
Abstract
Telomeres protect the ends of linear chromosomes against loss of genetic information and inappropriate processing as damaged DNA and are therefore crucial to the maintenance of chromosome integrity. In addition to providing a pathway for genome-wide DNA repair, homologous recombination (HR) plays a key role in telomere replication and capping. Consistent with this, the genomic instability characteristic of HR-deficient cells and tumours is driven in part by telomere dysfunction. Here, we discuss the mechanisms by which HR modulates the response to intrinsic cellular challenges that arise during telomere replication, as well as its impact on the assembly of telomere protective structures. How normal and tumour cells differ in their ability to maintain telomeres is deeply relevant to the search for treatments that would selectively eliminate cells whose capacity for HR-mediated repair has been compromised.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Telomeres are protein-DNA complexes which cap the ends of linear chromosomes. Although the existence of end protective structures was suggested by Barbara McClintock’s first visualization of chromosomal fusions in maize in the early 1940s (McClintock 1941), it was not until the 1960s that telomeres were assigned a role in cellular ageing. The concept that linear chromosome ends could not be fully replicated by the Watson and Crick classical replication pathway (Watson and Crick 1953) causing progressive erosion of terminal DNA sequences and proliferative arrest (the end-replication problem) was later recognized as the basis of Hayflick’s limited lifespan theory (Hayflick 1965), which stated that cells in culture divide only a finite number of times. Telomeric DNA was first identified in Tetrahymena thermophila (Szostak and Blackburn 1982) as sequences that can stabilize a yeast artificial chromosome. Pioneering work subsequently led to the discovery of telomerase, an enzyme which adds sequentially telomeric DNA repeats onto chromosome ends, thus counteracting the end-replication problem (Greider and Blackburn 1985).
Concomitantly, with the understanding of chromosome end structure and maintenance, the concept of DNA recombination became a focal point of scientific investigation. In 1964, Robin Holliday suggested a DNA recombination mechanism to explain the independent segregation of fungal genes located on the same chromosome (Holliday 1964). This model linked for the first the genetic recombination to DNA repair and identified the cross-stranded structure that physically connects two DNA molecules, the Holliday junction (for a review see Liu and West 2004). In 1983, Jack Szostak and colleagues proposed that recombination could be initiated by a DNA double-strand break (DSB). Their ‘double-strand-break repair model for recombination’ (Szostak et al. 1983) (Fig. 1) predicted that end resection generates single-stranded DNA (ssDNA) tails capable of invading a homologous duplex DNA and leading to gene conversion events with or without crossing over. Although variations of this homologous recombination (HR) mechanism have meanwhile been envisaged, the basic model proposed by Szostak and colleagues remains valid today.

Model for DSB repair by HR. The DNA at a break site is resected to generate ssDNA tails, which represent substrates onto which RAD51 monomers are loaded in BRCA2- and RAD51 paralog-dependent manner. The nucleoprotein filament thus generated invades a homologous dsDNA and, following second end capture, a double Holliday junction structure is formed. Branch migration, which is dependent among others on RAD54, facilitates cleavage of Holliday junctions by GEN1 or MUS81-EME1 resolvases, or their dissolution dependent on the BLM-TOPIIIα-RMI1 complex. Crossover or non-crossover molecules are the final products of this DNA repair reaction
Telomere DNA with its 3′ ssDNA overhang on the G-rich strand (Blackburn 1984) was recognized as a potential substrate for HR reactions. At this time it was demonstrated in yeast that recombination reliant on RAD52 could occur between linear plasmids and chromosomal telomere-adjacent sequences (Dunn et al. 1984), indicating that HR could indeed provide a mechanism for telomere elongation. Further work in yeast (Pluta and Zakian 1989) also demonstrated the recombinogenic potential of telomeric sequences, both chromosome- and plasmid-positioned. Subsequently, it became more accepted that telomere-telomere recombination via canonical gene conversion reactions could provide an alternative mechanism of telomere elongation to telomerase (Wang and Zakian 1990).
Whilst telomerase activity is easily detectable in most model systems using robust in vitro assays, telomere-telomere recombination events are difficult to visualize using conventional methods due to the sequence similarity of the telomeric repeats. HR events have, however, been detected and quantified at single-telomere resolution in yeast, where the DNA sequences of particular telomeres are sufficiently variable to allow this (Teixeira et al. 2004). These recombination events between TG repeats, which elongated individual telomeres, occurred in a telomerase-deficient background at very low frequency (approximately 0.3 % per generation), suggesting that HR-mediated lengthening is a rare occurrence even when telomerase was inactivated. Telomerase-deficient yeast cells gradually lose their telomeres and enter senescence, until a fraction switches to Rad52-dependent recombination pathway for telomere elongation (Le et al. 1999; Lundblad and Blackburn 1993). These cells are known as post-senescence survivors. Spontaneous telomere HR reactions similar to those described in yeast have also been detected in human cells lacking telomerase activity (Dunham et al. 2000), where they provide a mechanism for alternative lengthening of telomeres (ALT; Conomos et al. 2013). Moreover, a recently reported mouse model containing an exogenous DNA tag inserted in one telomere unravelled that spontaneous ALT reactions take place at telomeres under physiologically normal conditions in various tissues (Neumann et al. 2013). This important work supports the concept that telomere-telomere recombination events, cryptic and possibly infrequent, occur even in telomerase-proficient cells.
Plants and insects can also rely on recombination-based mechanisms for telomere regulation. For example, Drosophila species lack telomerase and rely instead on telomere-specific retrotransposons to extend their chromosome ends. Indeed, three telomere-specific non-long-terminal-repeat retrotransposons have been described in Drosophila (HeT-A, TART and TAHRE), which together form protective head-to-tail arrays at the chromosome termini (Pardue and DeBaryshe 2011). In addition to transpositions, recombination and gene conversion events have also been detected at Drosophila telomeres (Kahn et al. 2000) and shown to mediate telomere elongation. The gene coding for heterochromatin protein 1 (HP1) is crucial for regulating these events in Drosophila, as its disruption caused elevated levels of both telomere transposition and recombination (Savitsky et al. 2002). Importantly, recombination is the prevalent mechanism for elongating chromosome ends in dipteran species including, the malaria vector Anopheles gambiae (Roth et al. 1997).
Protection against the loss of critical genetic information is now a well-established role of telomeres in the maintenance of genome stability. Additionally, telomeres block recognition of chromosome ends as DSBs, thereby preventing detrimental repair reactions mediated either by non-homologous end joining (NHEJ) or HR. NHEJ of unprotected telomeres generates end-to-end chromosomal fusions leading to ‘bridge-fusion-breakage’ cycles (Maser and DePinho 2002) and rampant genome instability. Likewise, illegitimate HR reactions at telomeres (e.g. sister chromatid exchanges) can have catastrophic consequences for genome integrity leading to excessive lengthening of one sister telomere at the expense of the other. Paradoxically, however, HR also acts to promote telomere integrity by facilitating telomere replication and by contributing to the remodelling of telomere DNA into protective secondary structures known as T-loops (Fig. 2). In this review, we discuss this apparent contradiction and the mechanisms that balance the opposing effects of HR at telomeres.

