
Abstract
Ilmenite-, magnetite- and clinopyroxene−melt trace element partition coefficients were investigated experimentally as a function of oxygen fugacity and melt composition in a range of synthetic ferrobasaltic bulk compositions. The experiments were performed at a constant temperature (1080 °C) and pressure (1 atm) over a range of oxygen fugacity (fO2) conditions from ca. 2 log units below to ca. 2 log units above the FMQ buffer. The partitioning behaviour of the divalent cations Zn, Mn, Co and Ni are found to be controlled by the degree of polymerisation of the coexisting melt; the partitioning behaviour of rare earth elements, Y and Sc can be explained well by the lattice strain model and the partitioning of the high-field strength elements Zr, Hf, Ta and Nb is influenced by the TiO2 content of the melt. Vanadium partitioning is strongly influenced by oxygen fugacity and a series of linear regression equations are presented to express the dependence of the mineral−melt partitioning behaviour of the multivalent cation V on oxygen fugacity. Furthermore, calibration of the partitioning of vanadium between magnetite–ilmenite pairs as an oxybarometer is proposed and applied to a ferrobasaltic layered intrusion—the Skaergaard intrusion—to provide an estimate of oxygen fugacity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Iron-rich basaltic magmas (ferrobasalts) form the link between basaltic and silicic magmatism. Natural examples include flood basalts (e.g. Xu et al. 2001), volcanic tholeiitic suites (e.g. Streck and Grunder 2012) and the parent magmas to layered igneous intrusions, e.g. Skaergaard and Sept Îles (Hunter and Sparks 1987; Toplis and Carroll 1996; McBirney 1998; Namur et al. 2010). The evolution of such basalts is characterised by early enrichment in TiO2 and FeO, followed by SiO2 enrichment and FeO and TiO2 depletion at higher degrees of differentiation, marked by the onset of Fe−Ti oxide crystallisation (Snyder et al. 1993; Toplis and Carroll 1996; Pownceby and Fisher-White 1999; Namur et al. 2011; Howarth and Prevec 2013). Iron–titanium oxides generally become liquidus phases just before or just after the saturation of clinopyroxene (Toplis and Carroll 1995; Namur et al. 2011). Clinopyroxene thus also strongly influences the evolution of melt composition, in terms of both major and trace elements (Toplis and Carroll 1996; Namur and Humphreys 2018).
Clinopyroxene is a key component in mafic to intermediate magmas (Putirka et al. 1996; Armienti et al. 2007; Putirka 2008; Neave and Putirka 2017; Mollo et al. 2020). Clinopyroxene contains a sixfold coordinated M1 site which typically hosts smaller cations and an eightfold coordinated M2 site which hosts the larger cations, such as rare earth elements (REE) (Blundy and Wood 2003; van Kan Parker et al. 2011; Sun and Liang 2012; Fabbrizio et al. 2021). Its stability across a range of conditions is derived from its ability to incorporate a range of cations in both octahedral and tetrahedral coordinations into the crystal structure and this sensitivity to intensive parameters, combined with its relatively slow lattice diffusion (Watson and Yan Liang 1995; Van Orman et al. 2001; Costa and Morgan 2010; Müller et al. 2013), means that clinopyroxene crystals hold a lot of information on pre-eruptive processes (Humphreys 2009; Ubide and Kamber 2018; Ubide et al. 2019; Di Flavio et al. 2020; Masotta et al. 2020). Trace elements in clinopyroxenes are particularly good records of magmatic processes as they are more sensitive to magmatic processes such as fractional crystallisation, magma recharge and mixing (Mollo et al. 2020) than major and minor elements and they are especially useful in layered intrusions where slowly diffusing trace elements record changes in the intercumulus liquid composition during differentiation, melt migration and infiltration and reactive dissolution (e.g. Humphreys 2009; Namur et al. 2013; Leuthold et al. 2014; Namur and Humphreys 2018).
Iron–titanium oxides are also common major and accessory phases in igneous rocks (O’Neill et al. 1988; Klemme et al. 2006) and typically form solid solutions between ulvöspinel and magnetite (Fe2TiO4–Fe3O4), ilmenite and hematite (FeTiO3–Fe2O3) and ferropseudobrookite and pseudobrookite (FeTi2O5–Fe2TiO5). Iron–titanium oxides are critical minerals in mafic layered intrusions and whilst their compositions provide information on magma chamber processes such as fractional crystallisation, magma replenishment and magma mixing (Frost and Lindsley 1992; Pang et al. 2008; Namur et al. 2010), they are also often used to constrain the temperature of crystallisation and oxygen fugacity (Lindsley and Frost 1992; Sauerzapf et al. 2008). This is because the Fe and Ti content of magnetite–ilmenite pairs is well known to be sensitive to magmatic redox conditions (Buddington and Lindsley 1964; Andersen and Lindsley 1988; Ghiorso and Sack 1991; Lattard et al. 2005; Ghiorso and Evans 2008).
Ilmenite in particular also plays an important role in the petrogenesis of lunar basalts (Hess et al. 1978; McKay et al. 1986), saturating during the later stages of crystallisation in the lunar magma ocean (Snyder et al. 1992; Dygert et al. 2013). Dense ilmenite-rich cumulates have implications for the overturn of the lunar mantle (Hess and Parmentier 1995; Thacker et al. 2009) and for the generation of high-Ti (up to 16 wt.% TiO2) lunar basalts (Longhi 1992; van Kan Parker et al. 2011; Krawczynski and Grove 2012). The structure of ilmenite is characterised by hexagonal, closely packed oxygen atoms with interstitial alternate layers of octahedrally-coordinated M sites; one site is typically occupied by Mg2+ or Fe2+, whilst the other is occupied by Ti4+ (Raymond and Wenk 1971; Dygert et al. 2013). Both lattice sites are sixfold coordinated (van Kan Parker et al. 2011). Magnetite ([Fe3+][Fe2+Fe3+]O4) has an inverse spinel structure with crystallographic sites in both tetrahedral and octahedral coordination, giving it the general formula B(AB)O4 where one B cation occupies the tetrahedral site and the remaining A and B cations fill the octahedral sites, thus easily facilitating incorporation of divalent and trivalent cations (O’Neill and Navrotsky 1984; Nielsen et al. 1994; Bosi et al. 2009). Tetravalent cations can be substituted readily into tetrahedral or octahedral sites, charge balanced by additional Fe2+ (Sievwright et al. 2017).
The importance of oxygen fugacity (fO2) on the composition and stability of Fe−Ti oxides and coexisting silicate phases (e.g. clinopyroxene) and the partitioning of elements between mineral and silicate melt, has been demonstrated by many authors (e.g. Lindsley and Frost 1992; Toplis and Carroll 1995; Toplis and Corgne 2002; Klemme et al. 2006; Mallmann and O’Neill 2009; Dygert et al. 2013; Leitzke et al. 2016; Arató and Audétat 2017a; Michely et al. 2017; Sievwright et al. 2020) following the pioneering work of Buddington and Lindsley (1964). In previous studies of Fe−Ti oxide and clinopyroxene trace element partitioning (e.g. Canil and Fedortchouk 2000; Toplis and Corgne 2002; Prowatke and Klemme 2006; Hill et al. 2011; Dygert et al. 2013, 2014; Michely et al. 2017; Sievwright et al. 2017, 2020), partition coefficients are strongly influenced by crystal and melt composition, temperature, pressure and fO2. Oxygen fugacity-driven valence state changes result in changes to mineral−melt partition coefficients (or Di); thus the partitioning of redox-sensitive trace elements may provide constraints on the redox conditions of a melt. Vanadium in particular is highly sensitive to redox conditions and can be present as V2+, V3+, V4+ or V5+ in silicates and oxides, covering the entire range of terrestrial redox conditions; this lends itself to the applicability of V as a universal redox indicator, or oxybarometer (Canil 1999; Toplis and Corgne 2002; Papike et al. 2005; Karner et al. 2006; Mallmann and O’Neill 2009, 2013). The partitioning behaviour of vanadium between Fe−Ti oxides and melt is of particular interest, as much of the economically important concentrations of vanadium are associated with iron-rich cumulates that are typically characteristic of layered intrusions e.g. Bushveld (Reynolds 1985; Yuan et al. 2017).
The aims of this study are to report the major element geochemical variability of the silicate and Fe−Ti oxide phases we produce under a range of oxygen fugacity and melt TiO2 conditions and to assess the effect of redox conditions and melt composition on the partitioning of trace elements in ilmenite, magnetite and clinopyroxene under isobaric and isothermal conditions. Partition coefficients are reported for divalent, trivalent and tetravalent cations in each phase over a range of redox conditions (FMQ −2 to FMQ + 2, with FMQ being the fayalite–magnetite–quartz equilibrium). The mineral−melt partitioning behaviour of vanadium in Fe−Ti oxides and clinopyroxene is used as a proxy for fO2 and a series of oxybarometers are calibrated that can be used to predict fO2 at atmospheric pressure in ferrobasaltic to dacitic melts. We also calibrate an oxybarometer based on the ratio of \(D_{{\text{V}}}^{{\text{mt - melt}}}\) to \(D_{{\text{V}}}^{{\text{ilm - melt}}}\) which can be applied to layered intrusions when the V content of the melt is unknown, but the average bulk DV is known.
Methods
Starting materials and experimental techniques
The initial starting composition used in the partitioning experiments is a synthetic analogue of an evolved ferrobasalt with a magnesium number [mg # = molar MgO/(MgO + FeO)] of 37 (composition ‘SC4’ from Toplis & Carroll 1995), which is assumed to be the residual, fractionated liquid of the parental magma of the Skaergaard intrusion (composition ‘SC1’, Toplis & Carroll 1995) after approximately 40% crystallisation. The material was synthesised from a mixture of reagent-grade oxides (SiO2, MgO, Al2O3 and Fe2O3) and carbonates (CaCO3, Na2CO3 and K2CO3) with variable TiO2 contents to force the saturation of a range of Fe−Ti oxides (~ 5, 10 and 15 wt.% TiO2) Additional iron was added as Fe2O3 to some starting compositions (Table 1). Phosphorus was added as H3PO4 to half of the preparation to investigate the effect of phosphorus on phase relationships (e.g. Toplis et al. 1994a). Starting materials were doped with 1000 ppm of V, Cr and Mn (as V2O5, K2Cr2O7 and MnO2) and 500 ppm of Co, Ni and Cu (as Co2O3, NiO and CuO) using high-purity oxide powders. Standard solutions were used to dope the preparation with 500 ppm of Zn, Y, Sc, Zr, Hf, Nb, Ta, Sn, Mo, W, Sr, Rb, Ba, Pb, U, Th and 200 ppm of REE (rare earth elements) (La, Ce, Nd, Sm, Eu, Gd, Dy, Er, Yb and Lu). Trace element doping in experimental series 8 to 11 (Table 2) was decreased to a fourth of these values, after finding that this initial concentration produced trace element levels that are not representative of natural Fe−Ti oxides. Subsequent to the doping procedure the starting compositions were re-melted twice in air at 1300 °C for 1.5 h, and the resultant glass was finely crushed in an agate mortar to a homogenous powder.
Experiments were carried out at atmospheric pressure in a Gero vertical drop-quench gas mixing furnace at the Observatoire Midi-Pyrénées (OMP), Toulouse and at the Department of Earth and Environmental Sciences, KU Leuven. To cover a range of redox conditions, experiments were conducted at five fO2 conditions between FMQ −2 and FMQ + 2 (FMQ = fayalite–magnetite–quartz buffer). Temperature was controlled using a Eurotherm 818 controller and oxygen fugacity (fO2) was controlled using high purity CO−CO2 gas mixtures regulated by two Tylan mass flow controllers. The oxygen fugacity was measured adjacent to the samples in the hotspot of the furnace with an yttrium-stabilized zirconia probe and recorded variations show that a precision of around ± 0.1 log units. A summary of the experimental run conditions is presented in Table 2.
