere-associated network) proteins at kinetochores in Chmp4c-deficient cells was similar to controls suggesting that Chmp4c-depletion does not cause a gross disassembly of the KMN or inner kinetochore complexes (Fig. 2d; Online Resource Fig. S1d-f). Depletion of Nuf2 by siRNA inside the 3′-untranslated region (siNuf2) diminished Chmp4c kinetochore staining compared with control cells (Fig. 2e, f; Online Resource Fig. S1 g). Depletion of Spc24 also reduced Chmp4c and Nuf2 localisation to kinetochores compared with control cells (Fig. 2e, f; Online Resource Fig. S1 h,i), which is consistent with the Spc24–Spc25 sub-complex being required for kinetochore localisation of Hec1–Nuf2 (Bharadwaj et al. 2004; Ciferri et al. 2005). These results suggest that Chmp4c and Hec1–Nuf2 localise to kinetochores in an interdependent manner in prometaphase cells.

Chmp4c-depletion reduces localisation of Hec1 and Nuf2 to prometaphase kinetochores. Localisation of Hec1 and Nuf2 (a–c), Nuf2:GFP (d), Spc24 and Spc25 (e–g), and Spc24:GFP (h) in cells transfected with negative siRNA (control) or Chmp4c siRNA (siChmp4c). Relative green/red fluorescence intensity is shown and values in control were set to 1. n = (number of cells; kinetochores). Points represent the average kinetochore intensity per cell. Error bars show the s.d. ***P < 0.001 compared with control. Student’s t test was used. Insets show magnified kinetochores. Scale bars, 5 μm

Expression of siRNA-resistant Chmp4c:GFP rescues Hec1-localisation to kinetochores in cells depleted of the endogenous Chmp4c. a, b Cells expressing GFP or Chmp4c:GFP resistant to Chmp4c siRNA (siChmp4c) mediated degradation were transfected with negative siRNA (control) or siChmp4c. c Western blot analysis of co-immunoprecipitated (co-IP) Nuf2 or Spc24 from mitotic-enriched cell extracts. Relative band intensity values are shown and values in control (third lane from left) were set to 1. Ab, antibody; NS, nonspecific; nocod, nocodazole. d Localisation of Knl1. e, f Localisation of Chmp4c in cells transfected with negative siRNA (control), Nuf2 siRNA (siNuf2), or Spc24 siRNA (siSpc24). Relative green/red fluorescence intensity is shown and values in control were set to 1. n = (number of cells; kinetochores). Points represent the average kinetochore intensity per cell. Error bars show the s.d. ***P < 0.001 compared with control. Statistical significant differences were determined by ANOVA and Student’s t test. Insets show magnified kinetochores. Scale bars, 5 μm
Chmp4c associates with Hec1 and Nuf2
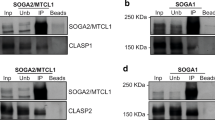
To investigate a possible interaction of Chmp4c with Hec1 and Nuf2, wild-type (WT) or truncated forms of Chmp4c fused to GST (Fig. 3a, b) were used in GST pull-down experiments. In mitosis-enriched cells, WT GST-Chmp4c predominantly associated with a phosphatase-sensitive (phosphorylated) form of Nuf2 (Fig. 3c; Online Resource Fig. S1 g) and the Chmp4c–Nuf2 interaction required the C-terminal (amino acids 78–233), but not the N-terminal (amino acids 1–77) sequence of Chmp4c (Fig. 3d). The N-terminal (1–77) region of Chmp4c contains 22 positively charged residues (net 14 positive charges at neutral pH) which, in terms of positive charges, is reminiscent of the Hec1 N-terminal tail (amino acids 1–80) that contains 15 positively charged amino acids (net 10 positive charges at neutral pH) and binds spindle microtubules (Miller et al. 2008). The C-terminal Chmp4c (78–233) sequence contains hydrophobic residues and an amphipathic helix inside the amino acids 216–233 that interacts with the spindle checkpoint protein ZW10 (Petsalaki et al. 2018). Further dividing the C-terminal Chmp4c (78–233) region into two fragments (78–163 and 164–233 amino acids) abolished Nuf2-binding (Fig. 3e) suggesting that the entire Chmp4c (78–233) region is required for interaction with Nuf2 or that the Nuf2 epitope was disrupted. Also, the endogenous Chmp4c co-precipitated with Nuf2 or Hec1 in mitosis-enriched cell extracts (Fig. 3f, g), but not in untreated cell extracts (Online Resource Fig. S2a). Taken together, we propose that Chmp4c interacts with Hec1 and Nuf2 at prometaphase kinetochores. These results suggest that Chmp4c is positioned at, or close to, the critical interface of microtubule attachment. However, purified Chmp4c did not bind to Nuf2 or Hec1 in vitro (Online Resource Fig. S2b-d) suggesting that in vivo interactions between Chmp4c and Hec1 or Nuf2 require additional components or modifications, or that our recombinant protein(s) is/are not properly folded.

