
Abstract
The mobility of trace elements in supercritical fluid-melt derived from pelite rich in volatiles has been studied experimentally at pressures from 3.0 to 7.8 GPa and temperatures from 750 to 1090 °С using the diamond trap method. The experiments simulate the conditions of warm and hot subduction, in which pelite either retains the whole inventory of volatiles or releases a fluid in three successive devolatilization steps. The 3.0 GPa and 750 °С runs with pelite rich in volatiles yield a supercritical fluid (SCF) which attains equilibrium with an eclogitic residue bearing phengite and accessory rutile, zircon, and monazite. At ≥5.5 GPa and ≥850 °С, above the second critical endpoint, the SCF transforms into a supercritical fluid-melt (SCFM) which acquires higher concentrations of almost all incompatible trace elements while the mineral assemblage of the equilibrium eclogitic residue remains the same but lacks monazite. The trace-element enrichment of SCFM is most prominent for Ba, Sr, LREE, Th, and U. At the hot subduction conditions, the fluid-melt likewise contains more K, Rb, Zr, and Hf, though LREE contents become lower. The negative Nb anomaly persists in all cases. SCFM has its trace-element composition generally similar to that of hydrous melt derived from oceanic sediments, but contains more REEs and water. Partitioning of LILE, HFSE, and LREE between the SCFM and residue phases mainly depends on the fluid-melt fraction and stability of host phengite, monazite, zircon, and rutile. Thus, sediment-derived SCFM can carry both fluid-mobile and sediment-melt elements to regions of arc- and back-arc magma generation and can translate the negative Nb anomaly inherited from sediment into the magmas. Early devolatilization of pelite increases the stability of monazite and phengite in the residue and provides efficient LREE, K and Rb transport to the mantle depths of ~ 250 km. Effective LREE and Th depletion of UHP metamorphic rocks is possible by SCFM release near peak metamorphic conditions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Fluids and/or melts formed in subduction zones have been traditionally considered as efficient carriers of elements from slabs to the mantle, which makes them key agents in mantle metasomatism and magma generation (Elliott 2003; Hermann et al. 2013; O’Reilly and Griffin 2013; Keppler 2017; Turner and Langmuir 2022). The flux from beneath arcs in subduction zones can be either a diluted aqueous fluid (supercritical) containing > 85 wt% H2O if temperatures are relatively low or a hydrous melt with < 35 wt% H2O at higher temperatures (Kessel et al. 2005a; Hermann and Spandler 2008; Hermann et al. 2013). Aqueous fluids poor in solutes but enriched in fluid-mobile elements (Rb, Ba, U, and Pb) presumably form by dehydration of altered oceanic crust (AOC) (Brenan et al. 1995; Elliott et al. 1997; Ayers 1998), whereas melting of sediments produces solute-rich hydrous melts carrying sediment-melt elements, such as Sr, LREE, and Th (Elliott 2003; Li et al. 2022; Turner and Langmuir 2022). However, the relative contributions of sediment melt and aqueous fluid released by AOC dehydration to the generation of arc- and back-arc basalts remain poorly constrained and open to discussions (Elliott et al. 1997; Ayers et al. 1997; Ayers 1998; Rustioni et al. 2019; 2021; Li et al. 2022; Turner and Langmuir 2022). It is often assumed that supercritical fluid-melt (SCFM) hardly can be involved in the generation of arc magma given that the second critical endpoint pressure for sediments and MORB is from 3.5 to 5.5 GPa (Kessel et al. 2005b; Hermann and Spandler 2008; Hermann et al. 2013; Ni et al. 2017). In this study, supercritical fluid (SCF), called also ‘fluid’ or ‘aqueous fluid’ herebelow, refers to a mobile phase formed above the first critical endpoint (L + V = SCF); correspondingly, the mobile phase formed above the second critical endpoint (SCF + Melt = SCFM) is referred to as supercritical fluid-melt (SCFM).
Importantly, the flux from subducting slabs changes notably from solute-poor aqueous fluid at arc depths (beneath the volcanic front) to solute-rich hydrous melt or SCFM compositions at rear-arc pressures (Pearce and Stern 2006; Portnyagin et al., 2007). The flux from subducted sediments and/or altered oceanic crust at depths ≥ 120 km mainly carries large-ion lithophile elements (LILE), light rare earths (LREE), Th, and U (Portnyagin et al. 2007; Spandler and Pirard 2013). Ultrahigh-pressure (UHP) rocks potentially represent the residue left after the removal of fluids and elements at 100–250 km depths in subduction zones, including continental settings (Shatsky et al. 1999, 2006, 2015; Bebout 2014; Hermann and Rubatto 2014; Zheng and Hermann 2014). The UHP rocks include diamond-bearing garnet-biotite gneiss, such as the garnet-pyroxene-quartz gneiss of the Kokchetav UHP metamorphic complex depleted in LREE and Th (Shatsky et al. 1999, 2006, 2015; Stepanov et al. 2014). On the other hand, products of anatectic melting in UHP rocks and multiphase solid inclusions in UHP minerals show significant LILE and LREE enrichment but are depleted in Nb, Ta, and HREE (Stepanov et al. 2014; Zheng and Hermann 2014). In general, the flux in rear-arc and back-arc magmas, products of anatectic melting in UHP rocks, and multiphase solid inclusions in UHP minerals share much similarity in trace-element patterns.
Experimental work is indispensable to reconstruct the properties of SCF, SCFM, and hydrous melts, and to detect the trace element signatures of subduction-zone flux. Earlier experiments at 2.5–7.5 GPa and 750–1120 °C (Schmidt et al. 2004; Hermann and Spandler 2008; Hermann and Rubatto 2009; Skora et al. 2015) constrained phase relations in deeply subducted sediments and the mobility of trace elements in hydrous melts. The results showed that the trace-element signatures of sediment melts in subduction zones were largely controlled by accessory phases (Hermann and Rubatto 2009). The role of supercritical fluid-melt in trace-element transport was reported to increase markedly at conditions common to the modern subduction zones at depths > 100–150 km (Schmidt et al. 2004; Kessel et al. 2005a, b; Hermann and Spandler 2008; Hermann and Rubatto 2009). Experiments with K-free MORB at 4.0–6.0 GPa and 800–1000 °С (Kessel et al. 2005a) provided the first evidence for the capability of SCFM to be efficient agents in transport of LILE, LREE, Th, and U. Later studies, likewise with K-free MORB (Rustioni et al. 2019, 2021), showed that Cl-rich aqueous fluids with a total solute content close to SCFM also can transport LILE, LREE, and U at 4.0–6.0 GPa and 700–800 °C.
Meanwhile, even the extensive available experimental data leave knowledge gaps as to the formation conditions of SCFM from slab sediment, as well as to the patterns of trace-element partitioning between SCFM and eclogite-like residue that contains stable mineral carriers of LILE, LREE, and HFSE. To bridge the gaps, we study the mobility of trace elements in fluids and eclogite-like residue derived from carbonate- and chlorine-bearing pelitic material and constrain the pressure- and temperature-dependent fluid-residue partitioning of LILE, HFSE, and REE by means of high-pressure (3.0-7.8 GPa) high-temperature (750–1090 °C) experiments with diamond traps. The new experimental data were used for (i) reconstructing the conditions of SCFM generation in subduction zones and (ii) picking the geochemical signatures that trace the role of SCFMs in the generation of mantle melts. The new results complement the previously reconstructed major-element compositions of fluids derived from the same pelite at the same P-T parameters (Sokol et al. 2023a, b).
Materials and methods
Materials
The procedures for sample preparation and experimental set up were described in detail in our recent publication (Sokol et al. 2023a). The experiments were performed by the diamond-trap method (Ryabchikov et al. 1989; Johnson and Plank 1999; Kessel et al. 2004, 2005a, b; Hermann and Spandler 2008) using pelitic marine sediment of the Middle Maykop Shale (Sokol et al. 2018) as starting material (Table 1). Pelite contained 8.6 wt% H2O and 2.0 wt% CO2, as well as authigenic pyrite, 0.7 wt% kerogen (Petrichenko 2000), and accessory rutile, alanite, monazite, and zircon as carriers of high field strength elements (HFSE) and rare earths (REE). Its trace-element contents (Table 1) differed only slightly from those in average global oceanic subducting sediment (GLOSS-II), oceanic sediments from Tonga, Marianas, and Cascadia (Plank and Langmuir 1998; Plank 2014).