Approximately 50 mg of starting material was pressed onto 0.2 mm diameter platinum wire loops. To prevent potential Fe loss, the Pt loops were pre-saturated with the experimental material for 3 days under the same experimental temperature and fO2 conditions reported in Table 2, then quenched and cleaned with HF. Up to five Pt loops were suspended at a time from the sample holder and loaded into the hotspot of the furnace next to an internal Pt−Pt10Rh thermocouple, calibrated against the melting point of gold (1064 °C). The samples were first held at 1200 or 1150 °C for 9 h, i.e. close enough to the liquidus to allow the equilibration of multivalent ions and fully melt the powder without hampering nucleation upon cooling, then cooled at a rate of 1 °C/h to facilitate the growth of crystals sufficiently large for analysis. The run temperature was continuously monitored and the temperature precision of this set up is better than ± 1 °C. After cooling to a final temperature of 1080 °C, which corresponds to the approximate point of oxide saturation in layered intrusions (Thy et al. 2009), the experimental charges were left to equilibrate for at least 72 h before drop-quenching into water. The quenched products were mounted in epoxy and polished with a series of SiC and diamond pastes for chemical analysis.
Analytical techniques
The concentration of major and minor elements in glass and crystal phases were acquired with a Cameca SX 100 electron microprobe microanalyser at the University of Clermont−Ferrand and a JEOL JXA-8530F wavelength dispersive spectrometer field emission gun electron probe microanalyser (WDS FEG-EPMA) at the Department of Material Engineering, KU Leuven. For glass analyses, a 15 kV accelerating voltage and 8 nA beam current were used, with a 10 µm incident beam diameter to minimise potential alkali migration. Conditions for mineral analyses were 15 kV and 15 nA with a 1 μm focused beam. Corrections for inter-elemental effects were made using the ZAF (Z, atomic number; A, absorption; F, fluorescence) procedure. Calibration standards used were forsterite (Mg), fayalite (Fe), wollastonite (Si, Ca), albite (Na), orthoclase (K), chromite (Cr), olivine (Ni), MnTiO3 (Mn, Ti) and Al2O3 (Al). To ensure internal consistency across multiple analytical sessions, EPMA results were normalised to international standards. For glass analyses, we used VG-2 basalt glass (NMNH 111240–52). For minerals, we used Kakanui augite (NMNH 122142, using preferred values) for clinopyroxene, orthopyroxene and plagioclase analyses and San Carlos olivine (NMNH 111312–44) for olivine analyses. Accuracy and precision were estimated by measuring the following standards during each session: A-99 (NMNH 113498) and BCR-2G (USGS) basaltic glasses and Lake County plagioclase (NMNH 115900) (Jarosewich et al. 1980; Jochum et al. 2005). We also repeatedly measured in-house and Smithsonian microbeam standards of hypersthene, diopside, plagioclase [An95; An = 100 Ca/(Ca + Na), mol. %] and olivine (Fo83 and Fo10). Major (> 1 wt. %) and minor (< 1 wt. %) element contents were determined with respective accuracies better than 2% and 10% and 1σ precisions better than 2% and 15%.
Trace element concentrations in glass and crystal phases were measured with an Agilent 7500cx inductively coupled plasma mass spectrometer (ICP-MS) coupled to a commercial femtosecond Ti:Sa laser (Amplitude Technologies Pulsar 10), based on the Chirped pulse amplification technique, at the University of Clermont-Ferrand and with an Agilent 8900 triple quadrupole ICP-MS coupled to an Analyte Excite Laser Ablation System at the KU Leuven. For glass and clinopyroxene measurements a frequency of 5 Hz and spot sizes ranging between 50 and 100 µm were used. For the Fe−Ti oxides, the laser conditions used were 7 Hz and 5 µm spot size. Calcium was used as an internal standard for glass and clinopyroxene, whilst Ti was used for Fe−Ti oxides. Calibration standards used were NIST610 and NIST612 and BHVO-2G was used as a secondary standard. Measurements of the BCR-2G glass standard indicate that most elemental analyses were accurate to better than 10% relative, with only P, Cr and Cu returning deviations > 10% relative.
Experimental results
Attainment of equilibrium
Evidence of equilibrium between mineral phases and the melt includes (i) run durations comparable to previous studies that reached equilibrium (Toplis and Carroll 1995; Toplis and Corgne 2002; Mallmann and O’Neill 2009; Dygert et al. 2013; Leitzke et al. 2016, 2017); (ii) phase assemblages similar to those reported by Toplis and Carroll (1995) for the same starting composition; (iii) low standard deviations across replicate analyses of each phase; (iv) melt-clinopyroxene Fe–Mg exchange coefficient (KD) values of 0.15–0.27 (Grove and Bryan 1983; Putirka 2008; Bédard 2010).
The partition coefficient \(K_{D}^{{\begin{array}{*{20}c} {\text{Cpx - Liq}} \\ {\text{Fe - Mg}} \\ \end{array} }}\) is used to describe the distribution of iron (as Fe2+) and magnesium between the clinopyroxene and the coexisting glass (Nielsen et al. 1992; Toplis and Carroll 1995). The KD values in this study range between 0.15 and 0.27 (Supplementary Table ST2), concordant with the results of Hoover & Irvine (1978) and Toplis & Carroll (1995), who produced similar values of 0.19–0.26 and 0.18–0.26, respectively.
Run products
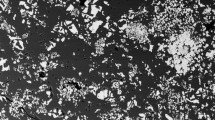
The phase relations and run conditions of each experiment are presented in Table 2. The observed crystalline phases are ilmenite (ilm), (Ti-)magnetite (mt), ulvöspinel (usp), ferropseudobrookite (psb), clinopyroxene (cpx) and plagioclase feldspar (pl). Phase proportions were calculated by mass balance. The aforementioned slow cooling rate of 1 °C per hour was chosen to produce crystals large enough for analysis (Hill et al. 2000; Leitzke et al. 2016); this was necessary for Fe−Ti oxide phases in particular which are typically very small, though some of the oxide phases produced still remained too small for analysis by LA-ICP-MS. The size of the Fe−Ti oxide crystals produced does, however, increase with the addition of TiO2; this may be a reflection of the depolymerising effect of TiO2 on silicate melts at higher TiO2 contents, as previously suggested by Leitzke et al. (2016). Run products of representative samples are illustrated in Fig. 1a and b. The abundance of mineral phases increases with increasing oxygen fugacity, e.g. from glass + one mineral phase in run 15Fe-11 (FMQ -2) to glass + five mineral phases in run 15Fe-10 (FMQ + 2). The melt proportion decreases overall with increasing fO2, from 80 to 93% melt at FMQ − 2 to 35 to 79% melt at FMQ + 2. The addition of P to the starting composition increases the melt proportion in each run, e.g. from 45% melt in run 5–7 to 79% melt in run 5P-7; this effect is reduced at lower fO2 and most pronounced at higher fO2. These observations are concordant with the study of Toplis et al. (1994a), who also observe an increase in melt proportion with the increased P2O5 content, similarly more pronounced under more oxidising conditions.

Back-scattered electron images of representative products and phase relations from experiments conducted at a FMQ + 2 and b FMQ − 2. Laser ablation pit labelled as ‘Laser pit’. Ilm ilmenite, Mt magnetite, Usp ulvöspinel, Psb Ferropseudobrookite, Cpx clinopyroxene, Pl plagioclase. Note the increased melt proportion at FMQ − 2, and the increased abundance of mineral phases at FMQ + 2
Major and minor element contents of glass and coexisting mineral phases are reported in Supplementary Table ST1. Select major element abundances in glasses are plotted as a function of MgO in Fig. 2. The glasses range from basaltic (46.3 wt. % SiO2) to dacitic (62.4 wt. % SiO2) and the run products of starting compositions doped with phosphorus (open symbols) are more primitive than their undoped counterparts, with lower SiO2 and higher MgO (Fig. 2a). The FeO content of the glass (Fig. 2d) increases under more reducing conditions, from 7.6 wt. % at FMQ + 2 to 18.7 wt. % at FMQ -2. The mg # of the glasses produced ranges from 25 to 37, with no strong correlation with fO2. Note the range in the TiO2 contents of the starting compositions (star symbols, Fig. 2b), though all experimental melts plot on a single trend against MgO regardless of this range in TiO2. Although the overall Al2O3 content of the glass decreases as MgO content increases, two trends are apparent which converge around MgO = 4 wt. % (Fig. 2c). The lower Al2O3 trend, which has a shallower slope and decreases from ~ 12 wt. % Al2O3 to ~ 11 wt. % Al2O3 and the higher Al2O3 trend which has a steeper slope, decreasing from ~ 15 wt. % to ~ 11 wt. %. Above the FMQ buffer, the lower Al2O3 trend may be explained by the crystallisation of plagioclase which incorporates more Al2O3 from the melt and therefore leaves the melt relatively depleted in Al2O3. In the higher Al2O3 trend, there are no plagioclase-bearing experiments therefore is less Al2O3 removed from the melt, resulting in relative Al2O3 enrichment. Below the FMQ buffer we observe no plagioclase in any experiments, likely due to the reduced stability field of plagioclase under more reducing conditions as described by Toplis and Carroll (1995). We do, however, observe that the higher Al2O3 trend below the FMQ buffer correlates to the crystallisation of clinopyroxene concomitant with Fe−Ti oxides, whilst clinopyroxene is absent in the experiments that are described by the lower Al2O3 trend.

Major element variation diagrams for experimental glasses, including starting compositions (stars), Phosphorus-bearing samples have open symbols, whilst P-free samples are filled. The P-free experiments conducted at FMQ + 1 (diamond symbols) and FMQ + 2 (circle symbols) are enriched in SiO2 and alkalis, and depleted in TiO2, FeO and MgO relative to their P-doped counterparts. Error bars = 1σ. Oxides are normalised to 100 wt. %
We find that the addition of P2O5 to the starting composition reduces the melt SiO2 content by up to ⁓ 5 wt.% in comparison to undoped runs, though this effect is less pronounced under more reducing conditions and the addition of P2O5 to starting composition 15Fe-9 produces a glass (15FeP-9) with a higher SiO2 content; 46.64 and 47.80 wt. %, respectively. Toplis et al. (1994b) have demonstrated that the addition of P2O5 has a significant effect on shear viscosity at a fixed temperature, directly influencing melt polymerisation. The degree of melt polymerisation can be quantified in terms of the ratio of non-bridging oxygens to tetrahedrally coordinated cations, or NBO/T (Mysen 1983). Between FMQ + 2and FMQ − 2(Fig. 2f) the NBO/T content of the glasses increases overall, reflecting depolymerisation of the melt under more reducing conditions. An alternative measure of melt structure is optical basicity (Λ), which distinguishes different cations with contrasting electron donor power (Duffy 1993). We find a positive correlation between NBO/T and optical basicity, suggesting that both parameters provide a representative description of melt composition.
Trace element contents of experimental glasses are reported in Supplementary Table ST2. No clear trend of trace element enrichment or depletion is observed as a function of the melt fraction in the experiments because various levels of trace element doping were used in our starting compositions.