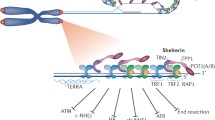
Model for HR-mediated telomere capping. In the ‘open’ telomere configuration, the G-rich ssDNA overhang is exposed and can potentially activate DNA damage responses, which arrest cell cycle progression. RAD51 is loaded onto the telomere ssDNA in BRCA2-dependent manner to promote a strand invasion reaction into the duplex DNA of the same telomere. This leads to formation of a D-loop and of a ‘three-way’ Holliday junction, which together define the T-loops capping chromosome ends. The shelterin complex binds telomeric DNA and stabilizes the ‘T-loop’ configuration
The structure of mammalian telomeres
In vertebrates, the telomere is minimally defined as the repetitive DNA tract ending in a 3′ G-rich overhang, together with the protein complex known as shelterin that binds specifically to telomere DNA. In addition, RNAs and proteins mediating DNA damage responses, repair, replication and transcription (Martínez and Blasco 2011; Schoeftner and Blasco 2009) associate with telomeres to maintain their function and integrity.
Telomere DNA
The double-stranded DNA (dsDNA) of mammalian telomeres consists of tandem repeats with the sequence TTAGGG, ranging in length between 10 and 15 kb in humans and 20 and 50 kb in mice. The 3′ ssDNA overhang on the G-rich telomere strand varies in length between 50 and 400 nucleotides and represents a key feature of telomere DNA thought to facilitate the assembly of the telomere protective structures known as T-loops (Fig. 2). The subtelomeric region located adjacent to the telomere DNA consists of tandem repeat arrays characterized by high sequence variability (Baird et al. 1995). Telomere replication and telomere-repeat containing RNA (TERRA) transcription both initiate at subtelomeric sites. TERRA is constitutively associated with telomeres and thus considered a core component of the telomere complex.
To ensure telomere functionality, the repetitive DNA tract must be maintained at a constant length. This is primarily achieved in most cell types through telomerase-mediated addition of telomeric repeats (Greider 1996). In cells with compromised telomerase activity, elongation requires the recombination-based ALT pathway. The recent discovery of ALT events in mouse somatic tissues (Neumann et al. 2013) suggests that the two mechanisms could coexist; however, their relative contribution to telomere homeostasis is not clear. Determining ALT frequency in telomerase-null mice may indicate to what extent this mechanism compensates for telomere shortening in the absence of telomerase. Additionally, suppression of ALT events in HR-deficient mouse models (e.g. Brca2 −/−) would support telomere recombination reactions as the mechanism for ALT occurrence.
The shelterin complex
The existence of proteins that bind to telomeric DNA with sequence specificity initially demonstrated in yeast and ciliates (Conrad et al. 1990; Fang and Cech 1993) was later reinforced by work that led to the identification of the first telomere dsDNA-binding proteins in mammals, TRF1 and TRF2 (Bilaud et al. 1997; Chong et al. 1995; van Steensel et al. 1998). RAP1 was subsequently identified (Li et al. 2000) as a factor recruited to telomeres by TRF2, whilst POT1 was characterized as a ssDNA-binding protein with specificity for telomeric sequences (Baumann and Cech 2001; Loayza and de Lange 2003). TRF1 and TRF2 are linked by TIN2 (Ye and de Lange 2004), which also connects POT1 to the telomere complex via TPP1 binding (Houghtaling et al. 2004; Liu et al. 2004a; Ye et al. 2004). The complex formed by these six proteins, referred to as shelterin, is stably associated with all telomeres throughout the cell cycle (Fig. 2; Palm and de Lange 2008). In addition, the CST (CTC1/Cdc13-STN1-TEN1) complex, a DNA polα.primase accessory factor, constitutively associates with telomeres via interaction with TPP1 (Wan et al. 2009) and plays important roles in replication fork re-start and post-replicative telomere processing (Miyake et al. 2009; Wu et al. 2012).
More recently, HOT1 was identified as a novel factor capable of binding telomere dsDNA with sequence specificity (Kappei et al. 2013). Unlike the core shelterin components, HOT1 is only present at a subset of telomeres and is also found in the Cajal bodies where active telomerase is assembled. HOT1 was therefore proposed to bring telomeres and telomerase in close proximity, facilitating telomere elongation.
The availability of conditional mouse models for individual shelterin components made it possible to better define their roles in telomere maintenance. Based on these studies, it is now established that TRF2 primarily promotes telomere capping and inhibits ataxia telangiectasia mutated (ATM)-dependent DNA damage responses (Celli and de Lange 2005; Denchi and de Lange 2007), whilst TRF1 is essential for telomere replication (Martinez et al. 2009; Sfeir et al. 2009). TPP1/POT1 act together to suppress ataxia telangiectasia mutated and Rad3-related (ATR)-dependent checkpoint activation through specific binding to the telomere ssDNA tails (Denchi and de Lange 2007; Wu et al. 2006). This function is critically mediated by TIN2, which tethers the TPP1/POT1 complex to telomeres (Takai et al. 2011). In addition, TPP1 recruits telomerase to chromosome ends (Abreu et al. 2010; Tejera et al. 2010). Importantly, RAP1, a TRF2-interacting partner and suppressor of illegitimate recombination events at telomeres (Martinez et al. 2010; Sfeir et al. 2010), was the first shelterin component known to perform extra-telomeric functions. Nuclear RAP1 associates with a subset of gene promoters and regulates gene transcription, similarly to its yeast counterpart (Martinez et al. 2010), whilst cytoplasmic RAP1 activates NF-kB signalling (Teo et al. 2010).
Thus, individual shelterin components play prominent roles in specific aspects of telomere maintenance, mediated by interactions with DNA damage sensors, repair, replication and transcription factors (for a review see Martínez and Blasco 2011). Yet, it appears that some functions are shared by several telomere proteins, for example, TRF1, TPP1 and RAP1 are together required to prevent telomere fragility. This suggests that the shelterin subunits act cohesively in order to ensure telomere protection, replication and length maintenance. Large-scale structural studies are required to solve the higher-order architecture of telomere-binding complexes and to identify the functional interactions essential for telomere integrity.
HR reactions in DSB repair
DSBs are considered the most deleterious form of DNA lesions, as failure to repair triggers cell cycle arrest and/or cell death. HR is the major error-free pathway for DSB repair in most cells and therefore essential for genome integrity and cell viability. Supporting this concept, HR abrogation leads either to lethal accumulation of DSBs or to illegitimate DSB repair, known to generate the chromosomal rearrangements accountable for onset and progression of tumorigenesis.
