
Abstract
Accumulating studies have implicated intracellular signaling through muscarinic acetylcholine receptors (mAChRs) in psychiatric illness. In the present study, carbamylcholine chloride (carbachol)-induced Gαi/o and Gαq/11 activation was identified in postmortem human prefrontal cortical membranes. The following two sample cohorts were used: subjects [1], consisting of 40 controls without neuropsychiatric disorders, and subjects [2], consisting of 20 with bipolar disorder (BP), 20 major depressive disorder (MDD), 20 schizophrenia, and 20 controls, strictly sex- and age-matched. Carbachol-stimulated [35S]GTPγS binding to human brain membranes was assessed by the two methods, i.e., conventional method using filtration techniques (Gαi/o activation coupled to M2/M4 mAChRs) applied to subjects [1], and [35S]GTPγS binding/immuno precipitation assay (Gαq/11 activation coupled to M1 mAChR) applied to subjects [1] and [2]. The concentration eliciting the half-maximal effect (EC50), maximum percent increase (%Emax), and slope factor were obtained from concentration–response curve of carbachol-induced Gαi/o and Gαq/11 activation. The pEC50 values of both carbachol-induced Gαi/o and Gαq/11 activations in subjects [1] were significantly correlated, though its implications or underlying molecular processes are unclear. The results of M1 mAChR-mediated Gαq/11 activation in subjects [2] indicated no significant disorder-specific alterations. However, the distribution patterns of the pEC50 values showed unequal variances among the groups. There was a significant inverse correlation between the %Emax values and the pEC50 values in subjects with schizophrenia, but not in those with BP or MDD, or controls. These data support the notion that schizophrenia patients consist of biologically heterogeneous subgroups with respect to M1 mAChR-mediated signaling pathways.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Acetylcholine (ACh) plays an important role as a neurotransmitter and neuromodulator in the central and peripheral nervous systems. ACh binds to two classes of receptors: metabotropic muscarinic receptors and ionotropic nicotinic receptors. In the central nervous system (CNS), there is evidence that muscarinic acetylcholine receptors (mAChRs) are involved in motor control, temperature regulation, cardiovascular regulation, and higher brain functions such as learning and memory. mAChRs are a family of seven-transmembrane domain receptors, consisting of five receptor subtypes (M1–M5) [1, 2]. As members of the G-protein coupled receptor (GPCR) superfamily, they associate with heterotrimeric G-proteins to translate extracellular signals into intracellular signals via transduction cascades. In general, M1, M3, and M5 mAChRs are coupled to the Gq/11 class of G-proteins, leading to activation of phospholipase C-β (PLC-β), which hydrolyses phosphatidylinositol 4,5-bisphosphate (PIP2) into second messengers, inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG). By contrast, M2 and M4 mAChRs are canonically coupled Gi/o class proteins, resulting in inhibition of adenylate cyclase, which is responsible for catalyzing the conversion of ATP to cyclic AMP [3].
A growing number of studies have implicated the ACh muscarinic system in psychiatric disorders and degenerative illnesses associated with dementia, such as Alzheimer’s disease [4]. Among the five mAChRs subtypes, M1 mAChR has been most strongly associated with cognition and psychotic disorders such as schizophrenia [5], possibly because the M1 subtype is most abundantly expressed in all major forebrain areas (including the cerebral cortex, hippocampus, and striatum), and has lower expression in the peripheral nervous system [6]. Additionally, there is some evidence to suggest that mAChRs are also involved in the pathological processes underlying mood disorders such as bipolar disorder (BP) and major depressive disorder (MDD) [4, 7, 8].
Initial investigations regarding mAChRs in postmortem human brain tissue from patients with mental disorders were performed using receptor binding assays with a non-selective radioligand, [3H]quinuclidinyl benzilate ([3H]QNB), and showed inconsistent results [9,10,11]. Since then, radioligand binding studies have been conducted with more selective radioligands such as [3H]pirenzepine [12,13,14,15,16,17,18,19,20,21], [3H]AF-DX384 [16,17,18, 22,23,24], and [3H]4-diphenylacetoxy-N-methylpiperidine methiodide (4-DAMP) ([3H]4-DAMP) [17, 18, 25].