Ilmenite and magnetite are the most common oxide phases in our experiments. Oxygen fugacity exerts a strong control on the stability field of magnetite, demonstrated by the decreased proportion and eventual absence of magnetite with decreasing fO2 in our experiments (Table 2) and corroborated by a range of experimental studies conducted on basalts and ferrobasalts at atmospheric pressure (e.g. Juster et al. 1989; Snyder et al. 1993; Toplis et al. 1994a; Toplis and Carroll 1995; Sievwright et al. 2017, 2020). Ilmenite is present only between FMQ − 2 and FMQ + 1, absent in the most oxidising runs at FMQ + 2. Although the saturation point of ilmenite is largely controlled by the TiO2 content of the melt (Toplis and Carroll 1995), the stability field of ilmenite is a function of fO2 and in contrast to magnetite, increases under more reducing conditions; this is linked to the destabilisation of magnetite and subsequent Fe-enrichment in the melt (Charlier et al. 2015).
Ilmenite crystals are typically elongate and needle-like and can be skeletal, often reaching up to 600 μm in length but only 5–20 μm wide. The composition of our experimental ilmenites, as expected, depends strongly on oxygen fugacity. The TiO2 content increases from 42.8 wt. % at FMQ + 1 to 54 wt. % at FMQ -2, whilst FeO decreases from 46.7 wt. % to 37.6 wt. % over the same range. As the FeO content of the ilmenites decreases, the MgO content increases from 3.5 to 6.6 wt. %. The Xilm component (Xilm = Fe2+ + [0.5*Al]), calculated following the QUILF algorithm of Andersen et al. (1993), increases from Ilm80 to Ilm100 between FMQ + 1 and FMQ − 2.
Magnetite crystals have a euhedral to subhedral equant form and can be skeletal, ranging between 5 and 50 μm in diameter. In our experiments performed at and below the FMQ buffer, magnetite is absent in runs doped with phosphorus (i.e. it is present in run 15Fe-9 but not run 15FeP-9), consistent with the study of Toplis et al. (1994a). The destabilisation of magnetite by the addition of phosphorus has been explained by Toplis et al. (1994a) as a result of the reduction in ferric iron and/or the formation of stable P−Fe3+ complexes. Between FMQ + 2 and FMQ − 2, the TiO2 and Al2O3 content of our experimental magnetites increases from 11.8 to 27.8 wt. % and from 2.1 to 4.3 wt. %, respectively. In the same range, the FeO content decreases from 74.5 to 52.2 wt. %. As the FeO content decreases, the MgO content of the magnetite increases from 3.1 to 4.5 wt. %. The Cr2O3 content is high, ranging between 0.2 and 6.9 wt. % and in general increases as oxygen fugacity decreases, however it should be noted that run 10P-7 (FMQ + 2) produces magnetite crystals with a significantly higher Cr2O3 content than other FMQ + 2 magnetites (5.7 wt.%) and run 15–9 (FMQ − 1) has a lower Cr2O3 content (2.9 wt. %) than other FMQ − 1 magnetites. The Xmt component (Xmt = 0.5*Fe3+/[Ti + (0.5*Fe3+) + (0.5*Al)]), calculated following the QUILF algorithm of Andersen et al. (1993), decreases under more reducing conditions from Mt64 at FMQ + 2 to Mt10 at FMQ − 2. The mole fraction of the ilmenite (Xilm) and magnetite (Xmt) end members are reported in Supplementary Table ST1 and illustrated as a function of fO2 in Fig. 3.

Ilmenite (a) and magnetite (b) composition as a function of oxygen fugacity (ΔFMQ), calculated following the QUILF algorithm of Andersen et al. (1993). The spread in the data at each fO2 is a result of the different starting compositions used. The influence of fO2 on Fe−Ti oxide composition is clear, particularly for magnetite in which a linear correlation between Xmt and ΔFMQ is observed
Minor oxide phases produced in our experiments are ulvöspinel (Fe2TiO4) and ferropseudobrookite (Fe2TiO5). Ulvöspinel is present only in runs at FMQ + 2 and crystals are elongate, between 5 and 15 μm in diameter. Ulvöspinel is characterised by higher TiO2 (32.7–38 wt. %) and lower FeO contents (51.1–56.7 wt. %) than coexisting magnetite and low Al2O3 contents < 0.80 wt. %. Ferropseudobrookite crystals, present at FMQ + 1 and FMQ + 2, are typically elongate and skeletal, reaching 40 μm in length but only 5 to 10 μm wide. Ferropseudobrookite is the oxide phase with the highest TiO2 content (50.3–57.4 wt. % TiO2) and lowest FeO content (30.1–38.6 wt. %). Minor components include Al2O3 (1.2–1.9 wt. %) and MgO (3.1–4.6 wt. %). Both ulvöspinel and ferropseudobrookite are unstable at low fO2 (e.g. Anovitz et al. 1985) appearing just below the hematite–magnetite buffer, which limits their incidence to FMQ > 1 in our experiments.
Clinopyroxene crystals are subhedral to euhedral, up to 250 μm in diameter. Clinopyroxene is present in all runs above the FMQ buffer, whilst below the FMQ buffer it is present only in starting compositions with the lowest Fe content (SC4/5 and SC4/10Fe). The TiO2 and MgO contents range between 1.8 and 3.1 wt. % and 11.9–13.8 wt. %, independent of fO2. The FeO contents range between 9.1 and 13.3 wt. % and the calcium component of clinopyroxene, expressed as wollastonite content (Wo) ranges between Wo37 and Wo48. The mg # ranges between 65 and 72. We observe no strong influence of fO2 on major element partitioning in clinopyroxene, which may be a result of the preferential incorporation of Fe3+ into the Fe−Ti oxides which always coexist with cpx.
Plagioclase feldspar, present at and above the FMQ buffer, is characterised by long, subhedral to euhedral elongate tabular grains which reach 600 μm in length and 10–75 μm in width. There is no significant influence of fO2 on the major element composition of plagioclase. The SiO2 contents range between 55.4 and 59.1 wt. %, Al2O3 between 25.0 and 27.2 wt. % and CaO between 9.2 and 10.7 wt. %. Anorthite contents in plagioclase range between An47 and An53 (Supplementary Table ST1).
Trace element contents for ilmenite, magnetite and clinopyroxene are reported in Supplementary Table ST3. We do not investigate trace element partitioning in plagioclase as this area has already been thoroughly studied (e.g. Bédard 2006; Aigner-Torres et al. 2007; Sun et al. 2017; Dygert et al. 2020) or in ferropseudobrookite as we have too few data on the latter to be statistically relevant. For ulvöspinel, only the largest crystals could be measured with the LA−ICP−MS, so trace element data is limited to only two experiments (runs 10P-7 and 5–10). This data will not be considered in the discussion but is reported in Supplementary Table ST3. It should also be noted that the Cr measured with LA-ICP-MS often gives a very large error in the magnetites with high Cr2O3 content although the EPMA data is much more consistent; therefore the EPMA measurements of Cr are reported in Supplementary Table ST3 and used for Cr partition coefficients in magnetite instead of LA−ICP−MS data.
Mineral−melt partition coefficients
Trace element mineral−melt partition coefficients \({(}D_{{\text{i}}} { = }c_{{{\text{mineral}}}}^{{\text{i}}} {/ }c_{{{\text{melt}}}}^{{\text{i}}}\)) for the phases of interest (ilmenite, magnetite, clinopyroxene) are presented in Supplementary Table ST3. The measured partition coefficients are in agreement with those in the literature that are measured under similar conditions (e.g. Toplis and Corgne 2002; Hill et al. 2011; Leitzke et al. 2016, 2017; Sievwright et al. 2017). As suggested by Prowatke and Klemme (2006) Henry’s Law is satisfied as the concentration of the trace element in the crystal is less than 1 wt. %. A summary of the partitioning data for ilmenite, magnetite and clinopyroxene is presented in Fig. 4.

Partitioning of trace elements between ilmenite−melt, magnetite−melt and clinopyroxene melt, illustrating the compatibility of each element. Divalent cations are dominantly compatible in all phases, whilst REE are incompatible. Error bars = 1σ. Nickel, V and Cr are the most compatible elements in all three phases. Shaded area represents detection limit which is < 0.1 for LREE and < 0.25 for HREE. Detection limit for Mo, Nb and Ta in cpx are 0.5, 1.2, and 1.7, respectively
The divalent cations Zn, Mn, Co and Ni are compatible in ilmenite and magnetite, whilst \(D_{{{\text{Zn}}}}^{{{\text{cpx}}}}\) and \(D_{{{\text{Mn}}}}^{{{\text{cpx}}}}\) are incompatible (D < 1) in some runs (though this does not appear to correlate with oxygen fugacity). Manganese is the least compatible divalent cation in each phase; \(D_{{{\text{Mn}}}}^{{{\text{ilm}}}}\) values range from 1.6 to 2.1, \(D_{{{\text{Mn}}}}^{{{\text{mt}}}}\) from 1.5 to 3.3 and \(D_{{{\text{Mn}}}}^{{{\text{cpx}}}}\) from 0.87 to 1.1, whilst Ni is the most compatible divalent cation (Fig. 4). Excluding Zn, which is present only in runs 5–10 and 10FeP-10, the partitioning behaviour of the divalent cations in magnetite appears to be influenced strongly by oxygen fugacity as they become more compatible with increasing fO2. Although 500 ppm of Zn was added to the starting composition in runs 7 and 12, and 250 ppm in runs 8 to 11, Zn is absent in every run except run 10. Zinc may have been lost from the experiments due to its low condensation temperature and volatility (Norris and Wood 2017); Sievwright et al. (2020) also report Zn loss from their experiments, attributed to bulk diffusion out of the crystal.
The rare earth elements (REE 3+) are incompatible (D < 1) in all three phases. The heavy REE (HREE: Gd-Lu + Y) are more abundant in ilmenite than the light REE (LREE: La-Eu), reflected in the partition coefficients (Fig. 4) which are close to or below the detection limit for the LREE and increase from 0.01 to 0.16 between DGd and DLu. The concentration of several REE in magnetite are also close to or below the detection limit, whilst clinopyroxene is the phase most enriched in REE, reflected in the higher DREE values. In contrast to ilmenite, the sharpest increase in DREE values for clinopyroxene is in the LREE which increase from 0.04 to 0.63 between DLa and DGd. Scandium is compatible (D > 1) in both ilmenite and clinopyroxene with DSc values ranging between 1.5 and 2.6 and 4.2 and 13.9, respectively, whilst it is incompatible in all but one run in magnetite. Overall, the partitioning of trivalent cations (REE, Y, Sc) shows no strong dependence on fO2 in the Fe−Ti oxides, whilst there is a weak negative correlation between clinopyroxene DREE and fO2. Additionally, below the FMQ buffer, clinopyroxene DEu is low relative to DSm and DGd.
Tantalum is the most compatible high-field strength (HFSE) cation studied in both ilmenite and magnetite, in agreement with the study of Sievwright et al. (2017), followed by Nb, Hf then Zr. In contrast, Hf is the most compatible HFSE cation in clinopyroxene, followed by Zr, Ta, then Nb. Although the HFSE Zr and Hf are incompatible in all three phases, Nb (D = 0.89–1.7) and Ta (D = 1.6–2.5) are compatible in ilmenite. There is no significant variation in DHFSE with fO2 for ilmenite and clinopyroxene, however oxygen fugacity exerts a strong control on DZr and DNb in magnetite which decrease with increasing fO2. A similar, albeit weaker trend is also observed for DTa in magnetite.