Chmp4c associates with Nuf2 and Hec1 in mitosis-enriched cell extracts. a A schematic of Chmp4c fragments used. The Chmp4c basic region is indicated by a grey box and the ZW10-binding region by a black box. Amino acid numbers are shown in brackets. b Coomassie staining of GST-Chmp4c proteins. c–e GST pull-down assays from mitosis-enriched cell extracts. Precipitated Nuf2 or GST was detected by western blotting. WT, wild-type. f, g Co-immunoprecipitation (co-IP) assays from mitosis-enriched cell extracts (left) or without cell extracts to demonstrate the bands originate from the extracts rather than the antibodies (right). Immunoprecipitated Nuf2, Hec1 and Chmp4c were detected by western blotting. Phosphorylated Nuf2 is indicated by an asterisk. Ab, antibody. Nocod, nocodazole. NS, nonspecific
Chmp4c directly contributes to kinetochore-microtubule stability
To investigate whether Chmp4c directly stabilises kinetochore-microtubules and minimise phenotypic contributions from perturbing Hec1 and Nuf2 localisation, we engineered constructs in which Nuf2 or Chmp4c are constitutively tethered to kinetochores by fusion to Mis12 and GFP (Nuf2:Mis12:GFP or Chmp4c:Mis12:GFP respectively). These fusion proteins were resistant to siNuf2 or siChmp4c-mediated degradation (Online Resource Fig. S2e) and exhibited similar localisation to kinetochores in cells depleted of the endogenous Chmp4c compared with controls (Online Resource Fig. S2f-h). Stably attached kinetochore microtubules are resistant to depolymerisation by low temperatures (Brinkley and Cartwright 1975; Sundin et al. 2011). After depletion of the endogenous Chmp4c, expression of Nuf2:Mis12:GFP fully restored localisation of the Hec1–Nuf2 complex to kinetochores as judged by Hec1-staining (Online Resource Fig. S3a,b). However, Nuf2:Mis12:GFP-expression only partially rescued cold-stable microtubule polymers in prometaphase compared with controls (Fig. 4a, b) suggesting that Chmp4c is required for optimal kinetochore-microtubule stability independently of Hec1 and Nuf2. This was likely not due to some effect of the Mis12:GFP tag because expression of Nuf2:Mis12:GFP fully restored cold-stable microtubule polymers in cells depleted of endogenous Nuf2 (Fig. 4a, b). Also, expression of Chmp4c:Mis12:GFP partially rescued cold-stable microtubule polymers in cells depleted of the endogenous Chmp4c and Nuf2 (Fig. 4c, d), despite an approximate 90% reduction in Hec1 kinetochore staining in these cells compared with controls (Online Resource Fig. S3c,d). Furthermore, in cells depleted of Nuf2 and treated with the proteasome inhibitor MG132 to block progression of mitosis beyond metaphase, expression of Chmp4c:Mis12:GFP partially rescued chromosome alignment compared with cells expressing GFP-only (Online Resource Fig. S3e,f). These results show that Chmp4c promotes formation of stable kinetochore-microtubule attachments independently of regulating Hec1 and Nuf2 localisation.

Expression of Chmp4c:Mis12:GFP partially rescues cold-stable microtubule polymers in Nuf2-deficient cells. a, b Cells expressing Nuf2:Mis12:GFP resistant to Nuf2 siRNA (siNuf2) mediated degradation or GFP were transfected with negative siRNA (control), Chmp4c siRNA (siChmp4c) or siNuf2 and treated with ice-cold medium for 15 min. c, d Cells expressing Chmp4c:Mis12:GFP resistant to siChmp4c-mediated degradation or GFP were transfected with siChmp4c, siNuf2 or a combination of siChmp4c and siNuf2 (siChmp4c + siNuf2), and treated with ice-cold medium for 15 min. Mean tubulin intensity is shown and values in control were set to 1. n = 90 cells from three independent experiments. Error bars show the s.d. ***P < 0.001. Statistical significant differences were determined by ANOVA and Student’s t test. Scale bars, 5 μm
Chmp4c binds and bundles microtubules
To investigate whether Chmp4c associates with tubulin, cells were enriched in prometaphase with an intact mitotic spindle. For this purpose, cells were treated with 300 nM nocodazole for 16 h, incubated with ice-cold medium for 1.5 h to completely depolymerise spindle microtubules and released into warm medium for 15 min to allow spindle formation (nocodazole and “release” protocol; Online Resource Fig. S4a). Afterwards, cells were lysed in ice-cold buffer that causes complete disassembly of microtubules. Both the endogenous Chmp4c and the wild-type GST-Chmp4c associated with α-tubulin in co-immunoprecipitation or GST pull-down experiments, respectively, suggesting that Chmp4c interacts with soluble tubulin in cell extracts (Fig. 5a and Online Resource Fig. S4b).