The experimental work included two series of diamond-trap experiments with different types of pelite: (i) rich in volatiles and (ii) devolatilized in several successive steps. In the first set of experiments, which was performed previously (Sokol et al. 2023a, b), platinum or gold capsules of 6–10 mm outer diameter and 0.2 mm wall thickness were charged with compressed discs of pelitic material above a layer of 14–20 μm ASM synthetic diamond powder (about 50 wt% of pelite). The pelitic samples and the diamond traps weighed from 70 to 500 mg and from 34 to 260 mg, respectively (Table 2). The amounts of pelite and fluid were large enough to yield a sufficient quantity of devolatilized residue to be used in further experiments and in analytical work (ICP-MS, EDS-SEM, and EMPA). In some cases, a small Au capsule (2 mm outer diameter and 0.2 mm wall thickness) was placed additionally as a control into the cell with 16–20 mg pelite, in the absence of a diamond trap. The prepared capsules were welded shut using a Lampert PUK 4U high-frequency arc welder and placed into a high-pressure cell upside down, with the diamond trap on the top. Most of residue and trap material for ICP-MS analyses (Table 2) was taken from previous experiments (Sokol et al.2023a).
In the second set of experiments (this study) the type of capsules and the method of their preparation were similar to those in the experiments with volatile-rich pelite (see Fig. S1 for the workflow sketch). Pelite was devolatilized under conditions simulating intermediate (warm) subduction in three successive steps: 3.0 GPa and 750 °С; 5.5 GPa and 850 °С; 7.8 GPa and 940 °С. The residue partly devolatilized at 3.0 GPa (run 1088_4_1) and dried at 100 °С for 24 h was then used in 5.5 GPa runs, with a new trap (Table 2, Fig. S1). At the following step, the residue devolatilized at 3.0 GPa and at 5.5 GPa was used in 7.8 GPa runs, again with a new trap. Meanwhile, stepwise devolatilization was impossible at hot subduction temperatures because the melt fraction reached 0.5 at 3.0 GPa and 900 °С (Sokol et al. 2023a).
High-pressure experiments
Experiments at 3.0, 5.5, and 7.8 GPa were carried out in a split-sphere multi-anvil high-pressure apparatus (Palyanov et al. 2017). The sizes of the high-pressure cell were 21.1 × 21.1 × 25.4 mm and 19 × 19 × 22 mm, respectively, in the 3.0-5.5 GPa and 7.8 GPa runs. The sizes of graphite heaters were 12.2 mm (inner diameters) and 18.5 mm (heights) in the 3.0-6.3 GPa runs, and 9.2 × 14.8 mm, respectively, in the 7.8 GPa experiments. Pressure was calibrated by recording the change in the resistance of Bi at 2.55 GPa and PbSe at 4.0 and 6.8 GPa at room temperature and by bracketing the quartz-coesite and the graphite-diamond equilibrium at 3.0, 5.5, and 7.8 GPa and high temperatures. Temperature was monitored in each experiment using a PtRh6/PtRh30 thermocouple calibrated at 6.3 GPa and 7.8 GPa using the melting points of Al, Ag, Ni and Pt. Pressure and temperature were measured to an accuracy of ± 0.1 GPa and ± 20 °C (Palyanov et al. 2017; Sokol et al. 2015). The charged capsules were quenched under isobaric conditions at a rate of 150–200 deg/s. The duration of runs with devolatilized pelite was extended from 40 to 150 h.
Although the initial pelite samples contained 2.6 wt% Fe2O3 (Table 1), the presence of the diamond trap buffered pelite fO2 about the CCO equilibrium. For the experimental P-T ranges, the CCO equilibrium imposed oxygen fugacity from (Ni-NiO) NNO + 0.46 to NNO + 1.0 log unit (Sokol et al. 2023a).
Opening of capsules and preparation of diamond trap and pelite residue for analysis
Currently, partitioning of major and trace elements between a mobile phase (melt, SCF, or SCFM) in the diamond trap and the residue has been studied using two main approaches. One approach was applied by Johnson and Plank (1999) who analyzed entrapped fluid solute and residue compositions by ICP-MS or ICP-AES following acid digestion, with mass-balance calculations applied to correct element concentrations for the dilution caused by diamond. The other approach was suggested in order to avoid losses of dissolved salts and dispersed quench phases with water leaked upon capsule opening (Kessel et al. 2005a, b). In the latter approach, the capsules are frozen with liquid nitrogen and kept frozen during opening and subsequent LA-ICP-MS analysis. The analyses of diamond trap are averaged using an ablation spot track. The absolute concentrations of elements and water content in the fluid phase (SCF or SCFM) are calculated with reference to highly incompatible Cs as an internal standard. Trace-element contents in the eclogitic phases are determined by LA-ICP-MS in experiments without a diamond trap. The bulk compositions of residue are calculated using the known fractions of minerals.
The experiments with both volatile-rich and stepwise devolatilized pelite in this study were performed with the former approach, which we modified slightly (Sokol et al. 2023a), using large capsules. The use of the approach proposed by Kessel et al. (2005a, b) was problematic in this work because it does not allow reconstructing the trace element composition of residue with accessory monazite, zircon, and rutile. In this respect, the modified approach of Johnson and Plank (1999) is advantageous as it provides (i) direct weighing of H2O + CO2 and solutes in the fluid or fluid-melt phase, (ii) high accuracy of trace-element concentrations analyzed by ICP-MS in solutions, due to a sufficient amount of fluid solute in the 20–50 mg diamond traps used for acid digestion, and (iii) direct determination of bulk element contents in the residue with phengite, monazite, rutile, and zircon. Note that an improved capsule opening procedure and subsequent drying at 100 °С minimized the loss of fluid solute.
After the experiments, the capsules were weighed, pierced, dried for 24 h at 100 °C when the release of gases and liquids had stopped, and weighed again (see Fig. S1 in Sokol et al. (2023a) for the workflow sketch). The difference measured the amount of liquid and gases in the capsule, mainly H2O and CO2, removed during fluid quenching. The procedures for estimating the amount of solutes in the diamond trap and reconstructing the bulk fluid composition were the same as in our previous study (Table S1 in Sokol et al. (2023a).
Energy dispersive spectroscopy (EDS) and electron probe microanalysis (EPMA)
The run products were cleaned and mounted in epoxy resin. After resin polymerization, the samples were polished under kerosene, without the use of water (final stage 1 μm Al2O3) and then examined on a Tescan MIRA 3 LMU scanning electron microscope (SEM). The solid phase compositions were analyzed using the Tescan MIRA 3 LMU SEM coupled with an INCA EDS 450 microanalysis system with an Oxford Instruments liquid nitrogen-free Large area EDS X-Max-80 Silicon Drift Detector. The instruments were operated at an accelerating voltage of 20 keV, a beam current of 1 nA, and a spot diameter of ~ 10 nm; the count time for spectra collection was 20 s. The EDS spectra were optimized for quantification using the standard XPP procedure built into the INCA Energy 450 software.
Element abundances were determined by electron probe microanalysis (EPMA) on a Jeol JXA-8100 microanalyzer at an accelerating voltage of 20 kV, a beam current of 30 nA, and beam diameter of 1 to 2 μm, in samples sputter-coated with 20 nm carbon. The data quality was checked against standards of pyrope for Si, Al, and Fe; diopside for Mg and Ca; albite for Na; orthoclase for K; ilmenite for Ti and Ba-glass for Ba. The uncertainty of the measurements was within 2 rel%.
Solution-based inductively coupled plasma mass spectrometry (ICP-MS)
The sediment residue and diamond trap material samples were analyzed by ICP-MS with acid digestion. Bulk samples (10–50 mg) were weighed in Savillex Teflon beakers and dissolved completely in a tri-acid mixture of HF ⁄HNO3⁄HClO4 (Ionov and Harmer 2002). The samples were dried down, redissolved in 3 N HCl, dried again, redissolved in 4 N HNO3, diluted with distilled-deionized water, and sonicated as in (Johnson and Plank 1999). The final dilution of 1/1000 (diamond traps) to 1/5000 (sediment residue) in ~ 2% HNO3 was further applied before measurements. The solution aliquots were analyzed for trace elements (Ba, Rb, Sr, Zr, Hf, Nb, Ta, Th, U, P, and REE) using a quadrupole Perkin-Elmer NexION 300D and a magnet-sector Thermo Finnigan Element 2 high-resolution mass spectrometers at the Center of Isotope and Geochemical Research (Vinogradov Institute of Geochemistry, Irkutsk), and a Perkin-Elmer NexION 300S mass spectrometer at the Institute of Geology and Geochemistry (Ekaterinburg). The analytical reproducibility was monitored using the BCR-2 and BHVO-2 standards. The detection limits for trace elements were in a range of 0.01–0.2 µg/l. The estimated precision and accuracy were within 5–10 rel%.