The partitioning behaviour of both Mo and V is strongly influenced by oxygen fugacity in all three phases. Molybdenum is least compatible in clinopyroxene, with DMo values ranging between 0.001 at FMQ + 2 to 0.04 at FMQ − 2. Similarly, with decreasing fO2 DMo increases from 0.25 to 1.1 in ilmenite, and from 0.03 to 0.49 in magnetite. Vanadium also becomes more compatible under more reducing conditions, reflected in the partition coefficients; with decreasing oxygen fugacity, \(D_{{\text{V}}}^{{{\text{cpx}}}}\) increases from 0.9 to 8.5, \(D_{{\text{V}}}^{{{\text{ilm}}}}\) from 7.0 to 17.4, and V is the most compatible in magnetite, ranging from 7.1 to 32.9. This can be explained by the flexible crystal structure of magnetite which permits the accommodation of a range of cations (Righter et al. 2006a; Arató and Audétat 2021); the similar ionic radii of V3+ (0.65 Å) and V4+ (0.61 Å) to Fe3+ (0.67 Å) and Ti4+ (0.64 Å) allows V to become strongly concentrated in magnetite (Arató and Audétat 2021). The relationship between DV and fO2 for our experimental data, corroborated by literature data, is illustrated in Fig. 5.

Partitioning of V between a ilmenite−melt, b magnetite−melt and c clinopyroxene melt as a function of fO2 for this study compared with previous work. Error bars = 1σ. A strong relationship between DV and fO2 is observed in all phases. Note that the experiments of Dygert et al. (2013, 2014) were conducted at pressures > 0.8 GPa
Chromium, likely in the form Cr3+ which is the most abundancy Cr valency in terrestrial basaltic melts (Papike et al. (2005) is the most compatible element in each phase with mineral−melt partition coefficients ranging between 25.3 and 153.3 for clinopyroxene, 42.6–120.3 in ilmenite and 196.4–2082.1 in magnetite. In contrast to Mo and V, DCr becomes more compatible with increasing fO2. Although there is an overall positive correlation between increasing fO2 and DCr in magnetite and clinopyroxene, there is a large spread in the data at FMQ + 2 with a cluster of anomalously low DCr values. Interestingly, the experimental run with the highest DCr for magnetite (run 10P-7) does not correspond to the run with the highest DCr for clinopyroxene (run 5–10). These observations suggest that fO2 is not the only control on Cr partitioning in magnetite and clinopyroxene.
Members of the magnetite–ulvöspinel solid solution have an inverse spinel structure and chromium is often a major component in spinels. Mixing along this solid solution is a potential contributing factor to the range in DCr values that we observe in magnetite. Chromium partition coefficients for our experimental magnetites and ulvöspinels are plotted as a function of mineral MgO content, alongside magnetite DCr from Toplis and Corgne (2002) for comparison (Supplementary Figure SF1). Although below 4 wt. % MgO there is a clear negative correlation between the MgO content and DCr of both spinel phases, there appears to be no obvious influence of fO2 on the relationship between mineral MgO content and DCr. Above 4 wt. % MgO, however, there are five magnetite data which show a lot of scatter and do not follow the same linear trend. The location of cations in the spinel structure is largely determined by ionic radii, valence and site preference energies which decrease in the octahedral site in the order Cr3+ > Al3+ ≥ Ti4+ > Fe2+ (Papike et al. 2005). It is therefore possible that substitution of Al3+ for Cr3+, for example, results in significantly higher chromium partition coefficients in these samples. This suggests that there may be mixing along the spinel solid solution, and the observed range in DCr values above 4 wt. % MgO may be evidence of disorder in the spinel structure.
Similarly, the range in clinopyroxene−melt DCr under more oxidising conditions is likely linked to the coupling of Cr3+ with Al3+ in the M1 site (Klemme & O’Neill 2000) as suggested by Mallmann and O’Neill (2009) for their own experimental clinopyroxenes. Site preference energies for the M1 site decrease in the order Al3+ > Fe3+ > Cr3+ > V3+ > Ti3+ (Papike et al. 2005).
Effect of melt composition
The degree of melt polymerisation and melt structure is here described by both NBO/T and optical basicity (Λ). For a comparison of the two measures as a function of melt MgO (wt. %), see Supplementary Figure SF2. The influence of NBO/T is strongest for the partitioning of divalent cations, which generally become less compatible as the melt becomes more depolymerised. Although there is no obvious correlation between DZn and NBO/T in any phase, which may be a result of its volatility as mentioned previously, there is a clear negative correlation between NBO/T and DMn, DCo and DNi for both ilmenite and magnetite (Fig. 6). The strongest correlation between NBO/T and \(D_{{\text{cpx}}}^{{{\text{2+}}}}\) is for DCo, whilst there is no clear correlation between NBO/T and DMn or DNi for clinopyroxene. In general, the slope that describes the relationship between NBO/T and D2+ is exponential, with a steeper slope at lower NBO/T tending towards a shallower slope with increasing depolymerisation of the melt. A similar trend has been described previously for several crystalline phases (Kohn and Schofield 1994; Toplis and Corgne 2002; Sievwright et al. 2017). One explanation for this trend is that more polymerised melts contain fewer potential sites onto which network-modifying cations can partition, however as pointed out by Sievwright et al. (2017) this does not explain why the relationship we observe is exponential, or why the relationship with NBO/T is observed only for divalent cations and not network-modifying cations with different valence states.

Mineral−melt partition coefficients of the divalent cations a zinc, b manganese, c cobalt and d nickel as a function of NBO/T. Runs at FMQ + 2, FMQ + 1, FMQ, FMQ -1 and FMQ -2 are denoted by circle, diamond, square, triangle and upside-down triangle symbols, respectively. Different phases are indicated by colour; ilmenite = blue, magnetite = green, clinopyroxene = purple. Error bars = 1σ
One of the drawbacks of using NBO/T is that it does not make a distinction between network-modifying cations. Optical basicity (Λ) provides an alternative measure of melt structure which distinguishes different cations with contrasting electron donor power (Duffy 1993) and therefore an advantage of Λ is that it differentiates between network-modifying cations like Ca, Mg that are cannot be distinguished in the calculation of NBO/T (Michely et al. 2017). Plotting D2+ against Λ produces a similarly exponential function of Λ (Supplementary Figure SF3) for DMn, DCo and DNi in magnetite and for DNi in clinopyroxene. This trend diverges into two paths at Λ = 0.54, producing two distinct populations; a similar trend is reported by Sievwright et al. (2017) for D2+ vs. optical basicity. This suggests that although melt composition has a clear effect, it is not the only control on divalent element partitioning. Furthermore, as mentioned previously we observe that decreasing oxygen fugacity results in melt depolymerisation; the Fe2+O content of the melt strongly influences NBO/T, therefore the observed correlation between D2+ and NBO/T may be an artefact of the changing fO2 conditions. For example, under more oxidising conditions the proportion of Fe−Ti oxides increases (Table 2) which decreases the concentration of melt FeO. The correlations that we observe between the melt structure—both optical basicity and NBO/T—and trace element partitioning are likely therefore driven by changes in fO2 and the relative abundance of Fe−Ti oxide phases that crystallise. As suggested by Sievwright et al. (2017), it is likely that the partitioning behaviour we observe is the result of the mineral−melt equilibria and associated changes in the Fe2+ and Fe3+ content of the melt.
Because TiO2 also has a depolymerising effect on silicate melts (Leitzke et al. 2016), the TiO2 content of the melt is also likely to influence trace element partitioning, though this is only important in studies with melts significantly enriched in TiO2, as basaltic TiO2 contents are low, around 2 wt. %. The effect of melt TiO2 in Ti-enriched melts has been demonstrated for DHFSE in ilmenite and clinopyroxene by Dygert et al. (2013, 2014) and Leitzke et al. (2016). Our ilm-melt, mt-melt and cpx-melt DHFSE partitioning data are plotted against melt TiO2 alongside literature data for comparison in Fig. 7 and Supplementary Figures SF4 and SF5, respectively. Ilmenite DHFSE are plotted as a function of melt TiO2 in Fig. 7 alongside literature data from Dygert et al. (2013) and Leitzke et al. (2016). We observe a negative correlation between ilmenite DHFSE and melt TiO2 content, particularly for DZr and DNb, corroborated by the experimental dataset of Dygert et al. (2013) over a similar melt TiO2 range. Although their melt compositions cover a broader range, between 0 and 10 wt. % TiO2 their ilmenite DHFSE follow the same trend that we observe in our own samples. We also find that there is a positive correlation between both DHf and DZr and the partitioning of Ti between ilmenite and melt. The relationship between melt TiO2 and magnetite-melt DHFSE is less significant (Supplementary Figure SF4), though there is a weak overall positive correlation. Literature data from Sievwright et al. (2020) are shown for comparison, however their data cover a smaller range of melt TiO2 and produce no clear trend. Our clinopyroxene-melt DHFSE data show an overall positive correlation with melt TiO2 for DZr and DHf (Supplementary Figure SF5), in agreement with Dygert et al. (2014), whilst there is no obvious correlation between DTa or DNb and melt TiO2. Melt TiO2 exerts the strongest influence on HFSE partitioning in ilmenite, concordant with the results of Dygert et al. (2013). They attribute this result to the association of HFSE with Fe−Ti–O complexes in the melt which are not constrained by a crystal lattice and thus accommodate cations with a range of sizes and charges, so highly charged cations like HFSE can be more easily accommodated by the melt which results in lower mineral−melt DHFSE with increasing melt TiO2.

Ilmenite−melt partition coefficients of the high-field strength elements a zircon, b hafnium, c tantalum and d niobium as a function of melt TiO2 (wt. %, normalised to 100%). Error bars = 1σ. Additional ilmenite−melt HFSE partitioning data from Dygert et al. 2013 and Leitzke et al. 2016 are shown for comparison. Similar figures for DHFSE in magnetite and clinopyroxene are given as Supplementary Figures SF4 and SF5
As mentioned previously, the effect that the addition of phosphorus to the starting compositions has on trace element partitioning is that it destabilises magnetite under more reducing conditions, resulting in limited trace element partitioning data at and below the FMQ buffer. At FMQ + 2, the divalent cations Mn, Co and Ni become less compatible with the addition of P, however this is a result of the affect that P has on NBO/T ratios rather than direct interactions between phosphorus and trace cations (Schmidt et al. 2006). A similar relationship is observed for clinopyroxene. Although there is similar effect of P on the divalent cations in ilmenite for starting composition SC4/10Fe and SC4/10FeP, the addition of P for starting composition SC4/15Fe results in an increase in Dilm2+. This effect may be explained by the influence that the addition of P has on the Fe3+/ΣFe ratios in the melt and the formation of P−Fe3+ melt complexes, which reduce the activity of Fe3+ and perhaps facilitate the substitution of divalent cations for Fe2+ in ilmenite.
Lattice strain model
Although partition coefficients for divalent cations and HFSE in ilmenite, magnetite and clinopyroxene are strongly dependent on melt composition, partition coefficients of REE, Y and Sc appear to be insensitive to melt composition. The mineral−melt partitioning of REE, Y and Sc can instead be quantified by the lattice strain model, Eq. 1, which relates the partition coefficient (Di) and ionic radius (ri, Å) of a particular element (i) to the ionic radius (r0) and partition coefficient (D0) of an idealised, optimal element (0) which enters the crystal lattice without straining it (Blundy and Wood 1994):
where E is the effective Young’s modulus of the site of interest in GPa; NA is Avogadro’s number; R is the gas constant and T is the temperature in Kelvin. The parameters D0 and r0 define the apex of an asymmetric parabola whilst E, the apparent Young’s modulus, measures the elastic response of the crystallographic site to the strain placed on the lattice structure by elements of a nonideal size (Blundy and Wood 1994). The greater the Young’s Modulus, the less elastic the site and the tighter the parabola (Blundy and Wood 2003; McDade et al. 2003; Fabbrizio et al. 2021). The lattice strain model has useful applications in estimating ideal partition coefficients for unmeasured elements, evaluating the quality of experimental datasets, and in determining the relationship between valency and oxygen fugacity (e.g. Toplis and Corgne 2002; Cartier et al. 2014). The model works particularly well for the REE, because there are no entropic or crystal field effects, and they comprise a continuous suite of ionic radii (e.g. Brice 1975; Beattie 1994; Burnham and O’Neill 2020). Lattice strain models were not fitted to divalent and tetravalent cations because of the possible mixed valence states for some elements, however the logarithm of D2+ for each phase are plotted as a function of ionic radius in Supplementary Figure SF6 and show that the partition coefficients in each phase decrease with increasing ionic radius.