As proposed by the classical model of HR-mediated repair (Szostak et al. 1983; Fig. 1), DSBs introduced in chromosomes are processed through extensive DNA resection on either side of the break, which generates recombinogenic ssDNA tails. This nucleolytic processing requires the MRE11-RAD50-NBS1 (MRN) complex and its binding partner, CtIP, to initiate resection. The concerted action of Bloom’s syndrome protein (BLM), WRN, EXO1 and DNA2 (Mimitou and Symington 2011; Nimonkar et al. 2011) further extends the resected tails. The ssDNA thus generated is coated by replication protein A (RPA), known to have a higher affinity for ssDNA than RAD51. Thus, the RAD51 recombinase must be actively loaded onto the resected ends in order to displace RPA and to form a nucleoprotein filament capable of invasion into homologous dsDNA.
The tumour suppressor BRCA2 acts as the loader of RAD51 at sites of DSBs. This function is mediated by the capacity of one BRCA2 molecule to bind several RAD51 monomers through its BRC motifs (Pellegrini et al. 2002) and to facilitate their assembly onto ssDNA, to which BRCA2 itself binds through its oligonucleotide/oligosaccharide-binding folds. The RAD51 paralogs (RAD51B, RAD51C, RAD51D, XRCC2 and XRCC3), a family of proteins related to each other and to RAD51 itself, also promote RAD51 accumulation at sites of DNA breaks, although the underlying molecular mechanism is not clear (Suwaki et al. 2011). RAD51-mediated strand invasion generates displacement loops (D-loops), well-characterized recombination intermediates that facilitate extension of the invading ssDNA by DNA polymerases. The second end of the break is ‘captured’ by annealing within the D-loop and formation of two Holliday junctions (West 2009), which are resolved either by GEN1-dependent symmetric cleavage or by asymmetric cleavage mediated by the MUS81/EME1 complex. These reactions lead to either crossover or non-crossover products (Fig. 1). Alternatively, Holliday junctions can be ‘dissolved’ through concerted Holliday junction migration catalysed by the BLM and decatenation, dependent on the topoisomerase TOPIIIα and its interacting partner, RMI1. This process gives rise only to non-crossover products, which explains the striking accumulation of crossovers in BLM-deficient cells.
HR-mediated DSB repair occurs in S and G2 phases of the cell cycle, when a sister chromatid, the most common form of duplex DNA used as a template for HR reactions, is available and positioned close to the broken DNA. However, cells can also repair DNA damage throughout the cell cycle using NHEJ, a process that joins together DNA ends regardless of sequence similarity and often inaccurately. Conventional thinking led to the assumption that HR repair is favoured when a sister chromatid is available due to its superior precision compared with NHEJ. This view, however, has been challenged recently by studies demonstrating that the choice between the two repair pathways is highly regulated through antagonistic actions on end resection by 53BP1 and BRCA1 (Bouwman et al. 2010; Bunting et al. 2010). Subsequent studies have shown competition for broken DNA ends by two distinct DNA binding modules, 53BP1/RIF1 and BRCA1/CtIP (Chapman et al. 2013; Di Virgilio et al. 2013; Escribano-Díaz et al. 2013), which repress or promote resection, respectively. Importantly, similar mechanisms regulate resection at telomeres artificially uncapped through shelterin removal (Zimmermann et al. 2013), thus extending this intriguing interplay between 53BP1 and BRCA1 to the chromosome end.
HR in telomere replication
Due to its G-quadruplex forming potential (Lipps and Rhodes 2009) (Fig. 3a), remodelling into protective T-loop structures (Fig. 2) and extensive heterochromatinization (Blasco 2007), telomere DNA poses a natural barrier to replication fork progression. More recently, RNA-DNA hybrids (R-loops) arising during TERRA transcription were shown to obstruct telomere replication in yeast (Pfeiffer et al. 2013). As origins of replication are thought to localize almost exclusively in subtelomeric regions, with rare initiation occurring within telomere DNA repeats (Drosopoulos et al. 2012; Gilson and Géli 2007), rescue of stalled forks by oncoming forks seems unlikely. However, replication initiation within the telomeric repeats could be more common than previously anticipated. The amino-terminal basic domain of TRF2 can recruit the origin recognition complex (ORC) to telomeres and this recruitment is essential for telomere integrity (Deng et al. 2007). Furthermore, TERRA interacts directly with the amino-terminal basic domain of TRF2, as well as with ORC forming a stable ternary complex required for telomere maintenance (Deng et al. 2009). Given the identification of this important interaction and the key role of ORC in origin firing, we should certainly be open to the possibility that initiation within the telomeric repeats could occur at a more significant level than has so far been detected.

HR facilitates telomere replication. a G-quadruplexes, highly stable DNA secondary structures that form spontaneously during replication, play opposing roles at telomeres. They impede replication fork progression causing DNA breakage whilst also concealing telomeric ssDNA overhang to prevent activation of DNA damage responses. DSBs introduced during telomere replication in the proximity of G-quadruplexes could be repaired by HR. b Template switching provides a mechanism to bypass replication fork barriers (e.g. G-quadruplexes) and re-start stalled replication forks. HR activities are required in this process
Recent studies demonstrated that telomere replication requires the coordinated action of structural telomere proteins and DNA damage repair pathways, including HR (Gilson and Géli 2007), to counteract the threat posed by collapsed forks. Consistent with this notion, shelterin component TRF1 promotes telomere replication (Martinez et al. 2009; Sfeir et al. 2009), most likely through recruitment of BLM and RTEL1 helicases, which remove secondary structures within telomere sequences. Likewise, TRF2 acts as a sensor of topological stress induced by T-loops and DNA supercoiling during telomere replication and facilitates fork progression through activation of nucleases and topoisomerases that dismantle these impediments (Ye et al. 2010).
In addition, HR provides a mechanism for effective telomere replication. Conceivably, HR reactions at telomeres promote re-start of stalled replication forks through template switching (Fig. 3b; Ciccia and Elledge 2010) and repair of replication-associated DSBs, similarly to other fork-stalling sites within the genome. However, the consequences of HR abrogation at telomeres are more severe, as unrepaired DSBs within terminal repeats can cause loss of telomeric DNA and abrupt telomere shortening. Moreover, indiscriminate joining of telomeres broken as a result of collapsed forks generates end-to-end fusions or interstitial telomere tracts, both known to spawn genomic instability. Supporting the role of HR in telomere replication, cells lacking HR factors RAD54 (Jaco et al. 2003), RAD51D (Tarsounas et al. 2004), RAD51C, BRCA2 and RAD51 (Badie et al. 2010) have short telomeres and elevated levels of multiple telomeric signals (MTS) (Badie et al. 2010), a hallmark of telomere fragility due to replication fork stalling and breakage. Moreover, these cells exhibit higher frequencies of chromosome fusions containing telomere DNA at the fusion site, compared to wild-type counterparts. Although likely to be caused by re-joining of replication-associated DSBs within telomeres, these could be also attributed to re-joining of telomeres that have lost protective structures by other means (Fig. 4).