Results
Experimental and analytical data
The mobility of trace elements in supercritical fluid-melt derived from carbonate- and chlorine-bearing pelite was investigated in diamond-trap experiments performed previously (Sokol et al. 2023a, b) and in this study. The quantity of samples was sufficient to prepare solutions for both ICP-AES and ICP-MS analytical procedures. The previous work (Sokol et al. 2023a, b) included only ICP-AES analyses, while this study has provided ICP-MS data for the same samples (Table 2, S2, S3). The new experiments were performed with stepwise devolatilization of pelite (Table 2, Fig. S1).
Phase relations
In the previous publications (Sokol et al. 2023a, b), we considered phase relations in samples obtained in the first set of experiments with starting pelite rich in volatiles. The run products included an eclogitic residue composed of garnet, coesite, ± phengite, clinopyroxene, and kyanite and accessory pyrite or pyrrhotite, ± monazite, ± carbonate, rutile, and zircon (Table 2), and a pelite-derived mobile phase (SCF or SCFM). The carbonate and monazite phases present in the starting pelite were partly dissolved in SCF at 3.0 GPa, 750–900 °C (Sokol et al. 2023b), and completely dissolved in SCFM in the 5.5 GPa, 850–1030 °C and 7.8 GPa, 940–1090 °C runs (Sokol et al. 2023a).
The runs at 3.0 GPa and 750 °C, near the second critical endpoint (2.CP), yielded a SCF mobile phase consisting of nearly equal amounts of solutes and H2O + CO2 (Sokol et al. 2023b). The products of the 3.0 GPa and 900°С run comprised up to 50 wt% of granitic melt (Sokol et al. 2023b) resulting from the peritectic reaction Phe + Coe + Cpx = Grt + L (Schmidt et al. 2004; Hermann and Spandler 2008; Schmidt and Poli 2014; Perchuk et al. 2020). Pelitic samples in the 5.5 and 7.8 GPa runs showed signatures of closed solvus between an aqueous fluid (SCF) and a hydrous silicate melt (Sokol et al. 2023a). The observed solvus closure signatures, composition trends, volatile/solute ratio, along with prominent morphological distinction between the quenched hydrous melt captured in the diamond trap (Hermann and Spandler 2008) and the quenched pelite-derived mobile phase (see Fig. 1e-i in Sokol et al. (2023) indicate that the pelite-derived mobile phase at 5.5–7.8 GPa and 850–1090 °C must be supercritical fluid-melt (SCFM). This inference agrees with the available data on the second critical endpoint pressure for sediments and MORB (Schmidt et al. 2004; Kessel et al. 2005a, b; Hermann and Spandler 2008). The pelite-derived SCFM contained 37–40 wt% H2O, 5–8 wt% CO2, and 0.4–0.5 wt% Cl on average (Sokol et al. 2023a).

Scanning electron micrographs of pelite residue samples. a: after 40-h run # 2239_2_1 (5.5 GPa and 850 °C); b: after 150-h run # 2304_2_1 (5.5 GPa and 850 °C); c: after 150-h run # 2306_2_4 (7.8 GPa and 940 °C). Symbols for phases are as in Table 2
Devolatilized pelite
The residue aggregates obtained in the second set of experiments with devolatilized pelite had a lower porosity than their counterparts in the case of the starting pelite (Sokol et al. 2023a). The content of volatiles in the residue, which was measured as difference between the initial amount and the amount released from pierced capsules, reduced successively: 4.2 wt% H2O + CO2 after the first devolatilization step at 3.0 GPa and 750 °С (Table 2); 2.7 wt% H2O + CO2 after the second step at 5.5 GPa and 850 °С (Fig. 1a, b); and 1.7 wt% H2O + CO2 at 7.8 GPa and 940 °С (Fig. 1c). Besides the volatiles, the residue lost solutes removed together with the trap, which was replaced by a new empty trap at each step (Fig. S1). The fraction of H2O + CO2 in the fluid/melt ranged from 44 wt% in the 3.0 GPa, 750 °С run to 21–24 wt% in the 5.5 GPa, 850 °С run with a devolatilized residue (Table 2).
The two-fold reduction of H2O + CO2 contents after the first devolatilization step led to the corresponding reduction of the fluid phase fraction in the products of the 5.5 GPa and 850 °С run. The devolatilized residue obtained in 40 h runs consisted of the same solids (garnet, coesite, phengite, clinopyroxene, kyanite, and pyrite) (Table 2; Fig. 1a) as that of the 5.5 GPa and 850 °С runs with pelite rich in volatiles (Sokol et al. 2023a), but contained visibly larger amounts of accessory zircon and rutile, as well as Mg-Fe carbonate and monazite. The solids appeared as ≤ 30 μm subhedral to euhedral garnet, up to 10 μm anhedral to subhedral phengitic muscovite (Fig. 2c), ≤ 20 μm anhedral coesite, up to 20 μm mainly subhedral or often isometric clinopyroxene, and pyrite grains occasionally exceeding 10 μm.

Mineral chemistry of garnet, clinopyroxene, and phengite obtained in 40-h runs with initial volatile-rich pelite (Sokol et al. 2023a) and in runs with devolatilized pelite (this study). a: garnet compositions plotted in molecular proportions of grossular (Grs), almandine (Alm), and pyrope (Prp). Contour lines are after Hermann and Spandler (2008). b: Na contents in clinopyroxene as a function of temperature and pressure. Compositions of clinopyroxene obtained at 3.5 and 4.5 GPa in the GLOSS system with higher CaO content (Hermann and Spandler 2008) are given for comparison. c: Si + Mg contents as a function of Al in phengite. Compositions of phengite synthesized at 4.5 GPa and 800–1000 °C in the GLOSS system (Hermann and Spandler 2008) are given for comparison
Longer runs (150 h at 5.5 GPa and 850 °С) yielded coarser solids: up to 50 μm coesite and clinopyroxene and ≤ 20 μm phengite grains (Table 2; Fig. 1b). The grains of accessory rutile and monazite were likewise slightly larger, while carbonates reduced to 5 μm. The final 150 h long 7.8 GPa and 940 °С run, in the absence of a trap, yielded a residue with a markedly smaller amount of fluid and finer grains in all solids except garnet (Fig. 1c). The eclogitic assemblage lacked kyanite and carbonate but included garnet, coesite, phengite, clinopyroxene, and all accessories (rutile, zirсon, and monazite).
Composition of solids
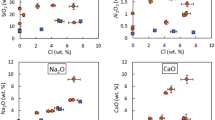
The composition of residue solids obtained in the first set of experiments with volatile-rich pelite was described previously (Table S4 in Sokol et al. (2023b). The residue included almandine-pyrope garnet, with the pyrope percentage increasing from 23 to 29% at 5.5 GPa and 850 °C to 40–47% at 7.8 GPa and 1090 °C (Fig. 2a), and clinopyroxene bearing 71–78% jadeite (Fig. 2b). The percentage of celadonite in phengite was quite high in the 5.5 GPa, 850–1030 °C runs (Fig. 2c) but lower at higher temperatures, whereby (Si + Mg) decreased from 4.0–4.2 to 3.9-4.0 apfu and Al increased from 1.6–1.8 to 1.8–1.9 apfu. The composition trends of garnet, clinopyroxene, and phengite derived from volatile-rich pelite at 5.5–7.8 GPa and 850–1090 °C extend the high-pressure branch of the trends reported previously (Hermann and Spandler 2008) for phases synthesized in the GLOSS system (Fig. 2).
The compositions of solids from the second set of experiments had some specific features. Garnet obtained in the 40-hr 5.5 GPa, 850 °С runs after the second devolatilization step had higher percentages of almandine and lesser amounts of pyrope than in the case of pelite rich in volatiles at the same P-T parameters, but its composition was similar to that formed at 3.0 GPa and 750 °С (Table S1, Fig. 2a) (Sokol et al. 2023a). Clinopyroxene obtained in these 40-hr runs was similar to that of the 3.5 GPa run in Na contents (Fig. 2b), while phengite plotted in a wide strip in the Si + Mg vs. Al (pfu) diagram, with overlapped compositions at 3.0 GPa, 750°С and 5.5 GPa, 850 °С (Fig. 2c).
The 150 h runs at 5.5 GPa and 850 °С, after the second devolatilization step, yielded garnet, clinopyroxene, and phengite compositions shifted to the levels of the respective phases derived from volatile-rich pelite of the first set experiments (Table 2, S1, Fig. 2). Garnet produced in the 150 h run at 7.8 GPa and 940 °С did not differ much from that after the second step but failed to approach the composition obtained with pelite rich in volatiles. At the same time, clinopyroxene in the residue after the final devolatilization step became almost identical to that derived from volatile-rich pelite. 2 Phengite obtained in the system with devolatlized pelite at 7.8 GPa and 940 °С comprised quite a high percentage of the celadonite component. Moreover, the fraction of phengite was notably larger than that in the runs with volatile-rich pelite.