Values for D0, r0 and E were obtained using a Monte Carlo-type approach following a method inspired from Cartier et al. (2014). This method consists of 100,000 randomly generated solutions in a defined range of lattice parameters. The optimum ionic radius, r0, was fixed to 0.69 Å for ilmenite and 0.63 Å for magnetite, based on the optimum r0 values determined by Dygert et al. (2013) and Sievwright et al. (2020), respectively, because all the elements of interest plot on one side of the parabola. The range in D0 values was restricted based on the visual observation of our Di values in each phase and the maximum E was set to 600 GPa for ilmenite and magnetite and 1200 GPa for clinopyroxene, based on the examples in the literature (Dygert et al. 2013; Liang et al. 2013; Sievwright et al. 2020). A total of 100,000 solutions were generated for each parameter, of which the best fit to measured Di values was determined using the Chi-squared test (χ2). This process was repeated 100 times in a loop using solutions within one standard deviation of the calculated Di values, and the mean of each parameter was substituted into Eq. 1 to produce best-fit parabolas. The fits are shown in Fig. 8a–c and values of D0 and E and their errors (reported as the standard deviation of 100 iterations) are provided in Supplementary Table ST4.

Onuma diagrams showing the the measured mineral−melt partition coefficients for REE, Sc and Y in a ilmenite, b magnetite and c clinopyroxene. Coefficients from the lattice strain model (Eq. 2) are used to plot the parabolas for each experiment, which describe the best fit for measured partition coefficients. Runs at different oxygen fugacity conditions are denoted by the different colours. Error bars = 1σ
The estimated D0 for the ilmenites produced in our experiments is higher than the D0 of 0.61 ± 0.19 reported by Dygert et al. (2013), ranging between 2.79 ± 0.78 and 3.53 ± 0.88. Young’s modulus values range between 376 ± 57 GPa and 425 ± 58 GPa. In magnetite, the D0 range between 1.96 ± 0.59 and 2.70 ± 0.18, whilst E values range between 299 ± 56 GPa and 434 ± 73 GPa. Only the right limb of the Onuma diagrams is present due to the possible mixed valence state of Cr and the incorporation of Al3+ and Cr3+ as major cations.
The REE and Y enter the M2 site in clinopyroxene, whilst Sc enters the M1 site. The REE and Y describe a parabola with an optimum ionic radius of 1.00 ± 0.02 to 1.02 ± 0.01, and Young’s moduli of 199 ± 32 GPa to 314 ± 40 GPa. The D0 values range between 0.38 ± 0.02 and 0.69 ± 0.04. The effect of fO2 on the REE and Y is reflected in the lattice strain parameters; at lower fO2, r0 and E are at the lowest end of the range and D0 is the highest. As scandium enters the M1 site of clinopyroxene, a parabola is not fitted through the DSc values. We find that there is no clear influence of oxygen fugacity on the partitioning behaviour of REE, Y and Sc described by the lattice strain model.
The usefulness of the lattice strain model for trivalent cations is that it can be used to predict the partition coefficient of an unknown trivalent element by fixing the parabola to a reference element, e.g. La, of which the partition coefficient is known. Re-writing Eq. (1) in terms of \(D_{{{\text{La}}}}^{{\text{ilm/melt}}}\) gives the following equation:
where \(r_{{0}}^{{{\text{ilm}}}}\) is fixed to 0.69 in ilmenite, \(D_{{{\text{La}}}}^{{\text{ilm/melt}}}\) and Eilm can be obtained from the modelled results at the required oxygen fugacity in Supplementary Table ST4, and ri defines the ionic radius of the trivalent element in question (sixfold coordination in ilmenite) which can be obtained from Shannon (1976), so that \(D_{{\text{i}}}^{{\text{ilm/melt}}}\) of a trivalent element of interest can be calculated.
Vanadium as an oxybarometer
Cations such as Fe, Ti, Cr and V exhibit a range of valence states over the broad spectrum of oxygen fugacity conditions experienced by planetary basalts; their typical oxidation states in oxides and silicates are Fe2+ and Fe3+, Ti3+ and Ti4+, Cr2+ and Cr3+, and V2+, V3+, V4+ and V5+ (Schreiber and Balzas 1982; Papike et al. 2005; Sutton et al. 2005; Mallmann and O’Neill 2009; Krawczynski and Grove 2012). In terrestrial magmas, vanadium is likely to be present as V3+, V4+ or V5+, however Toplis and Corgne (2002) have demonstrated that the V4+ is the dominant valence state between FMQ -2 and FMQ. Further decreasing oxygen fugacity results in a decrease in the relative proportion of V4+ in the melt and an increase in the proportion of V3+ (Schreiber and Balzas 1982). This may be explained by the large octahedral site preference energy of vanadium (Canil and Fedortchouk 2000). Based on the similarities between Fe3+ and Ti4+, respectively, it is assumed that both V3+ and V4+ may enter the M1 site of clinopyroxene (Shannon 1976; Wood and Blundy 1997; Toplis and Corgne 2002) whilst V is predominantly V3+ in magnetite and occupies only the octahedral site (Canil 1999; Toplis and Corgne 2002; Righter et al. 2006a). Previous experimental studies have shown that the partitioning behaviour of V is strongly dependent on oxygen fugacity (Schreiber and Balzas 1982; Canil 1999; Toplis and Corgne 2002; Sutton et al. 2005; Karner et al. 2006; Righter et al. 2006b, Mallmann & O’Neill 2009, 2013). The proportion of V3+ (i.e. the V3+/ΣV ratio) in the melt and thus the total DV (the sum of DV2+, DV3+, DV4+, DV5+) decreases with increasing oxygen fugacity. As such, the compatibility of vanadium is a function of the redox conditions of the magma (Canil 2002). The benefit of using vanadium in particular as a proxy for fO2 is that as long as the system remains closed, changes to the V3+/ΣV ratio after crystallisation have no effect on the redox record. The bulk vanadium mineral−melt partition coefficient can be defined as
And the 3 dominant V oxide components in terrestrial magmas can be related to each other through a series of redox reactions (Mallmann & O’Neill 2009):
Toplis and Corgne (2002) show that for starting composition SC4, V4+ is the dominant valence state at FMQ -2. With increasing fO2, the proportion of V5+ increases whereas that of V4+ decreases, until V5+ becomes the dominant valence state above FMQ + 2. The proportion of V3+ in the melt is negligible above FMQ -2. Based on these observations, we can infer that the relative proportion of V4+ and V5+ in the melt is approximately equal in our experiments at FMQ + 2, whilst V4+ becomes increasingly dominant as fO2 decreases.
As illustrated in Fig. 5, our data suggest that \(D_{{\text{V}}}^{{\text{ilm/liq}}}\) is less dependent on fO2 than \(D_{{\text{V}}}^{{\text{mt/liq}}}\). The TiO2 content of magnetite increases with decreasing fO2, which increases the possibility of potential Ti4+−V4+ substitutions (Toplis and Corgne 2002). Under the same conditions, proportion of V3+ increases in the melt and thus in associated magnetite; the combination of these effects increases the \({\text{D}}_{{\text{V}}}^{{\text{mt/liq}}}\). Ilmenite, on the other hand, easily concentrates highly charged cations such as Nb5+ and Ta5+. Niobium in particular has a similar ionic radius to Ti4+ and thus may easily partition into ilmenite by coupled substitution, charge balanced by trivalent cations such as Al3+ (e.g. Nb5+ + Al3+ = 2Ti4+) (Horng et al. 1999). Our data show that Nb and Ta are compatible in ilmenite (DNb > 1, DTa > 1.5) and incompatible in magnetite (DNb < 0.21, DTa < 0.33), corroborated by literature data (Green and Pearson 1987; Nielsen and Beard 2000; Dygert et al. 2013; Sievwright et al. 2020). This ability to accommodate highly charged cations may result in the insensitivity of ilmenite to different vanadium valence states and thus the reduced dependence of \(D_{{\text{V}}}^{{\text{ilm/liq}}}\) partitioning on fO2, relative to magnetite.
The overall dependence of DV on oxygen fugacity (ΔFMQ) and melt composition is expressed in a series of linear regression equations for ilmenite, magnetite and clinopyroxene:
Although we find no clear influence of melt composition on the partitioning of elements other than divalent cations, we include a parameter for NBO/T for the Fe−Ti oxides to account for potential interplay between Fe and P in the melt. Select literature data from Toplis and Corgne (2002) and Sievwright et al. (2020), conducted under similar conditions (1 bar, and 1068–1095 °C) were included in the regression model for ΔFMQ cpx and ΔFMQ mt. Additional literature data from the same authors are fit using Eqs. (6–8) and are generally well-predicted; the accuracy of these models are shown in Fig. 9a–c. The majority of data points fall on or close to the 1:1 line, attesting to the accuracy of the model.

Comparison between model predicted and experimentally measured log fO2 values for a ilmenite, b magnetite and c clinopyroxene. Dotted lines define 1:2 and 2:1 values; solid lines define 1:1 values. Predicted values were calculated using Eqs. 6–8. Literature data: Klemme et al. 2006; Sievwright et al. 2020; Toplis and Corgne 2002. Panel d shows the calibration of the of DVmt/melt / DVilm/melt oxybarometer. Regression equations for each fit are given in each panel
Calibration of a DV oxybarometer
Although our data show that there is a strong relationship between DV and fO2, the application of the above vanadium oxybarometers for Fe−Ti oxides in particular requires knowledge of the melt composition. Bulk mineral−liquid partition coefficients can be estimated for cumulate rocks, for example, by using appropriate (in terms of e.g. fO2, pressure, temperature and expected parental magma composition) experimentally derived partition coefficients, such as those reported in this study. Thus, the composition of the glass is not required. Various two-oxide oxybarometers have been developed (Ghiorso and Sack 1991; Lindsley and Frost 1992; Andersen et al. 1993; Lattard et al. 2005; Sauerzapf et al. 2008; Arató and Audétat 2017b) which are generally based on the compositions and cation proportions of coexisting Fe−Ti oxides, however there are few oxybarometers that take advantage of the strong fO2 dependence of vanadium partitioning in Fe−Ti oxides in melt-free compositions.