Chmp4c binds and bundles microtubules in vitro. a Co-immunoprecipitation (co-IP) assays from prometaphase-enriched cell extracts. Immunoprecipitated α-tubulin and Chmp4c were detected by western blotting. Ab, antibody. Nocod, nocodazole. b Microtubule GST pull-down assay. Purified wild-type (WT) GST-Chmp4c on agarose beads was incubated with taxol-stabilised microtubules (MTs). Precipitated tubulin or GST were analysed by western blotting. c Microtubule co-sedimentation assay. Wild-type GST-Chmp4c eluted from beads was incubated with or without 1 μM taxol-stabilised microtubules. After centrifugation, supernatant (S) and pellet (P) were analysed for tubulin or GST by western blotting. d Coomassie-stained gel of microtubule co-sedimentation assay. Three hundred and fifty nanograms of purified His-Chmp4c was incubated with 1 μM taxol-stabilised microtubules. e, f Co-sedimentation analysis of WT GST-Chmp4c using increased concentrations of taxol-stabilised microtubules. The percentage of WT GST-Chmp4c in the pellet fraction was plotted versus microtubule concentration. Data is from two independent experiments. Error bars show the s.d. g Chmp4c bundles microtubules. One micromolar of rhodamine-labelled microtubules were imaged in the presence of wild-type or truncated GST-Chmp4c. Scale bars, 40 μm. h, i Microtubule co-sedimentation assays. Truncated GST-Chmp4c eluted from beads were incubated with or without 1 μM taxol-stabilised microtubules and analysed as in (c)
To investigate whether Chmp4c directly binds to microtubules or forms an indirect connection through a binding partner, purified GST-Chmp4c was incubated with taxol-stabilised microtubules in vitro. WT GST-Chmp4c (but not GST protein) associated with increasing concentrations of stabilised microtubules in GST pull-down assays (Fig. 5b; Online Resource Fig. S4c). Also, WT GST-Chmp4c eluted from beads (but not GST alone) or purified His-Chmp4c co-sedimented with stabilised microtubules and did not pellet on their own (Fig. 5c–f; Online Resource Fig. S4d). Purified His-Chmp4c did not significantly alter the percentage of GST-Hec1 bound to microtubules in vitro (Online Resource Fig. S4e-g). GST-Chmp4c also bundled microtubules in vitro and, for 1 μM tubulin, an approximately equal concentration of WT Chmp4c was required to observe robust bundling (Fig. 5g). Microtubule bundling and binding activities required the N-terminal (1–77 amino acids), but not the C-terminal (78–233) region, of Chmp4c (Fig. 5g–i) and Chmp4c (1–77) fused to GFP associated with spindle microtubules in mitotic cells (Online Resource Fig. S4 h). We conclude that Chmp4c directly binds and bundles microtubules in vitro.
The N-terminal (1–77) region of Chmp4c is required for stable kinetochore-microtubule attachments
To investigate whether the in vitro microtubule binding activity of Chmp4c has functional relevance at the kinetochore, cells expressing truncated Chmp4c (78–233) fused to GFP (lacking the 1–77 amino acids microtubule binding domain of Chmp4c) were depleted of the endogenous Chmp4c by siRNA and treated with ice-cold medium for 15 min. These cells exhibited reduced cold-stable microtubule polymers in prometaphase (Fig. 6a, b) and increased chromosome misalignment in metaphase in the presence of the proteasome inhibitor MG132 (Fig. 6c, d), compared with cells expressing WT Chmp4c:GFP. However, expression of Chmp4c (78–233):GFP rescued ZW10-kinetochore localisation after depletion of the endogenous Chmp4c compared with GFP only (Online Resource Fig. S4i; Petsalaki et al. 2018). These results suggest that the N-terminal (amino acids 1–77) sequence of Chmp4c is required for optimal kinetochore-microtubule attachments and chromosome alignment.

Expression of Chmp4c (78–233):GFP diminishes cold-stable microtubules and increases chromosome misalignment. a, b Cold-stable microtubule polymers. Cells expressing GFP, wild-type (WT) or truncated Chmp4c:GFP resistant to Chmp4c siRNA (siChmp4c) mediated degradation, were transfected with siChmp4c and treated with ice-cold medium for 15 min. Mean tubulin intensity is shown and values in control were set to 1. n = 90 cells from three independent experiments. c, d Chromosome misalignment. Cells were transfected as in (a) and treated with MG132 for 1 h. n(GFP) = 133, n(WT Chmp4c:GFP) = 181, n(Chmp4c(78–233):GFP) = 65 cells from three independent experiments. Error bars show the s.d. ***P < 0.001. Statistical significant differences were determined by ANOVA and Student’s t test. Insets show magnified unaligned kinetochores exhibiting Chmp4c (78–233):GFP staining. Scale bars, 5 μm
Discussion
On the basis of those findings, we propose that the ESCRT protein Chmp4c localises to prometaphase kinetochores and promotes stable kinetochore-microtubule attachments by directly binding to spindle microtubules and stabilising the Hec1–Nuf2 kinetochore complex. We also propose that Chmp4c links kinetochore-microtubule attachment with spindle checkpoint signalling: In prometaphase, Chmp4c localises to unattached kinetochores, where it promotes kinetochore-microtubule interactions and RZZ-kinetochore targeting (Petsalaki et al. 2018). In metaphase, when stable kinetochore-microtubule attachments are formed, Chmp4c dissociates from kinetochores leading to dynein-dependent stripping of RZZ and Mad1–Mad2, and spindle-checkpoint silencing.
Ska1 binds to kinetochores following microtubule attachment and facilitates kinetochore binding to depolymerising microtubules during chromosome movement (Hanisch et al. 2006; Schmidt et al. 2012). Also, the Astrin-SKAP complex localises to properly bi-oriented sister kinetochores and stabilises correctly formed kinetochore-microtubule attachments (Friese et al. 2016; Kern et al. 2017). Because Chmp4c localises to unattached kinetochores and unaligned chromosomes but is reduced from kinetochores aligned at the metaphase plate (Petsalaki et al. 2018), perhaps Chmp4c facilitates kinetochore-microtubule interactions before mature kinetochore attachments are formed.