The accessories, with their compositions estimated by SEM-EDS, were:
-
sulfides appeared as pyrite at 5.5 GPa and 850 °C, as well as at 7.8 GPa and 940 °C, with a Fe/S atomic ratio of ∼ 0.5, but as pyrrhotite (Fe/S atomic ratio ~ 0.9) in higher- temperature runs of 5.5 GPa and 1030 °C and 7.8 GPa and 1090 °C. Both phases contained up to 0.5 at% Ni;
-
kyanite was Fe-bearing with up to 0.8 wt% FeO;
-
rutile with 1.8–2.3 wt% Al2O3, 0.8–1.2 wt% FeO and 0.6–0.9 wt% V2O5;
-
zircon with Hf (1.5–2.2 wt% HfO2) and FeO (0.5–0.6 wt%) as main impurities;
-
carbonate as a Fe–Mg solid solution with FeO/MgO ≈ 1, CaO = 1–2 wt%, and MnO < 1 wt%.
Monazite was Th- and Si-bearing (2.5–6.6 wt% ThO2 and up to 3 wt% SiO2) and thus should be attributed to the [LREEPO4] – [ThSiO4] (huttonite) solid-solution series. REE oxides in monazite varied as 31–34 wt% Ce2O3, 14–23 wt% La2O3, 6.2–12 wt% Nd2O3, 2.4–3.5 wt% Pr2O3, and 2.5–6.7 wt% ThO2. The monazite grains were often too small (≤ 5–7 μm) to reveal systematic SiO2 variations. The concentrations of other oxides at different devolatilization steps and P-T conditions varied as shown in Fig S2. Monazite acquired slightly higher La2O3 but lower Nd2O3, Pr2O3, and ThO2 during devolatilization.
Trace-element compositions of supercritical fluid-melt and residue
Trace element contents in SCF and SCFM equilibrated with the eclogitic residue are listed in Table 3 and S3, and the patterns normalized to the starting composition or chondrite (McDonough and Sun 1995) are plotted in Figs. 3 and 4. As it was shown previously (Sokol et al. 2023a, b), the runs at 3.0 GPa and 750 °С with the CO2- and Cl-bearing pelite yielded a solute-rich SCF near 2.CP and a SCFM at ≥5.5 GPa and ≥850 °С along the warm subduction P-T path. The composition of SCFM obtained at 5.5 GPa and 850 °С differed from SCF in notably greater enrichment with almost all incompatible trace elements (Table 3, S3, Figs. 3 and 4a). The elemental gain was the smallest for Sr and HFSE (Ti, Zr, Hf) and the largest for LILE, Th, U, and REE. As the P-T parameters increased further to 7.8 GPa and 1090 °C, the content of Rb and K in the SCFM increased, but LREE decreased. The SCFM obtained upon the change from warm to hot subduction temperatures (1030 °C at 5.5 and 1090 °C at 7.8 GPa) had a greater fraction and was more depleted in Li, Sr, Th, and LREE at greater Zr and Hf enrichment. The REE changes in the residue (Table S2, Fig. 3) relative to the starting composition consisted in systematic LREE depletion at enrichment in other REEs. REE abundances were the lowest in the SCF obtained at 3.0 GPa and 750 °C and the highest in the SCFM at 5.5 GPa and 850 °С. Compared to the residue composition, the SCF was enriched only in LREE, while the SCFM was depleted only in HREE.

Chondrite-normalized (McDonough and Sun 1995) REE abundances in starting pelite, SCF, SCFM, and eclogitic residue (± monazite). Here and throughout, the concentrations of elements in SCF and SCFM are quoted on hydrous basis
The concentrations of key minor and trace elements in subduction-zone fluids were shown previously (Klimm et al. 2008; Hermann and Rubatto 2009) to be controlled by accessory phases. Some minor and trace elements in pelite, within the applied P-T range, turned out to be main constituents of phengite (LILE), monazite (LREE), rutile (Nb and Ta), and zircon (Zr and Hf) in the residue. Thus, we infer that SCFM acquired higher LILE and LREE contents due to complete dissolution of monazite and phengite (at 5.5 and 7.8 GPa, respectively), while its concentrations of Nb and Ta, as well as Zr and Hf, depend on trace-element fractionation patterns in the presence of rutile and zircon which are stable at all applied pressures and temperatures.
The contents of Th, U, LREE, HREE, and Y in the pelite-derived SCFM at 5.5 GPa and 850–1030 °C (determined on hydrous basis and normalized to the initial composition) are only slightly higher than in the hydrous melt obtained at 4.5 GPa and 800–1050 °С in the GLOSS system (Fig. 4a) (Hermann and Rubatto 2009). Compared to hydrous melt at similar temperatures, the SCFM from the 7.8 GPa and 940–1090 °С run had higher K and Rb, similar Nb, and lower Ta concentrations. The higher HREE concentrations in SCFM may be due to lower CaO contents in pelite than in GLOSS and to the related lower percentage of grossular in garnet, main HREE carrier. However, this hypothesis is inconsistent with moderately variable garnet-melt HREE partitioning at 3.0 GPa and 1530–1565 °C reported by Westrenen et al. (1999).
The composition of SCFM (normalized to its concentration in the bulk system) obtained in a K-free MORB system at 6.0 GPa and 800–1000 °С showed notably greater enrichment in all key trace elements than the pelite-derived SCFM (Fig. 4b): at least ten-fold difference for some LILE, Th, U, LREE, Sr, as well as Nb and Ta, except for relatively small difference for Zr and Hf (at 800 °С), and HREE. This difference in the SCFM composition is evidence of the key role phengite and accessories play in the behavior of trace elements in the studied systems.

Trace-element fractionation
The contents of trace elements in the SCFM (determined on hydrous basis) and residue (Table S2, S3) phases were used to constrain fluid-residue partitioning (D = CSCFM/CResidue) in volatile-rich pelite at the P-T parameters of warm and hot subduction (Table 4, S4, Fig. 5). The consideration below is based on average values for specific pressures and temperatures, which are free from random errors and are thus more consistent than those for each run (Table S4). The main focus is on the behavior of elements that trace the participation of slab flux in the generation of arc and back-arc magmas and can furnish genetic information on the devolatilization of UHPM rocks.

Partitioning of trace elements between SCF or SCFM and residue. At pressures and temperatures increasing along the warm and hot subduction P-T paths, eclogitic residue derived from volatile-rich pelite first loses monazite and then phengite. However, monazite and phengite remain stable all over the applied P-T ranges in the case of devolatilized pelite
The residue contained Ba mainly in phengite but lacked potential hosts for significant amounts of Sr (the other element of similar compatibility in the Ba-Sr pair), though clinopyroxene and phengite can be subsidiary Sr carriers (Hermann and Rubatto 2014; Schmidt and Poli 2014). As temperature and pressure (3.0 to 5.5 GPa) rose along the warm subduction path, DBa and DSr increased from 2 to 7 and from 6 to 15, respectively (Table 4, S4, Fig. 6a). At the conditions of hot subduction, DBa and DSr were slightly lower at higher temperatures and greater SCFM fractions but smaller phengite percentages (Fig. 2a, b in Sokol et al. (2023a). Moreover, at 7.8 GPa and 1090 °С DBa exceeded DSr.

Partitioning of trace elements between SCF or SCFM and eclogite-like residue in the presence of accessory phases (± monazite and rutile) at pressure and temperature increasing along the warm and hot subduction P-T path. Element concentrations in SCF and SCFM are quoted on hydrous basis
Monazite, a major carrier of LREE, Th, and U, was stable in the first set of our experiments with volatile-rich pelite only at 3.0 GPa and 750 °С (Sokol et al.2023b) but became completely dissolved in the SCFM and disappeared from the residue obtained at P≥5.5 GPa and T≥850 °С (Sokol et al.2023a). The LREE, Th, and U partition coefficients increased correspondingly: e.g., DLa and DCe changed from 1.6 to 1.5, respectively, at 3.0 GPa and 750 °С to 14 and 11 at 5.5 GPa and 850 °С, though LREE incompatibility noticeably reduced in the DLa --> DNd series. As P-T rose along the warm and hot subduction paths, the SCFM fraction increased from 0.20 to 0.27 and the LREE partition coefficients became much lower (Tables 3 and 4, S4, Fig. 6c). DTh and DU increased as monazite was dissolved completely but decreased at higher P-T parameters at a greater SCFM fraction (Fig. 6b). The relative DU and DTh magnitudes are sensitive to monazite stability: DU > DTh at stable monazite and DTh> DU at unstable monazite as P-T increased.