To account for the different dependences of \(D_{{\text{V}}}^{{\text{mt/liq}}}\) and \(D_{{\text{V}}}^{{\text{ilm/liq}}}\) partitioning on fO2, we present an empirical approach in which the ratio of DV between magnetite and ilmenite pairs is used as a proxy for fO2, in the form of the equation:
in which the average DV of our ilmenite−magnetite pairs at fO2 = FMQ − 2.13, − 0.93 and 0.05 are used in the calibration of the equation, incorporating the standard deviation of DV at each fO2. The result of the calibration of the DVmt/ilm oxybarometer is shown in Fig. 9d. Although we produce magnetite–ilmenite pairs (of a sufficient size to measure) at only three fO2, we expect that we can predict fO2 as ΔFMQ within the range of fO2 in this study (i.e. fO2 = FMQ − 2.13 to FMQ + 0.05). To predict fO2 over a broader range than we cover in this study, further experiments are required to produce magnetite–ilmenite pairs covering more oxidising and reducing conditions. We do not use natural magnetite–ilmenite pairs to supplement our experimental data as the oxygen fugacity is already an estimate with error based on the alternative Fe−Ti oxide oxybarometers, however we do present tuff data from Arató and Audétat (2017a) and Greber et al. (2021) to illustrate that our oxybarometer may be extended to encompass more oxidising conditions. We also incorporate this natural data into our model in Supplementary Figure SF7 and provide an equation, but we do stress that calibration of reliable oxybarometers require precise knowledge of the fO2 conditions which can only be achieved in experimental studies.
Application to a ferrobasaltic layered intrusion
Widely considered the type example of a strongly differentiated layered intrusion, the Skaergaard intrusion is one of a series of intrusive complexes along the east coast of Greenland (Wager and Deer 1939; Wager and Brown 1967; Naslund 1984; McBirney 1989; Jang and Naslund 2001). The Skaergaard intrusion contains abundant ilmenite and magnetite, along with typical basaltic phases such as olivine, plagioclase and clinopyroxene; the occurrence of pyroxene, magnetite and olivine in particular is used to divide the layered series into zones. The layered series formed as a “cumulate”—a term introduced by Wager (1960) to describe the rocks of the Skaergaard intrusion—on the floor of the magma chamber.
One of the most important uses for estimated mineral/melt partition coefficients in layered intrusions such as the Skaergaard intrusion is to constrain differentiation processes and determine the distribution of economically important concentrations of elements such as PGE, Au and (e.g. Hunter and Sparks 1987; Jang and Naslund 2001; Jang et al. 2001; Tegner et al. 2009; Nielsen et al. 2014). Estimated vanadium/melt partition coefficients for the Skaergaard intrusion reported by McBirney (1998) are used to apply the above DVmt/ilm oxybarometer (Eq. 9) to the layered series of Skaergaard. We apply the equation to LZc of the Skaergaard intrusion, the horizon in which Fe−Ti oxides first become modally abundant (Jang et al. 2001), using estimated DVilm/liquid and DVmt/liquid values of 11.1 and 24.7, respectively from McBirney (1998). Application of the oxybarometer yields an oxygen fugacity of 0.18 log units above FMQ; this is in good agreement with the fO2 estimate of between 0.12 and 0.09 between LZb and LZc modelled by Thy et al. (2009). This model could be refined when further trace element analyses of magnetite and ilmenite from the Skaergaard intrusion are published.
Conclusions
We present partition coefficients for a suite of trace elements in ilmenite, magnetite and clinopyroxene under stable pressure and temperature conditions and over a range of redox conditions. The partitioning of the divalent cations Zn, Mn, Co and Ni, and the high-field strength elements Zr, Hf, Ta and Nb between mineral phases and coexisting silicate melt are shown to be influenced strongly by melt compositions, whilst the partitioning behaviour of REE, Y and Sc are described by cation radius and site elasticity by application of the lattice strain model. We find that the most compatible elements in all phases are the transition metals (e.g. Cr, V), which are also strongly influenced by oxygen fugacity. By exploiting the linear relationship that we observe between DV and oxygen fugacity, we present empirical equations to predict ΔFMQ in ilmenite, magnetite and clinopyroxene as a function of DV. We also calibrate an oxybarometer based on the partitioning of V between magnetite−melt and ilmenite−melt, and apply this oxybarometer to the Skaergaard intrusion to estimate the fO2 conditions at the horizon in which Fe−Ti oxides become modally abundant.
Our data span a range of fO2 and melt compositions and complement literature data, particularly for magnetite and clinopyroxene. As much of the previously published experimental data on ilmenite−melt partitioning is focused at higher pressures and temperatures analogous to the moon, future work may focus on the development of experiments at lower temperatures and pressures, similar to this study, over a broader range of oxygen fugacity conditions.
Availability of data and material
Additional data is available as supplementary electronic files.
References
Aigner-Torres M, Blundy J, Ulmer P, Pettke T (2007) Laser ablation ICPMS study of trace element partitioning between plagioclase and basaltic melts: an experimental approach. Contrib to Mineral Petrol 153:647–667. https://doi.org/10.1007/s00410-006-0168-2
Andersen DJ, Lindsley DH (1988) Internally consistent solution models for Fe-Mg-Mn-Ti oxides: Fe-Ti oxides. Am Min 73:714–726
Andersen DJ, Lindsley DH, Davidson PM (1993) QUILF: A pascal program to assess equilibria among FeMgMnTi oxides, pyroxenes, olivine, and quartz. Comput Geosci 19:1333–1350. https://doi.org/10.1016/0098-3004(93)90033-2
Anovitz LM, Treiman AH, Essene EJ et al (1985) The heat-capacity of ilmenite and phase equilibria in the system Fe-T-O. Geochim Cosmochim Acta 49:2027–2040. https://doi.org/10.1016/0016-7037(85)90061-4
Arató R, Audétat A (2017a) FeTiMM—a new oxybarometer for mafic to felsic magmas. Geochemical Perspect Lett 5:19–23. https://doi.org/10.7185/geochemlet.1740
Arató R, Audétat A (2017b) Experimental calibration of a new oxybarometer for silicic magmas based on vanadium partitioning between magnetite and silicate melt. Geochim Cosmochim Acta 209:284–295. https://doi.org/10.1016/j.gca.2017.04.020
Arató R, Audétat A (2021) Titanomagnetite—silicate melt oxybarometry. Magma redox geochemistry. Wiley, Hoboken, pp 369–380
Armienti P, Tonarini S, Innocenti F, D’Orazio M (2007) Mount Etna pyroxene as tracer of petrogenetic processes and dynamics of the feeding system. Spec Pap Geol Soc Am 418:265–276. https://doi.org/10.1130/2007.2418(13)
Beattie P (1994) Systematics and energetics of trace-element partitioning between olivine and silica melts: implications for the nature of mineral/melt partitioning. Chem Geol 17:57–71
Bédard JH (2006) Trace element partitioning in plagioclase feldspar. Geochim Cosmochim Acta 70:3717–3742. https://doi.org/10.1016/j.gca.2006.05.003
Bédard JH (2010) Parameterization of the Fe=Mg exchange coefficient (Kd) between clinopyroxene and silicate melts. Chem Geol 274:169–176. https://doi.org/10.1016/j.chemgeo.2010.04.003
Blundy J, Wood B (1994) Prediction of crystal-melt partition coefficients from elastic moduli. Nature 372:452–454
Blundy J, Wood B (2003) Partitioning of trace elements between crystals and melts. Earth Planet Sci Lett 210:383–397. https://doi.org/10.1016/S0012-821X(03)00129-8
Bosi F, Hålenius U, Skogby H (2009) Crystal chemistry of the magnetite-ulvöspinel series. Am Mineral 94:181–189. https://doi.org/10.2138/am.2009.3002
Brice JC (1975) Some thermodynamic aspects of the growth of strained crystals. J Cryst Growth 28:249–253. https://doi.org/10.1016/0022-0248(75)90241-9
Buddington AF, Lindsley DH (1964) Iron-titanium oxide minerals and synthetic equivalents. J Petrol 5:310–357. https://doi.org/10.1093/petrology/5.2.310
Burnham AD, O’Neill HSC (2020) Mineral–melt partition coefficients and the problem of multiple substitution mechanisms: insights from the rare earths in forsterite and protoenstatite. Contrib to Mineral Petrol 175:1–19. https://doi.org/10.1007/s00410-019-1636-9
Canil D (1999) Vanadium partitioning between orthopyroxene, spinel and silicate melt and the redox states of mantle source regions for primary magmas. Geochim Cosmochim Acta 63:557–572. https://doi.org/10.1016/S0016-7037(98)00287-7
Canil D (2002) Vanadium in peridotites mantle redox and tectonic environments: Archean to present. Earth and Planetary Science Letters 195(1–2):75–90. https://doi.org/10.1016/S0012-821X(01)00582-9
Canil D, Fedortchouk Y (2000) Clinopyroxene-liquid partitioning for vanadium and the oxygen fugacity during formation of cratonic and oceanic mantle lithosphere. J Geophys Res Solid Earth 105:26003–26016. https://doi.org/10.1029/2000jb900221
Cartier C, Hammouda T, Doucelance R et al (2014) Experimental study of trace element partitioning between enstatite and melt in enstatite chondrites at low oxygen fugacities and 5GPa. Geochim Cosmochim Acta 130:167–187. https://doi.org/10.1016/j.gca.2014.01.002
Charlier B, Namur O, Bolle O et al (2015) Fe-Ti-V-P ore deposits associated with Proterozoic massif-type anorthosites and related rocks. Earth-Science Rev 141:56–81. https://doi.org/10.1016/j.earscirev.2014.11.005
Costa F, Morgan D (2010) Time constraints from chemical equilibration in magmatic crystals. Wiley, Hoboken
Di Flavio S, Mollo S, Ubide T et al (2020) Mush cannibalism and disruption recorded by clinopyroxene phenocrysts at Stromboli volcano: New insights from recent 2003–2017 activity. Lithos. https://doi.org/10.1016/j.lithos.2020.105440
Duffy JA (1993) A review of optical basicity and its applications to oxidic systems. Geochim Cosmochim Acta 57:3961–3970. https://doi.org/10.1016/0016-7037(93)90346-X
Dygert N, Liang Y, Hess P (2013) The importance of melt TiO2 in affecting major and trace element partitioning between Fe-Ti oxides and lunar picritic glass melts. Geochim Cosmochim Acta 106:134–151. https://doi.org/10.1016/j.gca.2012.12.005
Dygert N, Liang Y, Sun C, Hess P (2014) An experimental study of trace element partitioning between augite and Fe-rich basalts. Geochim Cosmochim Acta 132:170–186. https://doi.org/10.1016/j.gca.2014.01.042
Dygert N, Draper DS, Rapp JF et al (2020) Experimental determinations of trace element partitioning between plagioclase, pigeonite, olivine, and lunar basaltic melts and an fO2 dependent model for plagioclase-melt Eu partitioning. Geochim Cosmochim Acta 279:258–280. https://doi.org/10.1016/j.gca.2020.03.037