By using [3H]pirenzepine as a radioligand for an autoradiographic study, Scarr et al. [20] have shown that a decrease in cortical M1 mAChRs is restricted to a subgroup of patients diagnosed with schizophrenia. The subgroup comprised approximately 25% of the schizophrenia group, and had on average 75% less M1 mAChRs than did control subjects or other subjects with schizophrenia.
A subsequent study using a guanosine-5′-O-(3-[35S]thio)triphosphate ([35S]GTPγS) binding assay combined with an immunocapture method revealed that activation of Gαq/11 coupled to M1 mAChR was also altered in a subgroup of schizophrenia patients, in whom M1 mAChRs were decreased in Brodmann’s area 9 compared to that in controls [26], suggesting biological heterogeneity of this disorder. This study used a method in which human brain tissues were pre-incubated with N-ethylmaleimide (NEM), an irreversible alkylating reagent, to reduce basal [35S]GTPγS binding [27].
Recently, we succeeded in establishing a method of [35S]GTPγS/immunoprecipitation to assess functional coupling between M1 mAChRs and Gαq/11 proteins in postmortem human brains [28]. This assay is a revised version of conventional immunoprecipitation techniques, the detailed technical notes of which have been described elsewhere [29]. This method allowed for the use of native brain membranes, with signal/noise ratio equivalent to that reported by Salah-Uddin et al. [26, 27]. Absence of NEM pre-treatment may avoid possible damage to membranes. In the present study, this method was applied to dorsolateral prefrontal cortical membranes prepared from control and psychiatric subjects, consisting of BP, MDD, and schizophrenia.
Methods
Postmortem human brain samples
Postmortem human brain samples were obtained at autopsy in the Basque Institute of Legal Medicine (Bilbao, Spain). In typical conditions, the corpse is refrigerated at 4℃ within 3–5 h of death, until autopsy. Samples from the dorsolateral prefrontal cortex (Brodmann’s area 9) were dissected at the time of autopsy and immediately stored at − 70 ℃. The screening of neuropathological disorders was performed in all the cases used in this study by expert pathologists. The presence of neuropathological or neurodegenerative alterations was an exclusion criteria in this study. Collection procedures were performed in accordance with the protocol for postmortem human brain research of the Basque Institute of Legal Medicine. The study was approved by the Research and Ethics Boards of both the University of the Basque Country, Spain and the Faculty of Medicine, Saitama Medical University, Japan.
In the present study, the following samples were used: subjects [1]; 40 subjects (24 males and 16 females, aged 16 to 80 years old) with no known history of neurological or psychiatric disorders, and subject [2]; 80 subjects comprising 4 subgroups: BP, MDD, schizophrenia and controls. Four samples (BP, MDD, schizophrenia, and control) included in one experimental procedure were strictly matched based on sex (15 males and 5 females for each cohort) and age (≤ 6 years difference). The means ± SD age for each cohort were 52.0 ± 11.8, 52.0 ± 10.8, 51.8 ± 11.6, and 51.8 ± 11.3 years for BP, MDD, schizophrenia, and control, respectively. It is especially important to match age, because we previously showed that carbamylcholine chloride (carbachol)-stimulated [35S]GTPγS binding to Gαq/11 is decreased with aging, particularly in females [28]. The postmortem delay (PMD) could not be matched as strictly, but the resultant means ± SD (17.2 ± 9.5, 18.0 ± 7.6, 16.4 ± 7.7, and 20.9 ± 9.8 h for BP, MDD, schizophrenia, and control, respectively) were not significantly different. Detailed information concerning cause of death and drugs detected in blood in subjects [1] is described in Table 1. The demographic data, cause of death, and drugs detected in blood in subjects [2] are listed in Table 2. The [35S]GTPγS/immunoprecipitation assays were performed using four samples from each experimental group in parallel, in order to minimize inter-assay errors.