Niobium and tantalum were considered to be “geochemical twins” in their fractionation behavior in subduction zones (Schmidt et al. 2004; Klimm et al. 2008; Hermann and Rubatto 2009), but our results showed slightly different behavior of these elements in the presence of rutile. DNb and DTa changed, respectively, from 0.4 to 0.5 at 3.0 GPa and 750 °С to 1.6 and 1.9 at 5.5 GPa and 850 °С (Fig. 6d). However, further P-T increase led to only minor change of DNb, while DTa decreased considerably at higher temperatures, both at 5.5 GPa and 7.8 GPa.
The mobility of trace elements in SCFM was compared using concentrations normalized to those in the starting pelite composition, as in Hermann and Rubatto (2009). The SrN/NdN ratio reached 2.7 (SrN=SrSCF/Srstarting pelite) at 3.0 GPa and 750 °С, in the absence of main Sr hosts and at stable monazite (carrier of LREE), but reduced to ~ 1 (i.e., fractionation stopped), when monazite became unstable (Fig. S4a). Unlike the partitioning behavior of Sr and Nd, the BaN/LaN ratio in SCFM was about 1 and thus did not differ much from that in the starting pelite (Fig. S4b). This behavior is easily explained for the products of the 7.8 GPa and 1090 °С run which lacked both phengite and monazite to control Ba and La, respectively. However, both phases were stable at 3.0 GPa and 750 °С. Since La is a major component in monazite structure (Raoult’s Law behavior), its fractionation does not change with the fraction of SCF or SCFM present in the experiment. At the same time, Ba is a passive trace component in phengite (Henry’s Law behavior), and its partitioning depends on SCF or SCFM and phengite fractions.
The ZrN/HfN ratio fluctuated about 1 in the presence of zircon in the residue, i.e., there was no preferential Hf enrichment in pelite-derived SCFM (Fig. S4c). The ThN/UN was more variable: 0.8 at 3.0 GPa and 750 °С (preferential U enrichment in SCF); 1.5 at 5.5 GPa and 850 °С, at unstable monazite; ~1 at greater P-T parameters and SCFM fraction (Fig. S4d).
The NbN/TaN ratio in SCF and SCFM demonstrated temperature sensitivity in the presence of rutile in the residue: <1 at warm subduction conditions but 1.6 to 2.1 at the conditions of hot subduction (Fig. S4f). In the experiments of Hermann and Rubatto (2009), Nb and Ta fractionation in hydrous melt was the highest (NbN/TaN = 1.7) at 800 °C and 4.5 GPa but lower (NbN/TaN = 1) as the temperature reached 1050 °С. According to our data, Nb and Ta from volatile-rich pelite mainly incorporate into SCFM and into residual rutile, respectively, at the hot subduction conditions.
The ThN/LaN ratio was slightly below 1 at all applied P-T parameters, which corresponds to higher mobility of La in SCFM compared to Th (Fig. 6). On the other hand, La showed much greater mobility than Nb at the conditions of warm subduction, judging by NbN/LaN <0.4, though the ratio was slightly greater (NbN/LaN = 0.6) at 7.8 GPa and hot subduction temperatures (Fig. S4h).
Discussion
Approach to equilibrium
The fractionation of trace elements between SCF or SCFM and residue was investigated in two series of experiments, using pelite rich in volatiles in the first set of 15 runs and pelite subjected to stepwise devolatilization in the second set of 5 experiments (Table 2). The system approached equilibrium at all applied P-T conditions in the first set of 40-hr runs with volatile-rich pelite (Sokol et al.2023a, b), judging by the equilibrated textures of phases, as well as by composition similarity of garnet, clinopyroxene, and phengite in our pelite-derived run products to the products of 75 to 260 h long runs obtained in the GLOSS system (Herman and Spandler, 2008; Herman and Rubatto, 2009). K, Ti, and Zr, which are essential components in buffering phases such as phengite, rutile, and zircon, demonstrate consistent decrease of their concentration in SCF and SCFM as temperature decreases (Fig. S3). The observed trends in element concentrations are typical of hydrous melts saturated with accessory phases (Hayden and Watson 2007; Klimm et al. 2008; Hermann and Rubatto 2009). Therefore, we can infer equilibrium fractionation of elements between SCF or SCFM and the residue containing rutile, zircon, and phengite. Monazite, the main concentrator of LREE, Th, and U, was found to be unstable at ≥5.5 and ≥7.8 GPa in the first set of experiments. Equilibration was facilitated by the presence of abundant volatiles in clay minerals which are unstable at high pressures and temperatures. The resulting aggregates of mineral phases in the residue were quite loose, with well pronounced intergranular space. Therefore, the volume of the fluid slightly exceeded the pore space in the diamond trap and maintained contact with the residue during the experiment. Thus, equilibrium fractionation was possible in experiments with volatile-rich pelite. The results of the first set of experiments were mainly used in this study when analyzing the fractionation of trace elements.
However, no equilibrium phase assemblage appeared yet in the 5.5 GPa and 850 °С 40-hr runs of the second set (2239_2_1 and 2237_2_3, Table 2) with devolatilized pelite, at markedly lower contents of volatiles in the eclogitic residue. The lack of equilibrium was indicated by non-equilibrated textures of the run products, as well as by unusual compositions of garnet, clinopyroxene, and phengite. The results of these experiments were not used to analyze trace elements partitioning. The residue acquired well-equilibrated textures and the solids approached the compositions obtained from volatile-rich pelite only in 150 h runs (2304_2_1 and 2304_2_6, Table 2; Fig. 1b). It was incomplete in the case of long (150 h) run duration at 7.8 GPa and 940 °С after the final devolatilization step (in the absence of a diamond trap) as garnet showed only minor composition changes, though clinopyroxene and phengite chemistry changed systematically (Figs. 1c and 2). Thus, only the product of the 2304_2_6 run in the second set of experiments was used to calculate the fractionation coefficients of trace elements (black line in Figs. 4 and 5; Table S2-S4).
Effect of pre-devolatilization on stability of phases and trace-element partitioning
Our previous (Sokol et al. 2023a, b) and present experiments simulated two devolatilization scenarios for slab sediments. Most of the experimental work was performed with natural pelite retaining the whole inventory of volatiles at subduction to 150–250 km depth, in order to reproduce the scenario of its single-step devolatilization. The experiments with stepwise devolatilization modeled the other scenario where the fluid phase (SCF or SCFM) released by dehydration and decarbonation is removed successively from the residue at the conditions of warm subduction, first at 3.0 GPa and then 5.5 GPa.
In the experiments simulating the first scenario, natural H2O-, CO2- and Cl-bearing pelite converted to an eclogitic residue, with phengite and a typical assemblage of accessory monazite, rutile, and zircon as residue solids and an SCF. Upon pressure and temperature increase along the warm and hot subduction paths, newly formed SCFM completely dissolved monazite and carbonate at 5.5 GPa and 850 °С and phengite at 7.8 GPa and 1090 °С (at 7.8 GPa and 940 °С, a few grains still remained); rutile and zircon kept stability over the whole P-T range.
Under the increasing P-T conditions, the mobile phase derived from dehydrated and decarbonated pelite changed from a solute-rich SCF (near the second critical endpoint) to an SCFM enriched in H2O, CO2, SiO2, Al2O3, and K2O, with lower Al2O3 and higher K2O at higher pressures and temperatures (Sokol et al. 2023a, b). The SCFM composition differs in higher concentrations of all geochemical tracers: slightly higher Sr and HFSE (Nb, Zr, Hf) and much higher LILE (K, Rb), REE, Th, and U at the warm subduction conditions but notably higher Zr and Hf and lower LREE at the hot subduction temperatures (Table 3, S3, Fig. 4a). Pelite-derived SCFM differs from hydrous melt obtained in the GLOSS system at 4.5 GPa and similar temperatures (Hermann and Spandler 2008; Hermann and Rubatto 2009) in higher contents of H2O and MgO + CaO + FeO at lower contents of SiO2 (on anhydrous basis) (Sokol et al. 2023a, b). Moreover, the quench products of SCFM and hydrous melts in diamond trap differ in the porosity-filling material: powder-like matter or aggregates of dendritic crystals and glass, respectively (Hermann and Spandler 2008; Sokol et al. 2023a, b). At the same time, SCFM and hydrous melt are similar in trace-element patterns, though SCFM contains more REEs. The results of this study show that the presence of 5–8 wt% CO2 and 0.4–0.5 wt% Cl in SCFM (Sokol et al. 2023a) causes no significant effect on trace-element partitioning in the pelite system.