Fabbrizio A, Schmidt MW, Petrelli M (2021) Effect of fO2 on Eu partitioning between clinopyroxene, orthopyroxene and basaltic melt: Development of a Eu3+/Eu2+ oxybarometer. Chem Geol 559:119967. https://doi.org/10.1016/j.chemgeo.2020.119967
Ghiorso MS, Evans BW (2008) Thermodynamics of rhombohedral oxide solid solutions and a revision of the Fe-Ti two-oxide geothermometer and oxygen-barometer. Am J Sci 308:957–1039. https://doi.org/10.2475/09.2008.01
Ghiorso MS, Sack RO (1991) Mineralogy and Fe - Ti oxide geothermometry : thermodynamic formulation and the estimation of intensive variables in silicic magmas. Contrib Miner Pet 108:485–510
Greber ND, Pettke T, Vilela N et al (2021) Titanium isotopic compositions of bulk rocks and mineral separates from the Kos magmatic suite: Insights into fractional crystallization and magma mixing processes. Chem Geol 578:120303. https://doi.org/10.1016/j.chemgeo.2021.120303
Green TH, Pearson NJ (1987) An experimental study of Nb and Ta partitioning between Ti-rich minerals and silicate liquids at high pressure and temperature. Geochim Cosmochim Acta 51:55–62. https://doi.org/10.1016/0016-7037(87)90006-8
Grove TL, Bryan WB (1983) Fractionation of pyroxene-phyric MORB at low pressure: an experimental study. Contrib to Mineral Petrol 84:293–309. https://doi.org/10.1007/BF01160283
Hess PC, Parmentier EM (1995) A model for the thermal and chemical evolution of the Moon’s interior: implications for the onset of mare volcanism. Earth Planet Sci Lett 134:501–514. https://doi.org/10.1016/0012-821X(95)00138-3
Hess P, Rutherford M, Campbell H (1978) Ilmenite crystallization in nonmare basalt - Genesis of KREEP and high-Ti mare basalt. Proc Lunar Sci Conf 9:705–724
Hill E, Wood BJ, Blundy JD (2000) The effect of Ca-Tschermaks component on trace element partitioning between clinopyroxene and silicate melt. Lithos 53:203–215
Hill E, Blundy JD, Wood BJ (2011) Clinopyroxene-melt trace element partitioning and the development of a predictive model for HFSE and Sc. Contrib to Mineral Petrol 161:423–438. https://doi.org/10.1007/s00410-010-0540-0
Horng W-S, Hess P, Gan H (1999) The interactions between M+5 cations (Nb+5, Ta+5, or P+5) and anhydrous haplogranite melts. Geochim Cosmochim Acta 63:2419–2428
Howarth GH, Prevec SA (2013) Hydration vs. oxidation: Modelling implications for Fe-Ti oxide crystallisation in mafic intrusions, with specific reference to the Panzhihua intrusion. SW China Geosci Front 4:555–569. https://doi.org/10.1016/j.gsf.2013.03.002
Hoover JD, Irvine TN (1978) Liquidus relations and Mg–Fe partitioning on part of the system Mg2SiO4–Fe2SiO4–CaMgSi2O6–CaFeSi2O6–KAlSi3O8–SiO2. Carnegie Institution of Washington Yearbook 77:774–784
Humphreys MCS (2009) Chemical evolution of intercumulus liquid, as recorded in plagioclase overgrowth rims from the Skaergaard intrusion. J Petrol 50:127–145. https://doi.org/10.1093/petrology/egn076
Hunter RH, Sparks RSJ (1987) The differentiation of the Skaergaard Intrusion. Contrib to Mineral Petrol 95:451–461. https://doi.org/10.1007/BF00402205
Jang YD, Naslund HR (2001) Major and trace element composition of Skaergaard plagioclase; geochemical evidence for changes in magma dynamics during the ® nal stage of crystallization of the Skaergaard intrusion. Contrib Miner Pet 140:441–457
Jang YD, Naslund HR, Mcbirney AR (2001) The differentiation trend of the Skaergaard intrusion and the timing of magnetite crystallization : iron enrichment revisited. Earth Planet Sci Lett 189:189–196
Jarosewich E, Nelen J, Norberg J (1980) Reference samples for electron microprobe analysis. Geostand Geoanalytical Res 4:43–47. https://doi.org/10.1111/j.1751-908X.1980.tb00273.x
Jochum KP, Willbold M, Raczek I et al (2005) Chemical characterisation of the USGS reference glasses GSA-1G, GSC-1G, GSD-1G, GSE-1G, BCR-2G, BHVO-2G and BIR-1G using EPMA, ID-TIMS, ID-ICP-MS and LA-ICP-MS. Geostand Geoanalytical Res 29:285–302. https://doi.org/10.1111/j.1751-908x.2005.tb00901.x
Juster TC, Grove TL, Perfit MR (1989) Experimental constraints on the generation of FeTi basalts, andesites, and rhyodacites at the Galapagos Spreading Center, 85°W and 95°W. J Geophys Res 94:9251–9274. https://doi.org/10.1029/JB094iB07p09251
Karner JM, Sutton SR, Papike JJ et al (2006) Application of a new vanadium valence oxybarometer to basaltic glasses from the Earth, Moon, and Mars. Am Mineral 91:270–277. https://doi.org/10.2138/am.2006.1830
Klemme S, Günther D, Hametner K et al (2006) The partitioning of trace elements between ilmenite, ulvospinel, armalcolite and silicate melts with implications for the early differentiation of the moon. Chem Geol 234:251–263. https://doi.org/10.1016/j.chemgeo.2006.05.005
Klemme Hugh S, O'Neill S (2000) The effect of Cr on the solubility of Al in orthopyroxene: experiments and thermodynamic modelling. Contrib Mineral Petrol 140(1):84–98. https://doi.org/10.1007/s004100000140
Kohn SC, Schofield PF (1994) The importance of melt composition in controlling trace-element behaviour: an experimental study of Mn and Zn partitioning between forsterite and silicate melts. Chem Geol 117:73–87. https://doi.org/10.1016/0009-2541(94)90122-8
Krawczynski MJ, Grove TL (2012) Experimental investigation of the influence of oxygen fugacity on the source depths for high titanium lunar ultramafic magmas. Geochim Cosmochim Acta 79:1–19. https://doi.org/10.1016/j.gca.2011.10.043
Lattard D, Sauerzapf U, Käsemann M (2005) New calibration data for the Fe-Ti oxide thermo-oxybarometers from experiments in the Fe-Ti-O system at 1 bar, 1,000–1,300°C and a large range of oxygen fugacities. Contrib to Mineral Petrol 149:735–754. https://doi.org/10.1007/s00410-005-0679-2
Leitzke FP, Fonseca ROC, Michely LT et al (2016) The effect of titanium on the partitioning behavior of high-field strength elements between silicates, oxides and lunar basaltic melts with applications to the origin of mare basalts. Chem Geol 440:219–238. https://doi.org/10.1016/j.chemgeo.2016.07.011
Leitzke FP, Fonseca ROC, Sprung P et al (2017) Redox dependent behaviour of molybdenum during magmatic processes in the terrestrial and lunar mantle : Implications for the Mo / W of the bulk silicate Moon. Earth Planet Sci Lett. https://doi.org/10.1016/j.epsl.2017.07.009
Leuthold J, Blundy JD, Holness MB, Sides R (2014) Successive episodes of reactive liquid flow through a layered intrusion (Unit 9, Rum Eastern Layered Intrusion, Scotland). Contrib Miner Pet. https://doi.org/10.1007/s00410-014-1021-7
Liang Y, Sun C, Yao L (2013) A REE-in-two-pyroxene thermometer for mafic and ultramafic rocks. Geochim Cosmochim Acta 102:246–260. https://doi.org/10.1016/j.gca.2012.10.035
Lindsley DH, Frost BR (1992) Equilibria among Fe-Ti oxides, pyroxenes, olivine, and quartz: part I. Theory Am Mineral 77:987–1003
Longhi J (1992) Experimental petrology and petrogenesis of mare volcanics. Geochim Cosmochim Acta 56:2235–2251
Mallmann G, O’Neill HSC (2009) The crystal/melt partitioning of V during mantle melting as a function of oxygen fugacity compared with some other elements (Al, P, Ca, Sc, Ti, Cr, Fe, Ga, Y, Zr and Nb). J Petrol 50:1765–1794. https://doi.org/10.1093/petrology/egp053
Mallmann G, O’Neill HSC (2013) Calibration of an empiricalthermometer and oxybarometer based on the partitioning of sc, Y and V between olivine and silicate melt. J Petrol 54:933–949. https://doi.org/10.1093/petrology/egt001
Masotta M, Pontesilli A, Mollo S et al (2020) The role of undercooling during clinopyroxene growth in trachybasaltic magmas: Insights on magma decompression and cooling at Mt. Etna Volcano Geochim Cosmochim Acta 268:258–276. https://doi.org/10.1016/j.gca.2019.10.009
McBirney AR (1989) The Skaergaard Layered Series: I Structure and Average Compositions. J Petrol 30:363–397
McBirney AR (1998) The skaergaard layered series. Part VI. Included Trace Elements J Petrol 39:255–276. https://doi.org/10.1093/petrology/43.3.535
McDade P, Blundy JD, Wood BJ (2003) Trace element partitioning on the Tinaquillo solidus at 1.5 GPa. Phys Earth Planet Inter 139:129–147. https://doi.org/10.1016/S0031-9201(03)00149-3
McKay G, Wagstaff J, Yang S-R (1986) Zirconium hafnium and rare earth element partition coefficients for ilmenite and other minerals in high-Ti lunar mare basalts: an experimental study. J Geophys Res: Solid Earth 91(B4):229–237. https://doi.org/10.1029/JB091iB04p0D229
Michely LT, Leitzke FP, Speelmanns IM, Fonseca ROC (2017) Competing effects of crystal chemistry and silicate melt composition on trace element behavior in magmatic systems: insights from crystal/silicate melt partitioning of the REE, HFSE, Sn, In, Ga, Ba, Pt and Rh. Contrib Miner Petrol. https://doi.org/10.1007/s00410-017-1353-1
Mollo S, Ubide T, Di F et al (2020) Polybaric/polythermal magma transport and trace element partitioning recorded in single crystals: a case study of a zoned clinopyroxene from Mt. Etna Lithos. https://doi.org/10.1016/j.lithos.2020.105382
Müller T, Dohmen R, Becker HW et al (2013) Fe-Mg interdiffusion rates in clinopyroxene: Experimental data and implications for Fe-Mg exchange geothermometers. Contrib to Mineral Petrol 166:1563–1576. https://doi.org/10.1007/s00410-013-0941-y
Mysen B (1983) The structure of silicate melts. Ann Rev Earth Planet Sci 11:75–97. https://doi.org/10.1090/gsm/146/03
Namur O, Humphreys MCS (2018) Trace element constraints on the differentiation and crystal mush solidification in the Skaergaard intrusion, Greenland. J Petrol 59:387–418. https://doi.org/10.1093/petrology/egy032
Namur O, Charlier B, Toplis MJ et al (2010) Crystallization sequence and magma chamber processes in the ferrobasaltic sept Iles layered intrusion, Canada. J Petrol 51:1203–1236. https://doi.org/10.1093/petrology/egq016
Namur O, Charlier B, Toplis MJ et al (2011) Differentiation of tholeiitic basalt to a—type granite in the sept Iles layered intrusion, Canada. J Petrol 52:487–539. https://doi.org/10.1093/petrology/egq088
Namur O, Humphreys MCS, Holness MB (2013) Lateral reactive infiltration in avertical gabbroic crystal mush, skaergaard intrusion, east greenland. J Petrol 54:985–1016. https://doi.org/10.1093/petrology/egt003
Naslund HR (1984) Petrology of the upper border series of the Skaergaard Intrusion. J Petrol 25:185–212
Neave DA, Putirka KD (2017) A new clinopyroxene-liquid barometer, and implications for magma storage pressures under Icelandic rift zones. Am Mineral 102:777–794. https://doi.org/10.2138/am-2017-5968
Nielsen RL, Beard JS (2000) Magnetite-melt HFSE partitioning. Chem Geol 164:21–34. https://doi.org/10.1016/S0009-2541(99)00139-4
Nielsen RL, Gallahan WE, Newberger F (1992) Experimentally determined mineral-melt partition coefficients for Sc, Y and REE for olivine, orthopyroxene, pigeonite, magnetite and ilmenite. Contrib to Mineral Petrol 110:488–499
Nielsen RL, Forsythe LM, Gallahan WE, Fisk MR (1994) Major- and trace-element magnetite-melt equilibria. Chem Geol 117:167–191