Purified GST-Chmp4c exhibits relatively high microtubule binding affinity in vitro; however, our analysis may be limited by the GST tag. Future experiments using GST-cleaved Chmp4c and a reconstituted Ndc80 complex are required to more accurately assess the relative affinity of Chmp4c and Ndc80 for binding microtubules.
We also show that Chmp4c is required for the integrity of the NDC80 complex in prometaphase. This was perhaps unexpected because the NDC80 is stable in vitro and can be reconstituted by expression of its individual subunits in bacteria (Cheeseman et al. 2006; Ciferri et al. 2005; Wei et al. 2005). However, additional proteins may be required for optimal NDC80 stability in cultured cells. The Spc24–Spc25 and Hec1–Nuf2 dimers are held together by overlapping α-helical coiled coil domains located at the C-terminus of the Hec1–Nuf2 dimer and the N-terminus of the Spc24–Spc25 dimer (Ciferri et al. 2005; Varma and Salmon 2012; Wei et al. 2005). One possibility is that Chmp4c (or an unidentified Chmp4c-interacting protein) binds at the interface of the Hec1–Nuf2 and Spc24–Spc25 heterodimers to stabilise the NDC80 heterotetramer. Interestingly, recent structural work on the “dwarf” NDC80 complex suggested that unknown cellular factors might bind at this interface and Chmp4c might be (or recruit) such a molecule (Valverde et al. 2016). However, Chmp4c is not required for NDC80 assembly in metaphase as Hec1–Nuf2 levels are not reduced after evacuation of Chmp4c from kinetochores in control cells. One possibility is that a different protein stabilises the NDC80 complex in metaphase after Chmp4c is dissociated from kinetochores. Alternatively, binding of kinetochore microtubules may stabilise the NDC80 complex in metaphase in the absence of Chmp4c.
Chmp4c-deficient cells can still align the majority of their chromosomes despite an approximate 50% reduction in Hec1–Nuf2 kinetochore localisation. Vertebrate kinetochores have excess copies of NDC80 complexes: there are approximately 14 NDC80 complexes per kinetochore microtubule in human cells and 30 NDC80 complexes per kinetochore microtubule in Xenopus, compared with just 8 NDC80 complexes per microtubule binding site in budding yeast (Emanuele et al. 2005; Joglekar et al. 2006; Suzuki et al. 2015). Presumably, the remaining Hec1–Nuf2 molecules in Chmp4c-deficient cells can still establish the majority of kinetochore-microtubule contacts, but this leads to delayed anaphase onset, misaligned and missegregated chromosomes (Petsalaki et al. 2018). In conclusion, our study identifies a novel protein that regulates kinetochore-microtubule attachments to promote accurate chromosome segregation in human cells.
Materials and methods
Antibodies
Mouse monoclonal antibody against GST (B-14) and rabbit polyclonal antibodies against CENP-B (H-65), His (H-15) and GFP (sc-8334) were from Santa Cruz Biotechnology. A mouse monoclonal anti-Chmp4c (ab168205) used for western blotting in Figs 3f, 5a and S2a was from Abcam. Rabbit polyclonal anti-Chmp4c (ab155668), anti-Mis12 (ab70843), anti-Knl1 (Casc5, ab70537), anti-Nuf2 (ab122962) and anti-Spc24 (EPR11548B, ab169786) antibodies were also from Abcam. Mouse monoclonal antibodies against Hec1 (9G3.23, GTX70268) and CENP-A (3–19, GTX13939) and rabbit polyclonal antibody against Spc25 (GTX111305) were from Genetex. Mouse monoclonal antibodies against α-tubulin (DM1A) and actin (AC-40) were from Sigma and a rabbit polyclonal antibody against CENP-H (PD031) was from MBL. Rabbit polyclonal antibody against ZW10 was a gift from Andrea Musacchio (Max Planck Institute of Molecular Physiology, Dortmund, Germany).