The SCF-residue and SCFM-residue partition coefficients of Ba, Sr, LREE, Th, and U are the highest at 5.5 GPa and 850 °С (Figs. 5 and 6a-c). This partitioning of LREE and the change from compatible to incompatible behavior are possible when the host monazite becomes unstable. The solubility of monazite in SCFM is obviously higher than in SCF (Fig. 4b). Our natural pelite samples had almost same LREE, Th, and U concentrations as GLOSS-II (Table 1), and the amount of stable monazite should correspond to that in average subducted sediment material at the respective P-T parameters. Thus, the LREE partitioning behavior inferred from the experiments corresponds to that in real sediments subducted to 100–250 km depths. The partition coefficients of trace elements decreased as the SCFM fraction increased from 0.20 at 850 °С to 0.24 at 1030 °С in 5.5 GPa runs and from 0.24 at 940 °С to 0.27 at 1090 °С in 7.8 GPa runs (Table 3; Figs. 5 and 6a-d) (Sokol et al.2023a). Phengite in the residue became unstable, the partition coefficients increased for K and Rb but decreased for Ba, Sr, LREE, and Th (Table 4, S4, Fig. 5), while those for U did not change much and remained notably above 1 at both 5.5 and 7.8 GPa. A similar change from compatible to incompatible behavior of LILE upon phengite destabilization was observed in experiments with red clay reported by Johnson and Plank (1999). Note that the Zr and Hf partition coefficients increased in the presence of stable zircon in the residue at the hot subduction conditions.
The stepwise devolatilization of pelite led to loss of fluid-mobile elements. According to our estimates, the 5.5 GPa and 850 °С run with the use of pelite pre-devolatilized at 3.0 GPa and 750 °С yielded a melt-like phase, at a fraction of 0.14. As pelite was devolatilized, monazite in the residue at 5.5 GPa and 850 °С became stable, while the LREEs content in the melt-like phase decreased and their melt-residue partition coefficients (Fig. 5) became lower than those between SCFM and residue. Monazite and phengite remained stable at 7.8 GPa and 940 °С, after the final devolatilization step. Unfortunately, the amount of twice devolatilized residue was insufficient to use a diamond trap and to measure LREE, K, and Rb in the mobile phase. Nevertheless, our results suggest reduced mobility of LREE, K, and Rb in devolatilized slab material subducted to ~ 250 km.
As it was noted previously (Shatsky et al. 1999, 2006, 2015; Stepanov et al. 2014), diamond-bearing garnet-pyroxene-quartz and garnet-kyanite-quartz UHP metamorphic rocks in the Kokchetav HP-UHP complex, depleted in LREE and Th, can represent a residue material after the loss of fluid and elements at 100–250 km depths in a continental subduction zone (Shatsky et al. 1999, 2006, 2015; Bebout 2014; Hermann and Rubatto 2014; Zheng and Hermann 2014). Natural UHP rocks, including the Kokchetav gneiss, commonly contain accessory monazite as a main carrier of LREE, Th, and U (Massonne et al. 2007; Hermann and Rubatto 2009, 2014), which is in some cases preserved only as an inclusion phase in prograde-to-peak robust phases like garnet (Shatsky et al. 2015). Our results indicate that efficient LREE and Th depletion of UHP rocks is possible when slab material subducted to SCFM generation depths (≥150 km) retains its volatiles. SCFM derived from pelite rich in volatiles at 5.5 GPa (Table 2) can dissolve all monazite, while subsequent outflow of SCFM can lead to LREE and Th depletion of the residue (Table S2, S3, Fig. 3). On the other hand, the loss of 50 wt% H2O + CO2 at 3.0 GPa and 750 °С, which was simulated in our experiments with stepwise devolatilization of pelite, led to greater stability of monazite and considerable decrease in SCFM-residue partition coefficients of LREE and Th at 5.5 GPa. Thus, the presence of monazite in the Kokchetav UHP gneiss can be due to early partial loss of volatiles. Supercritical fluid-melt could be responsible for loss of LREE and Th from rocks rich in volatiles near the peak of metamorphism.
Markers of slab-derived SCFM in arc and back-arc magmas
The composition of natural pelite, at the two scenarios of devolatilization within the applied pressure and temperature ranges, appears quite well representative of slab-top material subducted to 100–250 km. Our consideration proceeded from the assumption that the mobile phase formed in subduction zone at the back-arc depths can re-circulate back to the front of the arc (Hicks et al. 2023). Therefore, the data on trace-element mobility in pelite-derived SCFM have bearing on its role in the generation of arc- and back-arc magmas.
The contents of Ba, La, and Ce in arc basalt are mainly inherited from slab sediments (Manning 2004; Spandler and Pirard 2013; Keppler 2017; Plank and Manning 2019). In this respect, Ba/La and H2O/Ce ratios in arc basalt are traditionally used to discriminate between the aqueous fluid (high Ba/La and H2O/Ce) and hydrous melt (low Ba/La and H2O/Ce) types of slab flux (Elliott et al. 1997; Pearce et al. 2005; Li et al. 2022). Ba and La in experiments with pelite showed similar incompatible behavior, and DBa was only slightly lower than DLa. Correspondingly, Ba/La in SCFM varied moderately (8 to 13) over the entire range of P-T parameters (Fig. 7a), but the variability was greater (9 to 24) in hydrous melt in the presence of phengite and monazite in the residue. The pelite-derived SCFM, with its H2O contents intermediate between fluid and melt compositions, plots between the GLOSS-derived melt and aqueous fluid phases in the Ba/La vs. H2O/Ce diagram, close to SCFM from AOC (Kessel et al. 2005a). Note that the content of incompatible Ba in the pelite samples we used was about twice smaller than that in GLOSS (Table 1).

Trace element compositions of global arc basalt (Ruscitto et al. 2012) and experimentally obtained GLOSS aqueous fluid (AF i.e., SCF) and melt, AOC melt, AOC saline AF, AOC melt and supercritical fluid-melt (SCFM), pelite-derived SCF and SCFM. a: Ba/La vs. H2O/Ce; b: Nd/Sr vs. H2O/Ce. 1: after Kessel et al. (2005a); 2: after Klimm et al. (2008); 3: after Carter et al. (2015); 4–7: after Rustioni et al. (2019), NaCl content from 0 (4) to 7 wt% (7); 8: after Zheng and Hermann (2014); 9: after Hermann and Rubatto (2009); 10: after Skora and Blundy (2010); 11: pelite-derived SCF and SCFM obtained in this study
Nd and Sr in basalt may come either from sediment or from AOC melts (Manning 2004; Spandler and Pirard 2013; Plank and Manning 2019; Li et al. 2022). The 0.06 to 0.15 range of Nd/Sr we obtained for SCFM overlaps with that for GLOSS melt (0.03 to 0.19) (Hermann and Rubatto 2009), while the content of incompatible Sr in our pelite is twice smaller than in GLOSS (Table 1). Thus, pelite-derived SCFM and GLOSS hydrous melt are similar in the Ba/La and Nd/Sr ratios but differ notably in the content of water and related H2O/Ce.
The DNb partition coefficient between the mobile phase and the rutile-bearing residue derived from natural pelite with nearly GLOSS contents of HFSE increases from < 0.4 for SCF to 1.0-1.6 for SCFM at the P-T ranges of 3.0-7.8 GPa and 750–1090 °С (Table 4). Importantly, similar fluid/melt-residue Nb partitioning behavior was observed in several other systems, in experiments with different amounts of doped trace elements. It is namely Nb partitioning between aqueous fluid and basaltic or pelitic residues at 3 GPa and 650–700 °C (Green and Adam 2003) and 2.2 GPa and 600–650 °C (Spandler et al. 2007), respectively, and between hydrous melt and these two residue types at 2.5-3.0 GPa and 750–1000 °C (Klimm et al. 2008; Carter et al. 2015) and 2.5–4.5 GPa and 750–1050 °C (Hermann and Rubatto 2009), respectively. Thus, fractionation of Nb does not affect the size of negative anomaly in the mobile phase (fluid/melt/SCFM) in the experimental systems simulating slab materials.
The Ba/Nb vs. Th/Nb plot was used (Pearce and Stern 2006) to investigate the relative contributions of shallow and deep subduction components in back-arc basins, with Ba/Nb and Th/Nb being the proxies for total subduction flux and deep subduction inputs in back-arc magmas, respectively. Most back-arc basins predictably follow deep subduction trends toward the high Th/Nb end of the local arc-array (Fig. 8a), which was confirmed by our results. In the experiments with pelite, DTh increases considerably, from 1.1 to 6.9–7.8 upon the transition from SCF to SCFM at higher pressures, while the DNb increase is only from 0.4 to 1.6 (Table 4, S4). The Th/Nb values we obtained for pelite-derived SCFM are slightly higher than those for deep subduction components in back-arc basins.