Impact of dysfunctional HR on telomere integrity. Replication fork stalling at telomeres occurs frequently in HR-deficient cells, leading to DSBs and telomere shortening through loss of terminal DNA repeats. Broken telomeres lack capping structures and engage in ligation reactions with other telomeres or with break sites along the chromosome. The resulting chromosome fusions trigger genome instability and tumorigenesis onset in cells with compromised HR repair capacity
Notably, in cells lacking the core HR activities, RAD51 or BRCA2 replication intermediates become substrates for MRN-dependent resection (Hashimoto et al. 2010; Schlacher et al. 2011). This leads to ssDNA accumulation and checkpoint activation (Carlos et al. 2013). It is not known whether replication forks stalled within the telomeres of HR-deficient cells are susceptible to MRN-mediated degradation, similarly to other sites in the genome (Hashimoto et al. 2010; Schlacher et al. 2011). Whether MRN inactivation, genetically or with chemical compounds, could rescue the telomere fragility and excessive shortening characteristic of HR-deficient cells will be informative in this respect.
The stable secondary structures in the DNA such as G-quadruplexes and R-loops are likely to obstruct telomere replication. Some of the mechanisms currently known to unwind such structures are discussed below. Whether core HR activities are also required to bypass such telomere-specific barriers or to repair DSBs arising in their proximity has not yet been demonstrated.
G-quadruplexes
Telomeric G-rich ssDNA can adopt alternative secondary structures known as G-quadruplexes (Fig. 3a). Four guanine bases form a square planar arrangement via Hoogsteen base pairing, a non-conventional form of hydrogen bonding, and two or more of these can stack into a G-quadruplex structure characterized by high thermostability in vitro. Since the first proposal of their assembly at Oxytricha telomeres (Fang and Cech 1993), a large body of evidence has accumulated to support the existence of these structures in vivo in several other organisms (Tarsounas and Tijsterman 2013). Importantly, the 3′ G-rich telomere overhang could fold spontaneously into a G-quadruplex configuration, providing a capping modality additional to or alternative to T-loops (Gilson and Géli 2007). Lending support to this hypothesis, studies in yeast have shown that G-quadruplexes stabilized genetically or with chemical compounds can partially reverse uncapping caused by Cdc13 inactivation (Smith et al. 2011). Whether a similar effect is achievable at shelterin-free mammalian telomeres is not known.
On the other hand, the telomeric G-rich strand displaced during fork progression could also assemble spontaneously into G-quadruplex structures, which impede replication (Fig. 3a). These barriers to telomere replication are thought to be dismantled in vivo by PIF1, WRN, BLM or RTEL1 helicases, known to unwind G-quadruplexes in vitro (Ding et al. 2004; Fry and Loeb 1999; Huber et al. 2006; Opresko et al. 2003; Ribeyre et al. 2009; Sun et al. 1998). WRN binds TRF2 (Opresko et al. 2002) and promotes replication of G-rich telomeric strand (Crabbe et al. 2004), whilst BLM and RTEL1 presumably interact with TRF1 to suppress telomere fragility (Sfeir et al. 2009). RTEL1, the best characterized of these helicases, facilitates replication fork progression by dissolving both G-quadruplexes and T-loop structures (Vannier et al. 2012). Sequences with G-quadruplex forming potential are particularly susceptible to breakage in RTEL1-deficient cells, as suggested by the increased telomere fragility observed in these cells upon treatment with compounds that bind and stabilize G-quadruplex structures. Moreover, RTEL1 interacts directly with PCNA through a PIP-box domain (a signature PCNA-binding motif), which provided mechanistic insight into the role of this helicase during replication. Suppressing this interaction leads to high telomere fragility and accelerated tumorigenesis in mice (Vannier et al. 2013), phenotypes that highlight the oncogenic potential of deregulated telomere replication.
Taken together, these observations argue that G-quadruplexes could provide an effective capping mechanism; however, they concomitantly pose an intrinsic challenge to telomere replication. Understanding how these seemingly contradictory aspects of G-quadruplex biology are balanced in vivo to maintain telomere integrity awaits further investigation.
R-loops
The discovery of the non-coding telomere RNA TERRA has drastically challenged the conventional view that telomeres are transcriptionally silent (Azzalin et al. 2007; Schoeftner and Blasco 2008). TERRA transcription initiates from subtelomeric sites and is dependent on shelterin component TRF1. TERRA is a potent inhibitor of telomerase activity in vitro (Redon et al. 2010; Schoeftner and Blasco 2008), and its elimination by nonsense-mediated messenger RNA (mRNA) decay prevents telomere loss. An essential function of TERRA is to displace RPA from telomeric ssDNA following replication, which allows capping structures to re-form and prevents checkpoint activation (Flynn et al. 2011).
R-loops, RNA/DNA hybrids arising at sites of collision between replication and transcription machineries (Bermejo et al. 2012), assemble spontaneously during telomere replication and TERRA transcription in yeast (Pfeiffer et al. 2013). Telomere R-loops accumulate in cells where mRNA processing is compromised, causing toxic replication defects. In mammals, elevated R-loop levels were recently detected in BRCA2-deficient cells (Bhatia et al. 2014). BRCA2 recruitment to RNA/DNA hybrids, mediated by an interaction with the RNA export complex TRX-2, suggests that BRCA2 is required to bypass this type of obstruction during replication. Whether this involves a canonical HR reaction remains unclear. It is also unknown whether R-loops occur during TERRA transcription at mammalian telomeres and whether they require BRCA2 for their resolution. The striking telomere fragility observed in BRCA2-deficient cells (Badie et al. 2010) could be triggered by fork-stalling events proximal to both G-quadruplexes and R-loop structures.
HR-mediated telomere capping and the T-loop model
By concealing the 3′ telomeric overhang via an elaborate DNA secondary structure, T-loops could provide effective protection against activation of DNA damage responses. How telomere repeats become remodelled into T-loops and how the transition between the ‘open’ and ‘T-loop’ configuration (Fig. 2) is regulated remain key questions in telomere biology.
Localization of HR factors at telomeres in mouse and human cells (Badie et al. 2010; Tarsounas et al. 2004; Verdun and Karlseder 2006), together with elevated levels of telomere fusions and telomere dysfunction-induced foci (TIFs) observed in Rad51d −/− (Tarsounas et al. 2004), Brca2 −/− and RAD51-depleted MEFs (Badie et al. 2010) support a role for HR reactions in telomere capping (Tarsounas and West 2005). In vitro D-loop assays using telomeric substrates, human cell extracts and immuno-depletion with specific antibodies (Verdun and Karlseder 2006) have provided additional support for the notion that HR could promote formation of telomere protective structures. In this system, HR activities of RAD52 and XRCC3, together with the ssDNA signalling factors ATR and RPA, as well as shelterin components TRF2 and TIN2 are required for telomeric D-loop formation. In vitro T-loop reconstitution assays using purified proteins will help define the precise molecular requirements and sequence of events leading to telomere capping.