Plasmids and cloning
Plasmid pCR3.1 GFP-EXN/chmp4c encoding human Chmp4c fused to GFP was from Paul Bieniasz and Trinity Zang (The Aaron Diamond AIDS Research Center, New York, USA; Jouvenet et al. 2011). To generate plasmid pEGFP-N1/hChmp4c(78–233) encoding human Chmp4c (78–233) fused to GFP, the Chmp4c cDNA sequence was amplified by PCR using pCR3.1 GFP-EXN/chmp4c as template and cloned into the pEGFP-N1 vector (Clontech) as XhoI/ BamHI fragment. To generate pGEX4T1/hChmp4c vectors encoding wild-type or truncated forms of GST-tagged human Chmp4c, Chmp4c cDNA sequences were amplified by PCR using pCR3.1 GFP-EXN/chmp4c as template and cloned into the pGEX4T1 vector (GE Healthcare) as BamHI/ XhoI fragments. For pET28/hChmp4c vector encoding His-tagged human Chmp4c, full length Chmp4c cDNA was amplified by PCR using pCR3.1 GFP-EXN/chmp4c as template and cloned into the pET28a(+) vector (EMD Millipore) with XhoI/ EcoRI. To generate pcDNA4/GFP/Mis12/hChmp4c coding for N-terminal fusion of wild-type human Chmp4c to GFP:Mis12 (amino acids 1–205 of human Mis12), Chmp4c sequences were amplified by PCR and cloned into the pcDNA4/GFP/Mis12 vector (a gift from Geert Kops, University Medical Centre Utrecht, Utrecht, the Netherlands) as XhoI/ EcoRI fragments. Plasmids pEGFP N1/Nuf2 and pEGFP N1/Hec1 coding for C-terminal fusions of human Nuf2 or Hec1 to GFP were from Jennifer DeLuca (University of California, Santa Barbara, USA). Plasmid pEGFP/Spc24 coding for N-terminal fusion of human Spc24 to GFP was made by cloning the Spc24 cDNA into the pcDNA3.1 + N-eGFP vector with KpnI/BamHI (Genscript). To generate pcDNA4/GFP/Mis12/hNuf2 plasmid, human Nuf2 was PCR-amplified from the pEGFP N1/Nuf2 vector and cloned into pcDNA4/GFP/Mis12 with XhoI/ EcoRV. For pGEX4T1/hNuf2 coding for full length GST-tagged human Nuf2, the Nuf2 cDNA was amplified by PCR using pEGFP N1/Nuf2 as template and cloned into the pGEX4T1 vector with BamHI/ XhoI. Also, to make pGEX4T1/hHec1 coding for full length GST-tagged human Hec1, the Hec1 cDNA was amplified by PCR using pEGFP N1/Hec1 as template and inserted into pGEX4T1 vector with XhoI. To make the pGFP/Chmp4c (1–77) vector coding for N-terminal fusion of human Chmp4c (1–77) to GFP, we excised Mis12 (amino acids 1–205 of human Mis12) from the pcDNA4/GFP/Mis12/hChmp4c (1–77) plasmid (please see below) with NotI/ XhoI and filled in the 5′ overhangs with T4 DNA polymerase (New England Biolabs). The pcDNA4/GFP/Mis12/hChmp4c (1–77) plasmid (Petsalaki et al. 2018) was made by amplifying the Chmp4c cDNA sequence by PCR and cloning it into the pcDNA4/GFP/Mis12 vector with XhoI/ EcoRI. All plasmids were completely sequenced.
siRNA sequences
Negative siRNA (a pool of four different siRNAs: UAAGGCUAUGAAGAGAUAC, AUGUAUUGGCCUGUAUUAG, AUGAACGUGAAUUGCUCAA, UGGUUUACAUGUCGACUAA) were from Thermo Scientific. Human Chmp4c siRNA, previously described as Chmp4c-2 (Petsalaki et al. 2018), located in the 3′-untranslated region (GUAGAGGAGUCUUAUAUGA), Nuf2 siRNA located in the 3′-untranslated region (GAAGCGAAUGGAAGUAUCA) and Spc24 siRNA (a pool of three different siRNAs: UCACCAUGGAGAAGGAAGU, CAACUUUACCACCAAGUUA, CUCUCCAGGAAAUUCAUCA) were from Santa Cruz Biotechnology. Only the sense sequences of the siRNA duplexes are shown and all sequences are provided in 5′ → 3′ orientation.
Bacteria culture and protein purification
BL21 (DE3) cells (Agilent Technologies) were grown in 100–200 ml LB Broth (1% w/v tryptone, 0.5% w/v yeast extract, 171 mM NaCl) supplemented with the appropriate selection antibiotic. At OD600 = 0.5, cells were induced with 0.1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) at 16 °C for 16–18 h. Afterwards, cells were spun down at 1000 g for 10 min and the pellet was resuspended in 3 ml NETN (20 mM Tris pH = 8.0, 100 mM NaCl, 1 mM EDTA, 0.5% NP-40) for every 100 ml of culture. The suspension was sonicated for 3 × 10 s and cleared by centrifugation at 12,000 g for 10 min to produce the supernatant (crude extract) containing the GST fusion protein. To prepare GST fusion protein bound to glutathione agarose beads, 150 μl glutathione agarose slurry (sc-2009, Santa Cruz Biotechnology) was added for every 3 ml crude extract, mixed for 1 h at 4 °C using a spiramix, beads were spun down at 1000 g for 5 min, washed × 3with 500 μl NETN and an equal volume of NETN (150 μl) was added. GST proteins on glutathione agarose beads were stored at 4 °C for up to 1 week. To prepare GST proteins eluted from beads, GST proteins were isolated from the crude extract using the GST spintrap purification module (#28-9523-59, GE Healthcare) following the manufacturer’s instructions, aliquoted and stored at − 80 °C. His-tagged human Chmp4c was expressed in BL21 (DE3) cells as above and purified using the His-Bind purification kit (#70239-3, Millipore) following the manufacturer’s instructions. Eluted His-Chmp4c was dialysed in dialysis buffer (50 mM Tris pH = 7.5, 150 mM NaCl, 0.1 mM EGTA pH = 8.0) using Pur-A-lyzer 6000 dialysis tubes (PURX60015, Sigma), aliquoted and stored at − 80 °C.