a: Ba/Nb vs. Th/Nb plot illustrating relative contributions of shallow and deep subduction components in back-arc basins (Pearce and Stern 2006) and trace element compositions of SCF and SCFM experimentally obtained from pelite. The diagram shows that the fluids obtained in this study, like most back-arc basins, follow deep subduction vectors, toward the high Th/Nb end of the local arc-array. b: Th/La vs. Nb/La plot illustrating relative contributions of an end-member in the MORB array and a high Th/La end-member that is typically very similar to bulk sediment (Plank 2014). The South Sandwich arc mixes between MORB and a component with Nb/La similar to that obtained in experiments with rutile-saturated SCF and SCFM
Marine sediments inherently have high Th/La and low Nb/La ratios (Plank 2014). The concentrations of these elements in the pelite we used are similar to those in GLOSS-II (Table 1). The Th/La ratio in slabs known from different trenches worldwide can vary from < 0.1 in hydrogenetic clay to > 0.35 in terrigeneous sediments (Plank 2005), while the values in our pelite and pelite-derived SCFM are, respectively, 0.27 and ~ 0.2. In the Nb/La vs. Th/La diagram (Plank 2014), the SCFM plots in the end of the increasing Th/La trend from MORB to basalt from the Mariana and then S. Sandwich arcs (Fig. 8b). Therefore, SCFM involved in the generation of arc- and back-arc volcanic rocks can translate the slab Th/La variations. Our results confirm the explanation by Plank (2014) that there is no need in Nb fractionation from the other trace elements, while the arc- and back-arc magmas may simply inherit the Nb anomaly from sediments.
At 3.0-5.5 GPa and the temperatures of warm and hot subduction, Rb is more efficiently retained in residue phengite which becomes unstable while Rb becomes strongly incompatible (DRb up to 5.2–8.3) at 7.8 GPa only. The DZr and DHf values are below 1 at warm subduction temperatures and reach 2 at those of hot subduction over the entire pressure range we applied (Table 4; Fig. 5). The increase of DZr and DHf apparently records temperature-dependent increase of zircon solubility in SCFM. Thus, high K and Rb concentrations can mark a slab-derived SCFM formed at pressures above the phengite stability limit. On the other hand, high Zr and Hf are diagnostic of a SCFM formed at hot subduction temperatures.
In general, the SCFM we obtained at 5.5–7.8 GPa and 850–1090 °С (Table 3, S3, Fig. 4a) shares much trace-element similarity with GLOSS hydrous melts (Hermann and Rubatto 2009), though differs in notably higher H2O and REE contents. The presence of ≤ 5–8 wt% CO2 and 0.4–0.5 wt% Cl in SCFM (Sokol et al. 2023a) causes only minor influence on trace-element partitioning. Main fractionation trends of trace elements are controlled by the fluid fraction and the stability of the solid hosts. The fractionation of LREE, Th, and U apparently depends on the stability of monazite in equilibrium with SCFM.
Conclusions
The reported study led to several key inferences.
-
1.
A supercritical fluid (SCF) equilibrated with an eclogitic residue bearing phengite and accessory rutile, zircon, and monazite can change to a supercritical fluid-melt (SCFM) during P-T rise along the subduction paths from ~ 100 to ~ 150 km depths. Thereby the mobile phase acquires markedly higher contents of Ba, Sr, LREE, Th, and U at the warm subduction conditions and K, Rb, Zr, and Hf during hot subduction, though LREEs become lower in the latter case. SCFM can efficiently transport both fluid-mobile and sediment-melt elements to the zones of arc and back-arc magma generation.
-
2.
The SCFM-residue partitioning of trace elements is mainly controlled by the fraction of fluid and the stability of phengite, monazite, rutile, and zircon. As all monazite dissolves in the residue, the partition coefficients of LREE, Th, and U increase considerably. High K and Rb concentrations can mark pelite-derived SCFM formed at pressures above the phengite stability limit, while high Zr and Hf are diagnostic of SCFM formed at hot subduction temperatures. Pelite-derived SCFM is able to preserve the negative Nb anomaly and translate it into arc- and back-arc magmas.
-
3.
Slab material subject to early devolatilization can provide more effective transport of LREE, Th, U, K, and Rb to depths of ~ 150–250 km. Outflow of SCFM formed near peak metamorphic conditions causes notable LREE and Th depletion of UHP metamorphic rocks.
References
Ayers J (1998) Trace element modeling of aqueous fluid–peridotite interaction in the mantle wedge of subduction zones. Contrib Mineral Petrol 132:390–404
Ayers JC, Dittmer SK, Layne GD (1997) Partitioning of elements between peridotite and H2O at 2.0–3.0 GPa and 900–1100 C, and application to models of subduction zone processes. Earth Planet Sci Lett 150(3–4):381–398
Bebout GE (2014) Chemical and isotopic cycling in subduction zones. Treatise Geochem 4:703–747
Brenan J, Shaw H, Ryerson F, Phinney D (1995) Mineral-aqueous fluid partitioning of trace elements at 900◦C and 2.0 GPa: constraints on the trace element chemistry of mantle and deep crustal fluids. Geochim Cosmochim Ac 59:3331–3350
Carter LB, Skora S, Blundy J, De Hoog J, Elliott T (2015) An experimental study of trace element fluxes from subducted oceanic crust. J Petrol 56:1585–1606
Elliott T (2003) Tracers of the Slab. Geophys Monograph-American Geophys Union 138:23–46
Elliott T, Plank T, Zindler A, White W, Bourdon B (1997) Element transport from slab to volcanic front at the Mariana arc. J Geophys Res Solid Earth 102:14991–15019
Green TH, Adam J (2003) Experimentally-determined trace element characteristics of aqueous fluid from dehydrated mafic oceanic crust at 3.0 GPa, 650–700°C. Eur J Mineral 15:815–830
Hayden LA, Watson EB (2007) Rutile saturation in hydrous siliceous melts and its bearing on Ti-thermometry of quartz and zircon. Earth Planet Sci Lett 258(3–4):561–568
Hermann J, Rubatto D (2009) Accessory phase control on the trace element signature of sediment melts in subduction zones. Chem Geol 265:512–526. https://doi.org/10.1016/j.chemgeo.2009.05.018
Hermann J, Rubatto D (2014) Subduction of continental crust to mantle depth: geochemistry of ultrahigh-pressure rocks. In Treatise on Geochemistry, 2nd Edition. Elsevier
Hermann J, Spandler CJ (2008) Sediment melts at sub-arc depths: an experimental study. J of Petrol 49:717–740. https://doi.org/10.1093/petrology/egm073
Hermann J, Zheng YF, Rubatto D (2013) Deep fluids in subducted continental crust. Elements 9:281–287. https://doi.org/10.2113/gselements94281
Hicks SP, Bie L, Rychert CA, Harmon N, Goes S, Rietbrock A, VoiLA Working Group (2023) Slab to back-arc to arc: fluid and melt pathways through the mantle wedge beneath the Lesser Antilles. Sci Adv 9(5):eadd2143
Ionov D, Harmer RE (2002) Trace element distribution in calcite-dolomite carbonatites from Spitskop: inferences for differentiation of carbonatite magmas and the origin of carbonates in mantle xenoliths. Earth Planet Sci Let 198:495–510
Johnson MC, Plank T (1999) Dehydration and melting experiments constrain the fate of subducted sediments. Geochem Geophys Geosyst 1:1007. https://doi.org/10.1029/1999GC000014
Keppler H (2017) Fluids and trace element transport in subduction zones. Am Mineral 102:5–20. https://doi.org/10.2138/am-2017-5716
Kessel R, Ulmer P, Pettke T, Schmidt MW, Thompson AB (2004) A novel approach to determine high-pressure high-temperature fluid and melt compositions using diamond-trap experiments. Am Mineral 89(7):1078–1086
Kessel R, Schmidt MW, Pettke T, Ulmer P (2005a) Trace element signature of subduction-zone fluids melts and supercritical liquids at 120–180 km depth. Nature 437:724–727
Kessel R, Ulmer P, Pettke T, Schmidt MW, Thompson AB (2005b) The water-basalt system at 4 to 6 GPa: phase relations and the second critical endpoint in a K-free eclogite at 700–1400°C. Earth Planet Sci Lett 237:873–892