Membrane preparation
Membrane preparation was performed as previously described [28, 30]. Briefly, postmortem human brain tissues were homogenized in ice-cold TED buffer [5 mM Tris–HCl, 1 mM EDTA, 1 mM dithiothreitol (DTT); pH 7.4] containing 10% (w/v) sucrose. The nuclear fraction was removed by low-speed centrifugation, and membranes were prepared by sequential centrifugation and resuspension in buffer. Final aliquots were quickly frozen and stored at − 80 ℃ until the assay was performed.
Carbachol-stimulated [35S]GTPγS binding to Gαi/o
The [35S]GTPγS binding assay using conventional filtration techniques was performed as previously described [31]. Carbachol-stimulated [35S]GTPγS binding determined by this method is derived from Gαi/o proteins functionally coupled to M2/M4 subtypes of mAChRs. Briefly, thawed human brain membranes equivalent to 60 μg protein per tube were incubated at 30 ºC for 60 min in 500 μl of 50 mM Tris–HCl buffer (pH 7.4) containing 0.2 nM [35S]GTPγS, 5 mM MgCl2, 0.1 mM ethylenediaminetetraacetic acid, 0.2 mM ethylene glycolbis(2-aminoethylether)-N,N,N,N-tetraacetic acid (EGTA), 0.2 mM dithiothreitol, 100 mM NaCl, 50 μM GDP, and carbachol at the indicated concentrations. After the incubation period, the homogenates were filtered under vacuum through glass fiber filters (GF/B; Whatman International, Maidstone, UK) using a Brandel cell harvester with 2 × 5 ml washes with ice-cold 50 mM Tris–HCl buffer (pH 7.4). Nonspecific binding was determined in the presence of 100 μM unlabeled GTPγS. Radioactivity of [35S]GTPγS bound to the G-proteins retained on the filters was counted using a liquid scintillation spectrometer in 8 ml of Emulsifier Scintillator Plus cocktail (PerkinElmer, Waltham, MA, USA).
Carbachol-stimulated [35S]GTPγS binding to Gαq/11
The [35S]GTPγS binding/immunoprecipitation assay was performed as previously described [28, 29]. Thawed human brain membranes equivalent to 80 μg protein per tube were incubated at room temperature for 60 min in 200 μl of 50 mM Tris–HCl buffer (pH 7.4) containing 2.0 nM [35S]GTPγS, 20 mM MgCl2, 0.2 mM EGTA, 0.5 mM dithiothreitol, 100 mM NaCl, 10 nM GDP, and carbachol at the indicated concentrations. The tubes were incubated for 30 min after addition of Nonidet P40 substitute (0.3%). Finally, 25 μl of Dynabeads Protein A suspension coated with anti-Gαq/11 antibody (0.25 μg and 0.125 μg for Subjects [1] and [2], respectively), was added, and incubated for 60 min at room temperature with gentle occasional mixing. The Dynabeads Protein A were washed thoroughly with wash buffer (100 mM phosphate buffer containing 0.05% Tween 20, pH 7.4), and resuspended in 100 μl of wash buffer. The suspension was transferred into a scintillation mini vial, to which 4 ml of Emulsifier Scintillator Plus cocktail was added. The radioactivity of [35S]GTPγS bound to the Gαq/11 proteins, immunoprecipitated by Dynabeads Protein A coated with anti-Gαq/11 antibody, was determined with a liquid scintillation spectrometer. Non-specific binding was determined in the presence of 1 mM unlabeled GTPγS. As demonstrated previously [28], carbachol-stimulated [35S]GTPγS binding determined by this method is derived from Gαq/11 proteins functionally coupled to M1 mAChRs.