TRF2 is essential for the establishment of telomere capping as evidenced by the striking telomere fusion phenotype of cells lacking this shelterin factor (Celli and de Lange 2005). T-loop assays using TRF2 and telomere DNA substrates demonstrated that TRF2 binding alters DNA topology and stimulates spontaneous strand invasion and D-loop formation (Amiard et al. 2007). Whether this can be further enhanced by RAD51 loading onto the 3′ telomere overhang remains to be determined.
In a speculative model, RAD51 nucleoprotein filaments assemble on the 3′ telomere overhangs and invade the duplex telomeric DNA with formation of a D-loop and a Holliday junction, both well-characterized HR intermediates (Fig. 2). Holliday junctions are postulated to stabilize the T-loop structure, but equally they can pose a threat to telomere capping, as resolution or dissolution mechanisms similar to those acting in HR-mediated DSB repair (Fig. 1) could dismantle the T-loop with potential loss of telomeric DNA. Consistent with this, cleavage of the T-loop Holliday junction by an XRCC3-associated resolvase activity (Liu et al. 2004b) causes loss of telomeric sequences in the form of T-loop-size circles in cells lacking functional TRF2 (Wang et al. 2004). Conversely, functional TRF2 binds Holliday junctions with telomere sequences in vitro and prevents their resolution (Poulet et al. 2009). These observations argue that HR reactions promote telomere capping by facilitating T-loop formation, but equally can resolve these structures when TRF2-mediated protection fails.
To what extent HR contributes to telomere capping, as well as the interplay between HR and shelterin factors in this process, await further investigation. It cannot be excluded, however, that the abrupt telomere shortening in cells lacking HR could lead to shelterin dissociation and therefore an indirect effect on telomere capping. So far, conventional microscopy demonstrated that a subset of cellular telomeres are detectable as TIFs upon BRCA2 or RAD51 inactivation (Badie et al. 2010). These telomeres are therefore uncapped and likely to be rejoined with fusion formation. Replication fork collapse and DNA breakage within HR-depleted telomeres could also lead to loss of capping structures, making it difficult to define the precise mechanism underlying this phenotype.
The T-loop has become central to telomere biology as an elegant model for how the end capping could be achieved. However, the lack of robust methods for T-loop visualization still poses a major drawback to the study of such structures in living cells. Electron microscopy allowed the first visualization of T-loops in crosslinked telomere-enriched DNA isolated from human cells (Griffith et al. 1999). The main caveat of this approach was that crosslinking could stabilize transient folding of telomeric DNA, which would artificially generate loops in these DNA preparations. Also, a distinction could not be made using this technique between loops formed by G-rich overhang invasion into the same telomere or into interstitial telomeric repeats. The recently developed super-resolution fluorescence imaging of T-loops in mouse cells appears more promising (Doksani et al. 2013). Importantly, this approach allowed detection of significantly lower levels of T-loop assembly in TRF2-deleted compared to wild-type cells. Whether this new technology can be routinely used to detect T-loops in vivo remains to be established. Further studies will undoubtedly unravel the mechanisms underlying the transition between the linear telomere state, conducive for telomere replication and elongation, and the protected state which ensures telomere stability during most of the cell cycle.
Future perspectives
Surmounting evidence demonstrates that telomere dysfunction drives genomic alterations that facilitate tumour progression. It is now clear that maintaining telomere integrity is far more complex than originally anticipated and it involves telomere-specific complexes acting in concert with canonical replication, transcription, DNA damage signalling and repair machineries. In this context, significant progress has been made in defining the role of HR at telomeres, yet how HR is integrated with other aspects of telomere maintenance remains poorly understood.
The abrogation of core HR activities of BRCA2 or RAD51 is synthetically lethal with TRF1 deletion (Badie et al. 2010). This supports the concept that HR and shelterin provide independent pathways of telomere replication, promoting either repair of DSBs at collapsed forks within telomeres or resolution of impeding secondary DNA structures (e.g. G-quadruplexes). Importantly, concomitant abrogation of TRF1 and BRCA2 caused cell death, even when p53 function was abrogated. This is clinically relevant, as p53 inactivation occurs frequently in BRCA2-deficient tumours, where it provides a mechanism to sustain proliferation. Thus, interfering with telomere structure, for example, by stabilizing telomeric G-quadruplexes and thus preventing shelterin assembly (Tahara et al. 2006), may provide a basis for therapeutical targeting of HR-deficient tumours.
Mouse mammary tumours lacking Brca2 accumulate TIFs. Additionally, BRCA2-mutated human breast tumours have abnormally short telomeres (Badie et al. 2010). This demonstrates that the genomic instability characteristic of BRCA2-deficient mouse and human tumours is due in part to telomere dysfunction. In normal cells, telomere dysfunction is sufficient to limit proliferation; however, cancer cells develop mechanisms to overcome this barrier. Illegitimate joining of broken chromosome arms and uncapped telomeres (Fig. 4) could lead to inactivation of key checkpoint and tumour suppressor genes and thus facilitate clonal outgrowth of cells lacking HR. Alternatively, amplification of the hTERT locus and the ensuing telomerase activation could provide a mechanism for escape from telomere crisis, restoration of telomere length and cancer cell survival (Jones et al. 2014). Whether this could compensate for telomere shortening in BRCA2-defective tumours is not yet known. Telomere dysfunction detected using single-telomere amplification combined with DNA sequencing technologies provides a powerful prognostic signature for haematological and solid tumours (Lin et al. 2014; Roger et al. 2013), i.e. subpopulations of cells with short telomeres have a poor clinical outcome. Similar approaches could establish whether short dysfunctional telomeres have prognostic value for HR-deficient tumours and could potentially lead to the development of genomic instability markers and/or therapeutic targets for these types of tumours. Thus, understanding how telomeres can drive evolution of HR-compromised cancers could help develop novel prevention and therapeutic modalities.