Cell culture and treatments
Human colon carcinoma BE cells and cervical carcinoma HeLa cells were grown in DMEM (Gibco) containing 10% fetal bovine serum at 37 °C in 5% CO2 and treated with 3.32 μM or 300 nM nocodazole (Applichem), or 10 μg/ml MG132 (Calbiochem) as appropriate. siRNAs or GFP proteins were transfected into cells 24 h before analysis or drug treatment using Lipofectamine 2000 (Invitrogen). Experiments were generally performed in BE cells unless otherwise stated. Transient expression of GFP-tagged proteins was relatively more uniform in HeLa compared with BE cells; therefore, most rescue experiments were performed in HeLa cells as specified and cells exhibiting relatively low GFP kinetochore levels were analysed by fluorescence microscopy. All cell lines used exhibited consistent morphology and growth properties and were negative for mycoplasma contamination.
Enrichment of cells in mitosis (nocodazole and “shake-off”)
Cells were treated with 3.32 μΜ nocodazole for 16 h and mitotic cells isolated by “shake-off”. Microscopic examination had shown approximately 70–80% of cells were in prometaphase after this treatment compared with 2–3% in untreated cells.
Enrichment of cells in prometaphase with an intact mitotic spindle (nocodazole and “release”)
BE cells were treated with 300 nM nocodazole for 16 h, incubated with ice-cold-medium for 1.5 h to completely depolymerise spindle microtubules and released into warm medium for 15 min to allow spindle re-formation. Microscopic examination had shown approximately 55% of cells were in prometaphase after the 15-min release and exhibited a fully formed mitotic spindle (as judged by spindle morphology and the amount of α-tubulin incorporated in the mitotic spindle) compared with approximately 2% in untreated cells. Afterwards, cells were lysed in ice-cold immunoprecipitation/kinase buffer as described further below.
Indirect immunofluorescence microscopy
For Chmp4c, Nuf2, Spc24, Spc25, Knl1, Mis12, CENP-A or CENP-H staining, cells were fixed in ice-cold methanol for 5 min at − 20 °C, washed twice with PBS at room temperature, and immunostained. For ZW10, Hec1, α-tubulin or GFP staining, cells were fixed in 4% paraformaldehyde in cytoskeleton buffer (1.1 M Na2HPO4, 0.4 M KH2PO4, 137 mM NaCl, 5 mM KCl, 2 mM MgCl2, 2 mM EGTA, 5 mM Pipes, and 5 mM glucose, pH 6.1) for 5 min at 37 °C, permeabilized in 0.5% Triton X-100 in cytoskeleton buffer, washed twice with PBS at room temperature, and immunostained.
FITC- or rhodamine-TRITC–conjugated secondary antibodies (Jackson ImmunoResearch Laboratories, Inc.) were used as appropriate. DNA was stained with 10 μM TO-PRO-3 iodide (642/661 nm; Invitrogen) and cells were mounted in Vectashield medium (Vector Laboratories). Images were collected using a laser-scanning spectral confocal microscope (TCS SP2; Leica), LCS Lite software (Leica) and a 63Χ Apochromat 1.40 NA oil objective. The low fluorescence immersion oil (11513859; Leica) was used, and imaging was performed at room temperature. Average projections of image stacks were obtained using the LCS Lite software.
To analyse fluorescence intensities, background readings were subtracted and green/red fluorescence intensities quantified using LCS Lite. For tubulin fluorescence intensities (cold assays), background readings were subtracted and total tubulin-associated fluorescence for each mitotic cell was quantified by analysing an equal image area using ImageJ (National Institutes of Health).
Cold-induced microtubule depolymerisation
To induce microtubule disassembly of non-kinetochore microtubules, cell media was replaced with ice-cold media and cells were incubated on ice for 15 min. Subsequently, cells were fixed in 4% paraformaldehyde in cytoskeleton buffer, permeabilized in 0.5% Triton X-100, stained for α-tubulin and prometaphase cells were examined by fluorescence microscopy.
In vitro microtubule polymerisation
Microtubules were assembled from rhodamine-labelled tubulin (TL590M-A, Cytoskeleton) at 37 °C for 30 min in BRB80 buffer (80 mM PIPES, pH 6.9, 8 mM MgCl2, 0.5 mM EGTA, 1 mM GTP) and stabilised with 20 mM taxol (Applichem) for 5 min according to manufacturer’s instructions.
Microtubule pull-down, co-sedimentation and bundling assays
For microtubule pull-down assays, 0–5 μM of taxol-stabilised microtubules were incubated with 0.5 μg GST-Chmp4c or 1 μg GST on glutathione agarose beads (Santa Cruz Biotechnology) in BRB80 buffer at room temperature for 1 h, in 20 μl final volume. The beads were then washed three times with BRB80 buffer, boiled and analysed by SDS-polyacrylamide gel electrophoresis.