Klimm K, Blundy JD, Green TH (2008) Trace element partitioning and accessory phase saturation during H2O-saturated melting of basalt with implications for subduction zone chemical fluxes. J Petrol 49:523–553
Li H, Hermann J, Zhang L (2022) Melting of subducted slab dictates trace element recycling in global arcs. Sci Adv 8(2):eabh2166
Manning CE (2004) The chemistry of subduction-zone fluids. Earth Planet Sci Lett 223:1–16
Massonne HJ, Kennedy A, Nasdala L, Theye T (2007) Dating of zircon and monazite from diamondiferous quartzofeldspathic rocks of the Saxonian Erzgebirge – hints at burial and exhumation velocities. Mineral Mag 71:407–425
McDonough WF, Sun SS (1995) The composition of the Earth. Cheml Geol 120(3–4):223–253
Ni H, Zhang L, Xiong X, Mao Z, Wang J (2017) Supercritical fluids at subduction zones: evidence, formation condition, and physicochemical properties. Earth Sci Rev 167:62–71
Plank T, Langmuir CH (1998) The chemical composition of subducting sediment and its consequences for the crust and mantle. Chem Geol 145(3–4):325–394
O’Reilly SY, Griffin WL (2013) Mantle Metasomatism. In: Harlov DE, Austrheim H (eds) Metasomatism and the Chemical Transformation of Rock. Springer-, Berlin Heidelberg, pp 471–533
Palyanov YN, Kupriyanov IN, Khokhryakov AF, Borzdov YM (2017) High-pressure crystallization and properties of diamond from magnesium-based catalysts. CrystEngComm 19:4459–4475 https://doi.org/10.1039/C7CE01083D
Pearce JA, Stern RJ (2006) Origin of back-arc basin magmas: Trace element and isotope perspectives. Back‐arc spreading systems: geological, biological, chemical. Phys Interact 166:63–86
Pearce JA, Stern RJ, Bloomer SH, Fryer P (2005) Geochemical mapping of the Mariana arc-basin system: implications for the nature and distribution of subduction components. Geochem Geophys Geosyst 6:Q07006
Perchuk AL, Serdyuk AA, Zinovieva NG, Shur MY (2020) Melting and parageneses of global subducting water-enriched sedi- ment in closed and open systems: experiment and thermodynamic modeling. Russ Geol Geophys 61(5–6):571–591. https://doi.org/10.15372/RGG2019177
Petrichenko YA (2000) Geochemical characteristics of organic matter in the Maikop series of the Kerch-Taman trough. Mosc Univ Bull Ser 4:6:64–66 (in Russian)
Plank T (2014) The chemical composition of subducting sediments. in: Holland HD, Turekian KK (Eds) Treatise on Geochemistry, Elsevier, Amsterdam, pp 607–629. https://doi.org/10.1016/B978-0-08-095975-700319-3
Plank T, Manning CE (2019) Subducting carbon. Nature 574:343– 352. https://doi.org/10.1038/s41586-019-1643-z
Portnyagin M, Hoernle K, Plechov P, Mironov N, Khubunaya S (2007) Constraints on mantle melting and composition and nature of slab components in volcanic arcs from volatiles (H2O, S, cl, F) and trace elements in melt inclusions from the Kamchatka Arc. Earth Planet Sci Lett 255(1–2):53–69
Ruscitto DM, Wallace PJ, Cooper LB, Plank T (2012) Global variations in H2O/Ce: 2. Relationships to arc magma geochemistry and volatile fluxes. Geochem Geophys Geosyst 13:Q03025
Rustioni G, Audétat A, Keppler H (2019) Experimental evidence for fluid-induced melting in subduction zones. Geochem Persp Let 11:49–54
Rustioni G, Audetat A, Keppler H (2021) The composition of subduction zone fluids and the origin of the trace element enrichment in arc magmas. Contrib Mineral Petrol 176(7):51
Ryabchikov ID, Orlova GP, Kalenchuk GYu, Ganeev II, Udovkina NG, Nosik LP (1989) Interaction of spinel lherzolite with water–CO2 fluid at 20 kbar and 900°C. Geokhimiya 3:385–392 (in Russian)
Schmidt M, Poli S (2014) Devolatilization during subduction. In: Holland HD, Turekian KK (ed), Treatise on Geochemistry, Elsevier, Amsterdam, pp 669–701. https://doi.org/10.1016/B978-0-08-095975-700321-1
Schmidt MW, Vielzeuf D, Auzanneau E (2004) Melting and dissolution of subducting crust at high pressures: the key role of white mica. Earth Planet Sci Lett 228:65–84. https://doi.org/10.1016/j.epsl.2004.09.020
Shatsky VS, Jagoutz E, Sobolev NV, Kozmenko OA, Parkhomenko VS, Troesch M (1999) Geochemistry and age of ultrahigh pressure metamorphic rocks from the Kokchetav massif (Northern Kazakhstan). Contrib Mineral Petrol 137:185–205
Shatsky VS, Sitnikova ES, Koz’menko OA, Palessky SV, Nikolaeva IV, Zayachkovsky AA (2006) Behavior of incompatible elements during ultrahigh-pressure metamorphism (by the example of rocks of the Kokchetav massif). Russ Geol Geophys 47(4):485–498
Shatsky VS, Skuzovatov SYu, Ragozin AL, Sobolev NV (2015) Mobility of elements in a continental subduction zone: evidence from the UHP metamorphic complex of the Kokchetav massif. Rus Geol Geophys 56(7):1016–1034
Skora S, Blundy J (2010) High-pressure hydrous phase relations of radiolarian clay and implications for the involvement of subducted sediment in arc magmatism. J Petrol 51:2211–2243. https://doi.org/10.1093/petrology/egq054
Skora S, Blundy JD, Brooker RA, Green ECR, de Hoog JCM, Connolly JAD (2015) Hydrous phase relations and trace element partitioning behaviour in calcareous sediments at subduction-zone conditions. J Petrol 56:953–980
Sokol AG, Borzdov YM, Palyanov YN, Khokhryakov AF (2015) High-temperature calibration of a multi-anvil high pressure apparatus. High Pressure Res 35(2):139–147. https://doi.org/10.1080/0895795920151017819
Sokol E, Kokh S, Kozmenko O, Novikova S, Khvorov P, Nigmatulina E, Belogub E, Kirillov M (2018) Mineralogy and geochemistry of mud volcanic ejecta: a new look at old issues (a case study from the Bulganak field Northern Black Sea). Minerals 8(344). https://doi.org/10.3390/min8080344
Sokol AG, Kozmenko OA, Kruk AN (2023a) Composition of supercritical fluid in carbonate-and chlorine-bearing pelite at conditions of subduction zones. Contrib Mineral Petrol 178(12):90
Sokol AG, Koz’menko OA, Kruk AN, Nechepurenko SF (2023b) Composition of the fluid in carbonate- and chlorine-bearing pelite near the second critical point: results of diamond trap experiments. Russ Geol Geophys 64:1–13
Spandler C, Pirard C (2013) Element recycling from subducting slabs to arc crust: a review. Lithos 170:208–223
Spandler C, Mavrogenes J, Hermann J (2007) Experimental constraints on element mobility from subducted sediments using high-P synthetic fluid/ melt inclusions. Chem Geol 239:228–249
Stepanov AS, Hermann J, Korsakov AV, Rubatto D (2014) Geochemistry of ultrahigh-pressure anatexis: fractionation of elements in the Kokchetav gneisses during melting at diamond-facies conditions. Contrib Mineral Petrol 167:1002
Turner SJ, Langmuir CH (2022) Sediment and ocean crust both melt at subduction zones. Earth Planet Sci Lett 584:117424
Westrenen WV, Blundy J, Wood B (1999) Crystal-chemical controls on trace element partitioning between garnet and anhydrous silicate melt. Am Mineral 84(5–6):838–847
Zheng YF, Hermann J (2014) Geochemistry of continental subduction-zone fluids. Earth Planets and Space 66(1):1–16
Acknowledgements
We would like to thank Yu. Palyanov, Yu. Borzdov, and A. Khokhryakov for their assistance throughout the study. The manuscript profited much from thoughtful reviews by John C. Ayers and anonymous reviewer. The research was supported by the Russian Science Foundation (grant No. 22-17-00005). The EDS-SEM and EMPA studies of experimental samples were performed in the Analytical Center for Multi-element and Isotope Research (Novosibirsk). The ICP-MS studies at the Center for Isotopic and Geochemical Research (Irkutsk) were supported by the Russian Science Foundation (grant No. 21-77-10038, https://rscf.ru/project/21-77-10038) and the analytical work at the Zavaritsky Institute of Geology and Geochemistry (Ekaterinburg) was carried out as part of government assignment No. 123011800012-9.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Dante Canil.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Sokol, A., Kozmenko, O., Kruk, A. et al. Trace-element mobility in pelite-derived supercritical fluid-melt at subduction-zone conditions. Contrib Mineral Petrol 179, 50 (2024). https://doi.org/10.1007/s00410-024-02131-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00410-024-02131-2