References
Abreu E, Aritonovska E, Reichenbach P, Cristofari G, Culp B, Terns RM, Lingner J, Terns MP (2010) TIN2-tethered TPP1 recruits human telomerase to telomeres in vivo. Mol Cell Biol 30:2971–2982
Amiard S, Doudeau M, Pinte S, Poulet A, Lenain C, Faivre-Moskalenko C, Angelov D, Hug N, Vindigni A, Bouvet P et al (2007) A topological mechanism for TRF2-enhanced strand invasion. Nat Struct Mol Biol 14:147–154
Azzalin CM, Reichenbach P, Khoriauli L, Giulotto E, Lingner J (2007) Telomeric repeat containing RNA and RNA surveillance factors at mammalian chromosome ends. Science 318:798–801
Badie S, Escandell JM, Bouwman P, Carlos AR, Thanasoula M, Gallardo MM, Suram A, Jaco I, Benitez J, Herbig U et al (2010) BRCA2 acts as RAD51 loader to facilitate telomere replication and capping. Nat Struct Mol Biol 17:1461–1469
Baird DM, Jeffreys AJ, Royle NJ (1995) Mechanisms underlying telomere repeat turnover, revealed by hypervariable variant repeat distribution patterns in the human Xp/Yp telomere. EMBO J 14:5433–5443
Baumann P, Cech TR (2001) Pot1, the putative telomere end-binding protein in fission yeast and humans. Science 292:1171–1175
Bermejo R, Lai MS, Foiani M (2012) Preventing replication stress to maintain genome stability: resolving conflicts between replication and transcription. Mol Cell 45:710–718
Bhatia V, Barroso SI, García-Rubio ML, Tumini E, Herrera-Moyano E, Aguilera A (2014) BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature 511:362–365
Bilaud T, Brun C, Ancelin K, Koering CE, Laroche T, Gilson E (1997) Telomeric localization of TRF2, a novel human telobox protein. Nat Genet 17:236–239
Blackburn EH (1984) Telomeres: do the ends justify the means? Cell 37:7–8
Blasco MA (2007) The epigenetic regulation of mammalian telomeres. Nat Rev Genet 8:299–309
Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, van der Gulden H, Hiddingh S, Thanasoula M, Kulkarni A, Yang Q et al (2010) 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol 17:688–695
Bunting SF, Callén E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L et al (2010) 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 141:243–254
Carlos AR, Escandell JM, Kotsantis P, Suwaki N, Bouwman P, Badie S, Folio C, Benitez J, Gomez-Lopez G, Pisano D et al (2013) ARF triggers senescence in Brca2-deficient cells by altering the spectrum of p53 transcriptional targets. Nat Commun 4:2697
Celli GB, de Lange T (2005) DNA processing is not required for ATM-mediated telomere damage response after TRF2 deletion. Nat Cell Biol 7:712–718
Chapman JR, Barral P, Vannier JB, Borel V, Steger M, Tomas-Loba A, Sartori AA, Adams IR, Batista FD, Boulton SJ (2013) RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol Cell 49:858–871
Chong L, van Steensel B, Broccoli D, Erdjument-Bromage H, Hanish J, Tempst P, de Lange T (1995) A human telomeric protein. Science 270:1663–1667
Ciccia A, Elledge SJ (2010) The DNA damage response: making it safe to play with knives. Mol Cell 40:179–204
Conomos D, Pickett HA, Reddel RR (2013) Alternative lengthening of telomeres: remodeling the telomere architecture. Front Oncol 3:1–7
Conrad MN, Wright JH, Wolf AJ, Zakian VA (1990) RAP1 protein interacts with yeast telomeres in vivo: overproduction alters telomere structure and decreases chromosome stability. Cell 63:739–750
Crabbe L, Verdun RE, Haggblom CI, Karlseder J (2004) Defective telomere lagging strand synthesis in cells lacking WRN helicase activity. Science 306:1951–1953
Denchi EL, de Lange T (2007) Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature 448:1068–1071
Deng Z, Dheekollu J, Broccoli D, Dutta A, Lieberman PM (2007) The origin recognition complex localizes to telomere repeats and prevents telomere-circle formation. Curr Biol 17:1989–1995
Deng Z, Norseen J, Wiedmer A, Riethman H, Lieberman PM (2009) TERRA RNA binding to TRF2 facilitates heterochromatin formation and ORC recruitment at telomeres. Mol Cell 35:403–413
Di Virgilio M, Callen E, Yamane A, Zhang W, Jankovic M, Gitlin AD, Feldhahn N, Resch W, Oliveira TY, Chait BT et al (2013) Rif1 prevents resection of DNA breaks and promotes immunoglobulin class switching. Science 339:711–715
Ding H, Schertzer M, Wu X, Gertsenstein M, Selig S, Kammori M, Pourvali R, Poon S, Vulto I, Chavez E et al (2004) Cell 117:873–886
Doksani Y, Wu JY, de Lange T, Zhuang X (2013) Super-resolution fluorescence imaging of telomeres reveals TRF2-dependent T-loop formation. Cell 155:345–356
Drosopoulos WC, Kosiyatrakul ST, Yan Z, Calderano SG, Schildkraut CL (2012) Human telomeres replicate using chromosome-specific, rather than universal, replication programs. J Cell Biol 197:253–266
Dunham MA, Neumann AA, Fasching CL, Reddel RR (2000) Telomere maintenance by recombination in human cells. Nat Genet 26:447–450
Dunn B, Szauter P, Pardue ML, Szostak JW (1984) Transfer of yeast telomeres to linear plasmids by recombination. Cell 39:191–201
Escribano-Díaz C, Orthwein A, Fradet-Turcotte A, Xing M, Young JT, Tkáč J, Cook MA, Rosebrock AP, Munro M, Canny MD et al (2013) A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol Cell 49:872–883
Fang G, Cech TR (1993) The beta subunit of Oxytricha telomere-binding protein promotes G-quartet formation by telomeric DNA. Cell 74:875–885
Flynn RL, Centore RC, O’Sullivan RJ, Rai R, Tse A (2011) TERRA and hnRNPA1 orchestrate an RPA-to-POT1 switch on telomeric single-stranded DNA. Nature 471:532–536
Fry M, Loeb LA (1999) Human Werner syndrome DNA helicase unwinds tetrahelical structures of the fragile X syndrome repeat sequence d(CGG)n. J Biol Chem 274:12797–12802
Gilson E, Géli V (2007) How telomeres are replicated. Nat Rev Mol Cell Biol 8:825–838
Greider CW (1996) Telomere length regulation. Annu Rev Biochem 65:337–365
Greider CW, Blackburn EH (1985) Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell 43:405–413
Griffith JD, Comeau L, Rosenfield S, Stansel RM, Bianchi A, Moss H, de Lange T (1999) Mammalian telomeres end in a large duplex loop. Cell 97:503–514
Hashimoto Y, Chaudhuri AI, Lopez M, Costanzo V (2010) Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat Struct Mol Biol 17:1305–1311