Nielsen TFD, Andersen JCØ, Holness MB et al (2014) The Skaergaard PGE and gold deposit: the result of in situ fractionation, sulphide saturation, and magma chamber-scale precious metal redistribution by immiscible Fe-rich melt. J Petrol 56:1643–1676. https://doi.org/10.1093/petrology/egv049
Norris CA, Wood BJ (2017) Earth’s volatile contents established by melting and vaporization. Nature 549:507–510. https://doi.org/10.1038/nature23645
O’Neill HSC, Navrotsky A (1984) Cation distributions and thermodynamic properties of binary spinel solid solutions. Am Mineral 69:733–753
O’Neill HSC, Pownceby MI, Wall VJ (1988) Ilmenite-rutile-iron and ulvospinel-ilmenite-iron equilibria and the thermochemistry of ilmenite (FeTiO3) and ulvospinel (Fe2TiO4). Geochim Cosmochim Acta 52:2065–2072. https://doi.org/10.1016/0016-7037(88)90185-8
Pang KN, Zhou MF, Lindsley D et al (2008) Origin of Fe-Ti oxide ores in mafic intrusions: evidence from the Panzhihua intrusion, SW China. J Petrol 49:295–313. https://doi.org/10.1093/petrology/egm082
Papike JJ, Karner JM, Shearer CK (2005) Comparative planetary mineralogy: valence state partitioning of Cr, Fe, Ti, and V among crystallographic sites in olivine, pyroxene, and spinel from planetary basalts. Am Mineral 90:277–290. https://doi.org/10.2138/am.2005.1779
Pownceby MI, Fisher-white MJ (1999) Phase equilibria in the systems Fe2O3-MgO-TiO2 and FeO-MgO-TiO2 between 1173 and 1473 K, and Fe2+-Mg mixing properties of ilmenite, ferrous-pseudobrookite and ulvoÈspinel solid solutions. Contrib Miner Pet 135:198–211
Prowatke S, Klemme S (2006) Rare earth element partitioning between titanite and silicate melts : Henry’s law revisited. Geochim Cosmochim Acta 70:4997–5012. https://doi.org/10.1016/j.gca.2006.07.016
Putirka KD (2008) Thermometers and barometers for volcanic systems. Rev Mineral Geochemistry 69:61–120. https://doi.org/10.2138/rmg.2008.69.3
Putirka K, Johnson M, Kinzler R et al (1996) Thermobarometry of mafic igneous rocks based on clinopyroxene-liquid equilibria, 0–30 kbar. Contrib to Mineral Petrol 123:92–108. https://doi.org/10.1007/s004100050145
Raymond KN, Wenk HR (1971) Lunar ilmenite (Refinement of the crystal structure). Contrib to Mineral Petrol 30:135–140. https://doi.org/10.1007/BF00372254
Reynolds IM (1985) The nature and origin of titaniferous magnetite-rich layers in the upper zone of the Bushveld complex: a review and synthesis. Econ Geol 80:1089–1108. https://doi.org/10.2113/gsecongeo.80.4.1089
Righter K, Leeman WP, Hervig RL (2006a) Partitioning of Ni, Co and V between spinel-structured oxides and silicate melts: Importance of spinel composition. Chem Geol 227:1–25. https://doi.org/10.1016/j.chemgeo.2005.05.011
Righter K, Sutton SR, Newville M et al (2006b) An experimental study of the oxidation state of vanadium in spinel and basaltic melt with implications for the origin of planetary basalt. Am Mineral 91:1643–1656. https://doi.org/10.2138/am.2006.2111
Sauerzapf U, Lattard D, Burchard M, Engelmann R (2008) The titanomagnetite-ilmenite equilibrium: new experimental data and thermo-oxybarometric application to the crystallization of basic to intermediate rocks. J Petrol 49:1161–1185. https://doi.org/10.1093/petrology/egn021
Schmidt MW, Connolly JAD, Günther D, Bogaerts M (2006) Element partitioning: the role of melt structure and composition. Science 312:1646–1650. https://doi.org/10.1126/science.1126690
Schreiber H, Balzas G (1982) Vanadium as an oxygen geobarometer in basaltic magmas: The further development of a geochemical electromotive force series. In: Proceedings of the 13th Lunar and Planetary Science Conference. pp 692–693
Shannon RD (1976) Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr Sect A 32:751–767. https://doi.org/10.1107/S0567739476001551
Sievwright RH, Wilkinson JJ, O’Neill HSC, Berry AJ (2017) Thermodynamic controls on element partitioning between titanomagnetite and andesitic–dacitic silicate melts. Contrib Mineral Petrol 172:1–33. https://doi.org/10.1007/s00410-017-1385-6
Sievwright RH, O’Neill HSC, Tolley J et al (2020) Diffusion and partition coefficients of minor and trace elements in magnetite as a function of oxygen fugacity at 1150 °C. Contrib to Mineral Petrol. https://doi.org/10.1007/s00410-020-01679-z
Snyder GA, Taylor A, Neal CR (1992) A chemical model for generating the sources of mare basalts: combined equilibrium and fractional crystallization of the lunar magmasphere. Geochim Cosmochim Acta 56:3809–3823
Snyder D, Carmichael ISE, Wiebe RA (1993) Experimental study of liquid evolution in an Fe-rich, layered mafic intrusion: constraints of Fe-Ti oxide precipitation on the T-fO2 and T-ϱ paths of tholeiitic magmas. Contrib Mineral Petrol 113:73–86. https://doi.org/10.1007/BF00320832
Streck MJ, Grunder AL (2012) Temporal and crustal effects on differentiation of tholeiite to calcalkaline and ferro-trachytic suites, High Lava Plains, Oregon, USA. Geochem Geophys Geosystems. https://doi.org/10.1029/2012GC004237
Sun C, Liang Y (2012) Distribution of REE between clinopyroxene and basaltic melt along a mantle adiabat: effects of major element composition, water, and temperature. Contrib Mineral Petrol 163:807–823. https://doi.org/10.1007/s00410-011-0700-x
Sun C, Graff M, Liang Y (2017) Trace element partitioning between plagioclase and silicate melt: the importance of temperature and plagioclase composition, with implications for terrestrial and lunar magmatism. Geochim Cosmochim Acta 206:273–295. https://doi.org/10.1016/j.gca.2017.03.003
Sutton SR, Karner J, Papike J et al (2005) Vanadium K edge XANES of synthetic and natural basaltic glasses and application to microscale oxygen barometry. Geochim Cosmochim Acta 69:2333–2348. https://doi.org/10.1016/j.gca.2004.10.013
Tegner C, Thy P, Holness MB et al (2009) Differentiation and compaction in the Skaergaard Intrusion. J Petrol 50:813–840. https://doi.org/10.1093/petrology/egp020
Thacker C, Liang Y, Peng Q, Hess P (2009) The stability and major element partitioning of ilmenite and armalcolite during lunar cumulate mantle overturn. Geochim Cosmochim Acta 73:820–836. https://doi.org/10.1016/j.gca.2008.10.038
Thy P, Lesher CE, Tegner C (2009) The Skaergaard liquid line of descent revisited. Contrib to Mineral Petrol 157:735–747. https://doi.org/10.1007/s00410-008-0361-6
Toplis MJ, Carroll MR (1995) An experimental study of the influence of oxygen fugacity on fe-ti oxide stability, phase relations, and mineral-melt equilibria in ferro-basaltic systems. J Petrol 36:1137–1170. https://doi.org/10.1093/petrology/36.5.1137
Toplis MJ, Carroll MR (1996) Differentiation of ferro-basaltic magmas under conditions open and closed to oxygen: Implications for the skaergaard intrusion and other natural systems. J Petrol 37:837–858. https://doi.org/10.1093/petrology/37.4.837
Toplis MJ, Corgne A (2002) An experimental study of element partitioning between magnetite, clinopyroxene and iron-bearing silicate liquids with particular emphasis on vanadium. Contrib Miner Pet 144:22–37. https://doi.org/10.1007/s00410-002-0382-5
Toplis MJ, Dingwell DB, Libourel G (1994a) The effect of phosphorus on the iron redox ratio, viscosity, and density of an evolved ferro-basalt. Contrib Mineral Petrol 117:293–304. https://doi.org/10.1007/BF00310870
Toplis MJ, Libourel G, Carroll MR (1994b) The role of phosphorus in crystallisation processes of basalt: an experimental study. Geochim Cosmochim Acta 58:797–810. https://doi.org/10.1016/0016-7037(94)90506-1
Ubide T, Kamber BS (2018) Volcanic crystals as time capsules of eruption history. Nat Commun 9:1–12. https://doi.org/10.1038/s41467-017-02274-w
Ubide T, Mollo S, Zhao Xin J et al (2019) Sector-zoned clinopyroxene as a recorder of magma history, eruption triggers, and ascent rates. Geochim Cosmochim Acta 251:265–283. https://doi.org/10.1016/j.gca.2019.02.021
van Kan PM, Mason PRD, van Westrenen W (2011) Trace element partitioning between ilmenite, armalcolite and anhydrous silicate melt: Implications for the formation of lunar high-Ti mare basalts. Geochim Cosmochim Acta 75:4179–4193. https://doi.org/10.1016/j.gca.2011.04.031
Van Orman JA, Grove TL, Shimizu N (2001) Rare earth element diffusion in diopside: Influence of temperature, pressure, and ionic radius, and an elastic model fordiffusion in silicates. Contrib Mineral Petrol 141:687–703. https://doi.org/10.1007/s004100100269
Wager LR (1960) The major element variation of the layered series of the skaergaard intrusion and a re-estimation of the average composition of the hidden layered series and of the successive residual magmas. J Petrol 1:364–398. https://doi.org/10.1093/petrology/1.1.364
Wager LR, Brown G (1967) Layered igneous rocks. Freeman, San Francisco
Wager LR, Deer WA (1939) Wager, L.R., Deer, W.A., 1939. Geological investigations in East Greenland: part III. The petrology of the Skaergaard Intrusion, Kangerdlugssuaq. East Greenland Medd Groenl 105:1–352
Watson EB, Liang Y (1995) A simple model for sector zoning in slowly grown crystals: implications for growth rate and lattice diffusion, with emphasis on accessory minerals in crustal rocks. Am Miner 80:1179–1187. https://doi.org/10.2138/am-1995-11-1209
Wood BJ, Blundy JD (1997) A predictive model for rare earth element partitioning between clinopyroxene and anhydrous silicate melt. Contrib to Mineral Petrol 129:166–181. https://doi.org/10.1007/s004100050330
Xu Y, Chung SL, Jahn BM, Wu G (2001) Petrologic and geochemical constraints on the petrogenesis of Permian-Triassic Emeishan flood basalts in southwestern China. Lithos 58:145–168. https://doi.org/10.1016/S0024-4937(01)00055-X
Yuan Q, Namur O, Fischer LA et al (2017) Pulses of plagioclase-laden magmas and stratigraphic evolution in the upper zone of the Bushveld Complex, South Africa. J Petrol 58:1619–1643
Acknowledgements
We thank Jon Blundy, Raul Fonseca and an anonymous reviewer, and executive editor Othmar Müntener for their constructive reviews which significantly improved the quality of this manuscript. O. Namur and K. Shepherd acknowledge financial support from the FWO through an Odysseus grant to O. Namur. B. Charlier is a Research Associate of the Belgian Fund for Scientific Research-FNRS.
Funding
FWO, ZKD4437, Kat Shepherd.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Not applicable.
Additional information
Communicated by Othmar Müntener.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Shepherd, K., Namur, O., Toplis, M.J. et al. Trace element partitioning between clinopyroxene, magnetite, ilmenite and ferrobasaltic to dacitic magmas: an experimental study on the role of oxygen fugacity and melt composition. Contrib Mineral Petrol 177, 90 (2022). https://doi.org/10.1007/s00410-022-01957-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00410-022-01957-y