Data analysis
Data are presented as mean ± SEM of the indicated number of independent experiments, each performed in duplicate. The concentration-dependent increase in specific binding of [35S]GTPγS elicited by carbachol was expressed as a percentage of the basal unstimulated value, and analyzed using nonlinear regression with GraphPad Prism (GraphPad Software; LaJolla, CA, USA), to determine the concentration eliciting the half-maximal effect (EC50), maximum percent increase (%Emax), and slope factor. The EC50 values were transformed into pEC50 (− logEC50) to be analyzed. Pharmacological parameters among the four groups were analyzed using one-way analyses of variance (ANOVA) or Kruskal–Wallis tests according to the results of Bartlett's test for equal variances. Post-hoc comparisons were performed using Tukey’s test and Dunn's multiple comparison test, respectively. Linear regressions were calculated using the method of least squares and Pearson’s coefficient for simple correlation was calculated to test for possible associations between variables.
Materials
[35S]GTPγS (NEG030H, 1250 Ci/mmol) was purchased from PerkinElmer (Waltham, MA, USA). Carbachol, GDP, GTPγS, and Tween 20 were obtained from Sigma-Aldrich (St. Louis, MO, USA). Dynabeads Protein A were purchased from Life Technologies (Carlsbad, CA, USA). Anti-Gαq/11 rabbit polyclonal antibody sc-393 (E-17) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Non-ionic detergent Nonidet P40 substitute was obtained from Roche Diagnostics GmbH (Mannheim, Germany). All other chemicals used in this study were obtained from standard sources and were of the highest commercially available purity.
Results
Interrelationship between carbachol-stimulated Gαi/o and Gαq/11 functionality
The individual concentration–response curves of carbachol-induced Gαi/o activation determined in 38 samples from subjects [1] are depicted in Fig. 1a. The data for two subjects were lacking due to low sample volume. The %Emax, pEC50, and slope factors varied substantially, with a range of 11.6–72.9%, 3.77–5.24, and 0.46–1.24, respectively.

Stimulatory effects of carbachol on specific [35S]GTPγS binding to Gαi/o (a) and Gαq/11 (b) in postmortem human prefrontal cortical membranes. The thin lines represent individual concentration–response curves for 38 (a) and 40 (b) subjects without any neuropsychiatric disorder, expressed as a percentage increase over the basal, unstimulated specific binding. Open symbols represent the mean ± SEM of all samples, and the bold line depicts the concentration–response curve derived from these values. Figure 1b is replicated from [28]
The individual concentration–response curves of carbachol-induced Gαq/11 activation in 40 samples from subjects [1] have been reported elsewhere [28], which are replicatively presented in Fig. 1b.
There was no significant correlation with the %Emax values (Fig. 2a) or slope factors (not shown) determined by the two different assays. Interestingly, the pEC50 values determined for Gαq/11 were significantly correlated with those determined for Gαi/o, as presented in Fig. 2b (r = − 0.38, p < 0.05).