For microtubule co-sedimentation assays, 0.5 μg GST-Chmp4c (= 0.5 μM, approximately), 1 μg GST (= 2 μM), 0.6 μg GST-Hec1 (= 0.3 μM) eluted from beads or 250–350 ng purified His-Chmp4c (= 0.5–0.65 μM) were incubated with 0–2 μM taxol-stabilised microtubules in BRB80 buffer at room temperature for 1 h, in 20 μl final volume. The samples were then centrifuged at 20,000 g for 1.5 h at 30 °C. The supernatant and pellet were boiled and separated by SDS-polyacrylamide gel electrophoresis. In Figs 5f and S4 g, the percentage (x) of GST-Chmp4c (or GST-Hec1) bound to microtubules for every microtubule concentration was calculated as follows: x = [p/(s + p)] × 100%, where (s) and (p) are the intensities of the GST-Chmp4c (or GST-Hec1) bands in the supernatant and microtubule pellet fractions, respectively.
For microtubule bundling assays, 1 μM taxol-stabilised microtubules was incubated with 0–1 μM GST-Chmp4c in BRB80 buffer at room temperature for 15 min, in 20 μl final volume. Then, 5 μl were squashed in coverslips and observed by fluorescence microscopy.
Protein co-immunoprecipitations, GST pull-downs and in vitro binding assays
Cells were sonicated three times for 10 s in ice-cold immunoprecipitation/kinase buffer (50 mM Hepes, pH 7.5, 150 mM NaCl, 1 mM EDTA, 2.5 mM EGTA, 10% glycerol, 0.1% Tween 20, 0.1 mM PMSF, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 1 mM sodium fluoride, 10 mM sodium β-glycerophosphate and 0.1 mM sodium vanadate) and incubated for further 30 min on ice. For co-immunoprecipitations, 1 mg cell lysate was incubated with 1 μg antibody for 16 h followed by addition of 10 μl protein A/G PLUS–agarose beads for 1 h at 4 °C. For GST pull-downs or in vitro binding assays, 0.5 mg cell lysate or 0.5 μg purified His-Chmp4c respectively, was incubated with 1 μg GST protein on glutathione agarose beads for 4 h at 4 °C. Samples were spun down, washed three times with immunoprecipitation/kinase buffer and immunoprecipitated proteins on agarose beads analysed by SDS-PAGE.
Western blotting and phosphatase treatment
Cells were lysed in ice-cold whole cell extract buffer (20 mM Hepes, 5 mM EDTA, 10 mM EGTA, 0.4 M KCl, 0.4% Triton X-100, 10% glycerol, 5 mM NaF, 1 mM DTT, 5 μg/ml leupeptin, 50 μg/ml PMSF, 1 mM benzamidine, 5 μg/ml aprotinin and 1 mM Na3VO4) for 30 min on ice. Lysates were cleared by centrifugation at 15,000 g for 10 min, analysed by SDS-PAGE and transferred onto nitrocellulose membrane (Santa Cruz Biotechnology). For phosphatase assays, cells were lysed in whole cell extract buffer without phosphatase inhibitors and 30 μg cell extracts were incubated with 800 units lamda phosphatase (Santa Cruz Biotechnology), for 30 min at 30 °C prior to analysis by SDS-PAGE.
Densitometry
Densitometric analysis of bands was performed using ImageJ (National Institutes of Health).
Statistical analysis
For kinetochore fluorescence, several kinetochore pairs per cell from a minimum of five cells per experiment from at least two independent experiments were analysed per treatment and standard deviation (s.d.) was calculated. For tubulin fluorescence, 30 cells per experiment from three independent experiments were analysed for each treatment and s.d. was calculated. For chromosome misalignment, s.d. from the mean from three independent experiments was calculated and at least 20 cells per experiment and per treatment were analysed. Statistically significant differences between two groups were calculated using the Student’s t test. Statistically significant differences between three or more groups (Figs. 2b, f, 4b, d, 6b, d and S3d,f) were determined by one-way ANOVA followed by the Student’s t test between two groups. No statistical method was used to predetermine sample size.
Data availability
All data supporting the findings of this study are available within the paper and its supplementary information files.