Hayflick L (1965) The limited in vitro lifetime of human diploid cell strains. Exp Cell Res 37:614–636
Holliday R (1964) A mechanism for gene conversion in fungi. Genet Res 5:282–304
Houghtaling BR, Cuttonaro L, Chang W, Smith S (2004) A dynamic molecular link between the telomere length regulator TRF1 and the chromosome end protector TRF2. Curr Biol 14:1621–1631
Huber MD, Duquette ML, Shiels JC, Maizels N (2006) A conserved G4 DNA binding domain in RecQ family helicases. J Mol Biol 358:1071–1080
Jaco I, Munoz P, Goytisolo F, Wesoly J, Bailey S, Taccioli G, Blasco MA (2003) Role of mammalian Rad54 in telomere length maintenance. Mol Cell Biol 23:5572–5580
Jones RE, Oh S, Grimstead JW, Zimbric J, Roger L, Heppel NH, Ashelford KE, Liddiard K, Hendrickson EA, Baird DM (2014) Escape from telomere-driven crisis Is DNA ligase III dependent. Cell Rep 8:1063–1076
Kahn T, Savitsky M, Georgiev P (2000) Attachment of HeT-A sequences to chromosomal termini in Drosophila melanogaster may occur by different mechanisms. Mol Cell Biol 20:7634–7642
Kappei D, Butter F, Benda C, Scheibe M, Draskovic I, Stevense M, Novo CL, Basquin C, Araki M, Araki K et al (2013) HOT1 is a mammalian direct telomere repeat-binding protein contributing to telomerase recruitment. EMBO J 32:1681–1701
Le S, Moore JK, Haber JE, Greider CW (1999) RAD50 and RAD51 define two pathways that collaborate to maintain telomeres in the absence of telomerase. Genetics 152:143–152
Li BM, Oestreich S, de Lange T (2000) Identification of human RAP1: implications for telomere evolution. Cell 101:471–483
Lin TT, Norris K, Heppel NH, Pratt G, Allan JM, Allsup DJ, Bailey J, Cawkwell L, Hills R, Grimstead JW et al (2014) Telomere dysfunction accurately predicts clinical outcome in chronic lymphocytic leukaemia, even in patients with early stage disease. Br J Haematol. doi:10.1111/bjh.13023
Lipps HJ, Rhodes D (2009) G-quadruplex structures: in vivo evidence and function. Trends Cell Biol 19:414–422
Liu Y, West SC (2004) Happy Hollidays: 40th anniversary of the Holliday junction. Nat Rev Mol Cell Biol 5:937–946
Liu D, Safari A, O’Connor MS, Chan DW, Laegeler A, Qin J, Songyang Z (2004a) PTOP interacts with POT1 and regulates its localization to telomeres. Nat Cell Biol 6:673–680
Liu Y, Masson J-Y, Shah R, O’Regan P, West SC (2004b) RAD51C is required for Holliday junction processing in mammalian cells. Science 303:243–246
Loayza D, de Lange T (2003) POT1 as a terminal transducer of TRF1 telomere length control. Nature 423:1013–1018
Lundblad V, Blackburn EH (1993) An alternative pathway for yeast telomere maintenance rescues est1- senescence. Cell 73:347–360
Martínez P, Blasco MA (2011) Telomeric and extra-telomeric roles for telomerase and the telomere-binding proteins. Nat Rev Cancer 11:161–176
Martinez P, Thanasoula M, Munoz P, Liao C, Tejera A, McNees C, Flores JM, Fernandez-Capetillo O, Tarsounas M, Blasco MA (2009) Increased telomere fragility and fusions resulting from TRF1 deficiency lead to degenerative pathologies and increased cancer in mice. Genes Dev 23:2060–2075
Martinez P, Thanasoula M, Carlos AR, Gómez-López G, Tejera A, Schoeftner S, Dominguez O, Pisano D, Tarsounas M, Blasco MA (2010) Mammalian Rap1 controls telomere function and gene expression through binding to telomeric and extratelomeric sites. Nat Cell Biol 12:768–780
Maser RS, DePinho RA (2002) Connecting chromosomes, crisis, and cancer. Science 297:565–569
McClintock B (1941) The stability of broken ends of chromosomes in Zea mays. Genetics 26:234–282
Mimitou EP, Symington LS (2011) DNA end resection-unraveling the tail. DNA Repair (Amst) 10:344–348
Miyake Y, Nakamura M, Nabetani A, Shimamura S, Tamura M, Yonehara S, Saito M, Ishikawa F (2009) RPA-like mammalian Ctc1-Stn1-Ten1 complex binds to single-stranded DNA and protects telomeres independently of the Pot1 pathway. Mol Cell 36:193–206
Neumann AA, Watson CM, Noble JR, Pickett HA, Tam PP, Reddel RR (2013) Alternative lengthening of telomeres in normal mammalian somatic cells. Genes Dev 27:18–23
Nimonkar AV, Genschel J, Kinoshita E, Polaczek P, Campbell JL, Wyman C, Modrich P, Kowalczykowski SC (2011) BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev 25:350–362
Opresko PL, von Kobbe C, Laine JP, Harrigan J, Hickson ID, Bohr VA (2002) Telomere-binding protein TRF2 binds to and stimulates the Werner and Bloom syndrome helicases. J Biol Chem 277:41110–41119
Opresko PL, Cheng WH, von Kobbe C, Harrigan JA, Bohr VA (2003) Werner syndrome and the function of the Werner protein; what they can teach us about the molecular aging process. Carcinogenesis 24:791–802
Palm W, de Lange T (2008) How shelterin protects mammalian telomeres. Annu Rev Genet 42:16.11–16.34
Pardue ML, DeBaryshe PG (2011) Retrotransposons that maintain chromosome ends. Proc Natl Acad Sci U S A 108:20317–20324
Pellegrini L, Yu DS, Lo T, Anand S, Lee MY, Blundell TL, Venkitaraman AR (2002) Insights into DNA recombination from the structure of a RAD51-BRCA2 complex. Nature 420:287–293
Pfeiffer V, Crittin J, Grolimund L, Lingner J (2013) The THO complex component Thp2 counteracts telomeric R-loops and telomere shortening. EMBO J 32:2861–2871
Pluta AF, Zakian VA (1989) Recombination occurs during telomere formation in yeast. Nature 337:429–433
Poulet A, Buisson R, Faivre-Moskalenko C, Koelblen M, Amiard S, Montel F, Cuesta-Lopez S, Bornet O, Guerlesquin F, Godet T et al (2009) TRF2 promotes, remodels and protects telomeric Holliday junctions. EMBO J 28:641–651
Redon S, Reichenbach P, Lingner J (2010) The non-coding RNA TERRA is a natural ligand and direct inhibitor of human telomerase. Nucleic Acids Res 38:5797–5806
Ribeyre C, Lopes J, Boulé JB, Piazza A, Guédin A, Zakian VA, Mergny JL, Nicolas A (2009) The yeast Pif1 helicase prevents genomic instability caused by G-quadruplex-forming CEB1 sequences in vivo. PLoS Genet 5
Roger L, Jones RE, Heppel NH, Williams GT, Sampson JR, Baird DM (2013) Extensive telomere erosion in the initiation of colorectal adenomas and its association with chromosomal instability. J Natl Cancer Inst 105:1202–1211
Roth CW, Kobeski F, Walter MF, Biessmann H (1997) Chromosome end elongation by recombination in the mosquito Anopheles gambiae. Mol Cell Biol 17:5176–5183