Interrelationship between carbachol-stimulated Gαi/o and Gαq/11 functionality in 38 subjects without any neuropsychiatric disorder. a The symbols represent individual subjects with a %Emax value determined for M2/M4 mAChR-mediated Gαi/o activation (abscissa) and for M1 mAChR-mediated Gαq/11 activation (ordinate). b The symbols represent individual subjects with a pEC50 value determined for M2/M4 mAChR-mediated Gαi/o activation (abscissa) and for M1 mAChR-mediated Gαq/11 activation (ordinate). The regression line indicates a significant correlation (r = − 0.38, p < 0.05)
Carbachol-stimulated Gαq/11 functionality in subjects with psychiatric disorders
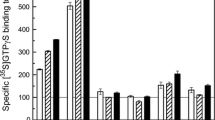
Carbachol-stimulated increase in specific [35S]GTPγS binding to Gαq/11 was determined in 80 individuals belonging to one of the four subgroups of subjects [2]. As shown in Fig. 3a, the %Emax values were not significantly different between groups, determined by one-way ANOVA [F(3,76) = 0.074, N.S.]. The results of Bartlett's test for equal variances indicated unequal distributions of pEC50 (p < 0.0001) and slope (p = 0.022). There was no significant difference in pEC50 values (Fig. 3b) or slope factors (Fig. 3c) among the groups, as determined by Kruskal–Wallis analysis. Since the psychiatric patients included some suicide victims (14/20 in BP, 17/20 in MDD, and 10/20 in schizophrenia), each group was further subdivided into suicide and non-suicide subjects. No significant difference was detected by Kruskal–Wallis analysis with respect to %Emax, pEC50, or slope factor among the groups (not shown). Further, some patients had been taking a variety of psychotropic drugs, as revealed by the toxicological data (Table 2). However, it is difficult to evaluate the effect of pharmacotherapy with psychotropic drugs, considering the multiplicity of modes of action of the medications used, and the possibility that lack of drug detection in toxicological screening does not infer lack of psychotropic medication prior to death. Nonetheless, the possible effects of antipsychotic agents in the schizophrenia group were examined, since these drugs have pharmacological properties common to dopamine D2 receptor antagonists. When divided into the two subgroups according to the toxicological data (antipsychotic (+) and antipsychotic (–)), the %Emax values were significantly different, as determined by one-way ANOVA [F(2,37) = 4.81, p < 0.05] (Fig. 4a). Post-hoc comparison using Tukey’s test indicated significant differences between antipsychotic (+) and antipsychotic (−) subgroups. Likewise, Kruskal–Wallis analysis indicated significant differences between the groups in the pEC50 values (p < 0.05), which was also ascribed to the difference between antipsychotic (+) and antipsychotic (−) subgroups as revealed by Dunn's multiple comparison test (p < 0.05) (Fig. 4b). Slope factor was not significantly different between the antipsychotic (+) and antipsychotic (−) subgroup, or the controls (Kruskal–Wallis test) (not shown).

Comparison of the stimulatory effects of carbachol on the specific [35S]GTPγS binding to Gαq/11 in postmortem human prefrontal cortical membranes among bipolar disorder (BP), major depressive disorder (MDD), schizophrenia, and control groups. a The symbols represent individual subjects with a %Emax value determined for M1 mAChR-mediated Gαq/11 activation in BP (empty circle), MDD (upward triangle), schizophrenia (downward triangle), and control (filled circle) group. The horizontal line with error bars indicates the mean ± SEM. b The symbols represent individual subjects with a pEC50 value determined for M1 mAChR-mediated Gαq/11 activation in BP (empty circle), MDD (upward triangle), schizophrenia (downward triangle), and control (filled circle) group. The horizontal line with error bars indicates the median ± interquartile range. c The symbols represent individual subjects with slope factor determined for M1 mAChR-mediated Gαq/11 activation in BP (empty circle), MDD (upward triangle), schizophrenia (downward triangle), and control (filled circle) group. The horizontal line with error bars indicates the median ± interquartile range

Comparison of the stimulatory effects of carbachol on the specific [35S]GTPγS binding to Gαq/11 in postmortem human prefrontal cortical membranes among the two schizophrenia subgroups [antipsychotic (+) and antipsychotic (−), divided according to the toxicological data] and controls. a The symbols represent individual subjects with a %Emax value determined for M1 mAChR-mediated Gαq/11 activation in antipsychotic (+) (empty circle), antipsychotic (−) (upward triangle), and control (filled circle) group. The horizontal line with error bars indicates the mean ± SEM. b The symbols represent individual subjects with a pEC50 value determined for M1 mAChR-mediated Gαq/11 activation in antipsychotic (+) (empty circle), antipsychotic (−) (upward triangle), and control (filled circle) group. The horizontal line with error bars indicates the median ± interquartile range. Significant differences revealed by post-hoc tests are indicated with asterisks (*p < 0.05)
Interrelationship between %Emax and pEC50 of carbachol-induced Gαq/11 activation
The correlations between the %Emax values and the pEC50 values determined for carbachol-induced [35S]GTPγS binding to Gαq/11 were investigated in each cohort. In subjects [1], there was no significant correlation between the two pharmacological parameters (Fig. 5a). Likewise, no significant correlations were detected in three of the cohorts (BP, MDD, and control) of subjects [2] (Fig. 5b). However, the %Emax values were significantly negatively correlated with the pEC50 values in the schizophrenia group (r = − 0.56, p < 0.05) (Fig. 5c).