References
Alushin GM, Ramey VH, Pasqualato S, Ball DA, Grigorieff N, Musacchio A, Nogales E (2010) The Ndc80 kinetochore complex forms oligomeric arrays along microtubules. Nature 467:805–810
Bharadwaj R, Qi W, Yu HT (2004) Identification of two novel components of the human NDC80 kinetochore complex. J Biol Chem 279:13076–13085
Brinkley BR, Cartwright J (1975) Cold-labile and cold-stable microtubules in mitotic spindle of mammalian cells. Ann N Y Acad Sci 253:428–439
Campsteijn C, Vietri M, Stenmark H (2016) Novel ESCRT functions in cell biology: spiraling out of control? CurrOpin Cell Biol 41:1–8
Cheeseman IM, Chappie JS, Wilson-Kubalek EM, Desai A (2006) The conserved KMN network constitutes the core microtubule-binding site of the kinetochore. Cell 127:983–997
Ciferri C, de Luca J, Monzani S, Ferrari KJ, Ristic D, Wyman C, Stark H, Kilmartin J, Salmon ED, Musacchio A (2005) Architecture of the human Ndc80-Hec1 complex, a critical constituent of the outer kinetochore. J Biol Chem 280:29088–29095
Ciferri C, Pasqualato S, Screpanti E, Varetti G, Santaguida S, Dos Reis G, Maiolica A, Polka J, de Luca JG, de Wulf P, Salek M, Rappsilber J, Moores CA, Salmon ED, Musacchio A (2008) Implications for kinetochore-microtubule attachment from the structure of an engineered Ndc80 complex. Cell 133:427–439
DeLuca JG, Moree B, Hickey JM, Kilmartin JV, Salmon ED (2002) hNuf2 inhibition blocks stable kinetochore-microtubule attachment and induces mitotic cell death in HeLa cells. J Cell Biol 159:549–555
DeLuca JG, Howell BJ, Canman JC, Hickey JM, Fang G, Salmon ED (2003) Nuf2 and Hec1 are required for retention of the checkpoint proteins Mad1 and Mad2 to kinetochores. Curr Biol 13:2103–2109
DeLuca JG, Gall WE, Ciferri C, Cimini D, Musacchio A, Salmon ED (2006) Kinetochore microtubule dynamics and attachment stability are regulated by Hec1. Cell 127:969–982
Emanuele MJ, McCleland ML, Satinover DL, Stukenberg PT (2005) Measuring the stoichiometry and physical interactions between components elucidates the architecture of the vertebrate kinetochore. Mol Biol Cell 16:4882–4892
Foley EA, Kapoor TM (2013) Microtubule attachment and spindle assembly checkpoint signalling at the kinetochore. Nat Rev Mol Cell Biol 14:25–37
Friese A, Faesen AC, Huis in ‘t Veld PJ, Fischböck J, Prumbaum D, Petrovic A, Raunser S, Herzog F, Musacchio A (2016) Molecular requirements for the inter-subunit interaction and kinetochore recruitment of SKAP and Astrin. Nat Commun 7:11407
Hanisch A, Sillje HH, Nigg EA (2006) Timely anaphase onset requires a novel spindle and kinetochore complex comprising Ska1 and Ska2. EMBO J 25:5504–5515
Hurley JH (2015) ESCRTs are everywhere. EMBO J 34:2398–2407
Joglekar AP, Bouck DC, Molk JN, Bloom KS, Salmon ED (2006) Molecular architecture of a kinetochore-microtubule attachment site. Nat Cell Biol 8:581–585
Jouvenet N, Zhadina M, Bieniasz PD, Simon SM (2011) Dynamics of ESCRT protein recruitment during retroviral assembly. Nat Cell Biol 13:394–401
Kern DM, Monda JK, Su KC, Wilson-Kubalek EM, Cheeseman IM (2017) Astrin-SKAP complex reconstitution reveals its kinetochore interaction with microtubule-bound Ndc80. Elife 6:26866
Maiato H, Rieder CL, Khodjakov A (2004) Kinetochore-driven formation of kinetochore fibers contributes to spindle assembly during animal mitosis. J Cell Biol 167:831–840
Miller SA, Johnson ML, Stukenberg PT (2008) Kinetochore attachments require an interaction between unstructured tails on microtubules and Ndc80(Hec1). Curr Biol 18:1785–1791
Perpelescu M, Fukagawa T (2011) The ABCs of CENPs. Chromosoma 120:425–446
Petsalaki E, Dandoulaki M, Zachos G (2018) The ESCRT protein Chmp4c regulates mitotic spindle checkpoint signaling. J Cell Biol 217:861–876
Schmidt JC, Arthanari H, Boeszoermenyi A, Dashkevich NM, Wilson-Kubalek EM, Monnier N, Markus M, Oberer M, Milligan RA, Bathe M, Wagner G, Grishchuk EL, Cheeseman IM (2012) The kinetochore-bound Ska1 complex tracks depolymerizing microtubules and binds to curved protofilaments. Dev Cell 23:968–980
Sundin LJ, Guimaraes GJ, Deluca JG (2011) The NDC80 complex proteins Nuf2 and Hec1 make distinct contributions to kinetochore-microtubule attachment in mitosis. Mol Biol Cell 22:759–768
Suzuki A, Badger BL, Salmon ED (2015) A quantitative description of Ndc80 complex linkage to human kinetochores. Nat Commun 6:8161
Valverde R, Ingram J, Harrison SC (2016) Conserved tetramer junction in the kinetochore Ndc80 complex. Cell Rep 17:1915–1922
Varma D, Salmon ED (2012) The KMN protein network—chief conductors of the kinetochore orchestra. J Cell Sci 125:5927–5936
Wei RR, Sorger PK, Harrison SC (2005) Molecular organization of the Ndc80 complex, an essential kinetochore component. Proc Natl Acad Sci U S A 102:5363–5367
Acknowledgments
We thank P. Bieniasz, J. DeLuca, G. Kops, A. Musacchio and T. Zang for generously sharing reagents.
Funding
This work was supported by the Worldwide Cancer Research (grant 15-0008) and by the Fondation Santé (cycle 2017–2018). E.P. was supported by a postdoctoral fellowship from the Bodossaki Foundation and M.D. by the Worldwide Cancer Research.
Author information
Authors and Affiliations
Contributions
E.P., M.D. and G.Z. performed experiments and analysed the results. G.Z. and E.P. designed the study and wrote the paper.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
ESM 1
(PDF 954 kb)
Rights and permissions
About this article

Cite this article
Petsalaki, E., Dandoulaki, M. & Zachos, G. Chmp4c is required for stable kinetochore-microtubule attachments. Chromosoma 127, 461–473 (2018). https://doi.org/10.1007/s00412-018-0675-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00412-018-0675-8