Savitsky M, Kravchuk O, Melnikova L, Georgiev P (2002) Heterochromatin protein 1 is involved in control of telomere elongation in Drosophila melanogaster. Mol Cell Biol 22:3204–3218
Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M (2011) Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145:529–542
Schoeftner S, Blasco MA (2008) Developmentally regulated transcription of mammalian telomeres by DNA-dependent RNA polymerase II. Nat Cell Biol 10:228–236
Schoeftner S, Blasco MA (2009) A ‘higher order’ of telomere regulation: telomere heterochromatin and telomeric RNAs. EMBO J 28:2323–2336
Sfeir A, Kosiyatrakul ST, Hockemeyer D, MacRae SL, Karlseder J, Schildkraut CL, de Lange T (2009) Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell 138:90–103
Sfeir A, Kabir S, van Overbeek M, Celli GB, de Lange T (2010) Loss of Rap1 induces telomere recombination in the absence of NHEJ or a DNA damage signal. Science 327:1657–1661
Smith JS, Chen Q, Yatsunyk LA, Nicoludis JM, Garcia MS, Kranaster R, Balasubramanian S, Monchaud D, Teulade-Fichou MP, Abramowitz L et al (2011) Rudimentary G-quadruplex-based telomere capping in Saccharomyces cerevisiae. Nat Struct Mol Biol 18:478–485
Sun H, Karow JK, Hickson ID, Maizels N (1998) The Bloom’s syndrome helicase unwinds G4 DNA. J Biol Chem 273:27587–27592
Suwaki N, Klare K, Tarsounas M (2011) RAD51 paralogs: roles in DNA damage signalling, recombinational repair and tumorigenesis. Semin Cell Dev Biol 22:898–905
Szostak JW, Blackburn EH (1982) Cloning yeast telomeres on linear plasmid vectors. Cell 29:245–255
Szostak JW, Orr-Weaver TL, Rothstein RJ, Stahl FW (1983) The double-strand-break repair model for recombination. Cell 33:25–35
Tahara H, Shin-Ya K, Seimiya H, Yamada H, Tsuruo T, Ide T (2006) G-Quadruplex stabilization by telomestatin induces TRF2 protein dissociation from telomeres and anaphase bridge formation accompanied by loss of the 3′ telomeric overhang in cancer cells. Oncogene 25:1955–1966
Takai KK, Kibe T, Donigian JR, Frescas D, de Lange T (2011) Telomere protection by TPP1/POT1 requires tethering to TIN2. Mol Cell 44:647–659
Tarsounas M, Tijsterman M (2013) Genomes and G-quadruplexes: for better or for worse. J Mol Biol 425:4782–4789
Tarsounas M, West SC (2005) Recombination at mammalian telomeres: an alternative mechanism for telomere protection and elongation. Cell Cycle 4:672–674
Tarsounas M, Muñoz P, Claas A, Smiraldo PG, Pittman DL, Blasco MA, West SC (2004) Telomere maintenance requires the RAD51D recombination/repair protein. Cell 117:337–347
Teixeira MT, Arneric M, Sperisen P, Lingner J (2004) Telomere length homeostasis is achieved via a switch between telomerase-extendible and -nonextendible states. Cell 117:323–335
Tejera A, Stagno d’Alcontres M, Thanasoula M, Martinez P, Liao C, Tarsounas M, Blasco MA (2010) TPP1 is required for TERT recruitment, telomere elongation during nuclear reprogramming, and normal skin development in mice. Dev Cell 18:775–789
Teo H, Ghosh S, Luesch H, Ghosh A, Wong ET, Malik N, Orth A, de Jesus P, Perry AS, Oliver JD et al (2010) Telomere-independent Rap1 is an IKK adaptor and regulates NF-kappaB-dependent gene expression. Nat Cell Biol 12:758–767
van Steensel B, Smogorzewska A, de Lange T (1998) TRF2 protects human telomeres from end-to-end fusions. Cell 92:401–413
Vannier JB, Pavicic-Kaltenbrunner V, Petalcorin MI, Ding H, Boulton SJ (2012) RTEL1 dismantles T loops and counteracts telomeric G4-DNA to maintain telomere integrity. Cell 149:795–806
Vannier J-B, Sandhu S, Petalcorin MI, Wu X, Nabi Z, Ding H, Boulton SJ (2013) RTEL1 is a replisome-associated helicase that promotes telomere and genome-wide replication. Science 342:239–242
Verdun RE, Karlseder J (2006) The DNA damage machinery and homologous recombination pathway act consecutively to protect human telomeres. Cell 127:709–720
Wan M, Qin J, Songyang Z, Liu D (2009) OB fold-containing protein 1 (OBFC1), a human homolog of yeast Stn1, associates with TPP1 and is implicated in telomere length regulation. J Biol Chem 284:26725–26731
Wang S-S, Zakian VA (1990) Telomere-telomere recombination provides an express pathway for telomere elongation. Nature 345:456–458
Wang RC, Smogorzewska A, de Lange T (2004) Homologous recombination generates T-loop-sized deletions at human telomeres. Cell 119:355–368
Watson JD, Crick FHC (1953) Genetical implications of the structure of deoxyribonucleic acid. Nature 171:964
West SC (2009) The search for a human Holliday junction resolvase. Biochem Soc Trans 37:519–526
Wu L, Multani AS, He H, Cosme-Blanco W, Deng Y, Deng JM, Bachilo O, Pathak S, Tahara H, Bailey SM et al (2006) Pot1 deficiency initiates DNA damage checkpoint activation and aberrant homologous recombination at telomeres. Cell 126:49–62
Wu P, Takai H, de Lange T (2012) Telomeric 3′ overhangs derive from resection by Exo1 and Apollo and fill-in by POT1b-associated CST. Cell 150:39–52
Ye JZ, de Lange T (2004) TIN2 is a tankyrase 1 PARP modulator in the TRF1 telomere length control complex. Nat Genet 36:618–623
Ye JZ, Hockemeyer D, Krutchinsky AN, Loayza D, Hooper SM, Chait BT, de Lange T (2004) POT1-interacting protein PIP1: a telomere length regulator that recruits POT1 to the TIN2/TRF1 complex. Genes Dev 18:1649–1654
Ye J, Lenain C, Bauwens S, Rizzo A, Saint-Léger A, Poulet A, Benarroch D, Magdinier F, Morere J, Amiard S et al (2010) TRF2 and apollo cooperate with topoisomerase 2alpha to protect human telomeres from replicative damage. Cell 142:230–242
Zimmermann M, Lottersberger F, Buonomo SB, Sfeir A, de Lange T (2013) 53BP1 regulates DSB repair using Rif1 to control 5′ end resection. Science 339:700–704
Acknowledgments
EMCT is funded by a Medical Research Council PhD Studentship. Work in MT laboratory is supported by Cancer Research UK, EMBO Young Investigator Award and The Royal Society.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Tacconi, E.M.C., Tarsounas, M. How homologous recombination maintains telomere integrity. Chromosoma 124, 119–130 (2015). https://doi.org/10.1007/s00412-014-0497-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00412-014-0497-2