Interrelationship between pEC50 and %Emax values determined for M1 mAChR-mediated Gαq/11 activation in postmortem human prefrontal cortical membranes. a The symbols represent individual subjects with a pEC50 value (abscissa) and a %Emax value (ordinate) determined for M1 mAChR-mediated Gαq/11 activation in 40 subjects without any neuropsychiatric disorder (subjects[1]). b The symbols represent individual subjects with a pEC50 value (abscissa) and a %Emax value (ordinate) determined for M1 mAChR-mediated Gαq/11 activation in bipolar disorder (BP) (empty circle), major depressive disorder (MDD) (upward triangle), and control (filled circle) group. c The symbols represent individual subjects with a pEC50 value (abscissa) and a %Emax value (ordinate) determined for M1 mAChR-mediated Gαq/11 activation in the schizophrenia group. The regression line indicates a significant correlation (r = − 0.56, p < 0.05). Three patients with extremely low pEC50 values are indicated with the experimental number (1, 8, and 17) (see Table 2)
Discussion
The muscarinic component of the central cholinergic system has been implicated in the pathophysiology of a number of neurological and psychiatric disorders associated with cognitive dysfunction. Accumulating evidence has revealed that cognitive deficits are one of the core symptoms of schizophrenia [32, 33]. Further, cognitive dysfunction is now considered an important feature of mood disorders including BP and MDD [34]. The main purpose of the current study was to elucidate possible alterations in M1 mAChR-mediated Gαq/11 signaling in psychiatric patients diagnosed as having BP, MDD, or schizophrenia prior to death, by assessing [35S]GTPγS binding/immunoprecipitation in prefrontal cortical membranes from postmortem brains. The method applied in this study was pharmacologically characterized previously [28]. That study showed the carbachol-stimulated increase in [35S]GTPγS binding to Gαq/11 in human prefrontal cortical membranes was potently inhibited by ( ±)-telenzepine, a selective M1 mAChR antagonist, in a competitive manner, with a pA2 value of 8.81. Although the involvement of other receptor subtypes such as M3 mAChR cannot be entirely excluded, the response appears to be derived mostly from Gαq/11 coupled to M1 mAChR, considering the relative density of mAChRs (M1 > > M3) in brain tissue [35, 36].
Although the pharmacological parameters of carbachol-stimulated Gαq/11 functionality (%Emax, pEC50, and slope factor) were not significantly different in any psychiatric disorder cohort (BP, MDD, or schizophrenia) compared to the control group, some interesting findings were obtained in the present study. In subjects [1], which consisted of 40 individuals without any neuropsychiatric disorder, both M1 mAChR-mediated Gαq/11 and M2/M4 mAChR-mediated Gαi/o activation were determined in 38 individuals. Neither the %Emax values nor slope factors determined by the two biochemical measures were not correlated, whereas the pEC50 values for M1 mAChR/Gαq/11 coupling were significantly negatively correlated to those for M2/M4 mAChR/Gαi/o coupling. Although the exact implications of these findings are unclear, it has been reported that mAChRs are regulated directly by, or are a consequence of, multiple internal changes due to different types of stressful stimuli such as physical, chemical, psychological/social, and cardiovascular system-disturbing events [37]. Multiple and complex molecular mechanisms have been reported to underlie alterations in mAChR-mediated signaling pathways [38]. Interestingly, crosstalk between Gαi- and Gαq-coupled receptors mediated by Gβγ exchange has been indicated, as exemplified by adenosine A1 and α2C adrenoceptor (Gαi-coupled), and bradykinin B2 and UTP-preferring P2Y receptor (Gαq-coupled) [39]. The data in the present study should be interpreted with caution, because the two determinations were obtained from the two independent experiments under different conditions, particularly regarding GDP concentrations (10 nM and 50 μM for M1 mAChR/Gαq/11 coupling and M2/M4 mAChR/Gαi/o coupling, respectively). Nevertheless, our results suggest there might also be some interactive regulatory processes between Gαi-coupled M2/M4 mAChRs and Gαq-coupled M1 mAChR via molecular mechanisms not yet elucidated.
Secondly, a significant inverse correlation between %Emax values and pEC50 values determined for M1 mAChR-mediated Gαq/11 activation was detected only in the schizophrenia group, but not in BP, MDD, or controls. The lack of a significant correlation was replicated in subjects [1], in which all subjects were free of neuropsychiatric illness. The pEC50 values for M1 mAChR-mediated Gαq/11 activation were distributed within a relatively narrow range in controls (4.48–5.08 in subjects [1], and 3.98–5.43 in subjects [2]), BP (4.22–5.07), and MDD (4.33–5.13, except for one subject with a particularly low value of 2.94). In contrast, the pEC50 values in the schizophrenia group ranged from 2.81 to 6.21, more than three orders of magnitude. The unequal distributions of pEC50 values were detected using Bartlett’s test for equal variances in subjects [2] (p < 0.0001), and the unique pattern of pEC50 distribution in the schizophrenia group appears to contribute to the significant negative correlation between pEC50 and %Emax values in this group.
The M1 mAChR-mediated Gαq/11 activation in postmortem human brains from schizophrenia patients has been reported by Salah-Uddin et al. [26]. They divided the schizophrenia subjects into two sub-populations based on [3H]pirenzepine binding, termed “muscarinic receptor-deficit schizophrenia (MRDS)” and “non-MRDS”. In addition to the lower M1 mAChR binding, the pEC50 values and maximal increases in oxotremorine-M-stimulated Gαq/11 determined in the MRDS group were significantly decreased and increased, respectively, compared to the control group. These findings suggest biological heterogeneity in schizophrenia, and the results of the present study support this hypothesis. To ascertain this, it is important to assess M1 mAChR expression levels in schizophrenic patients by radioligand binding assay and/or western blot in future studies.
Of 20 subjects in the schizophrenia group, three patients demonstrated extremely reduced potency of carbachol (Fig. 5c; 1, 8, and 17). These three patients had relatively high %Emax values, and the characteristics of these subjects underlie the significant negative correlation between pEC50 and %Emax values in this group. Interestingly, all of these patients belong to the antipsychotic (+) subgroup. Significant differences in %Emax and pEC50 values between antipsychotic (+) and antipsychotic (−) subjects may indicate that altered M1 mAChR/Gαq/11 coupling in schizophrenia patients is ascribed, at least in part, to pharmacotherapy with antipsychotics.
This significant negative correlation was restricted to the schizophrenia group, suggesting disorder-specific alterations in mAChR signaling. There is relatively little direct evidence to show perturbed central cholinergic activity in mood disorders [4, 7, 8]. Levels of M1 mAChRs in postmortem brains from patients with mood disorders have been reported similar to controls [17, 21, 25]. Our data are in line with these previous reports using radioligand binding techniques. On the other hand, several reports have indicated that Gαi/o-coupled mAChRs (M2 and/or M4 mAChR) are implicated in the pathogenesis of mood disorders [17, 23, 40]. As such, it is of interest to investigate possible alterations in mAChR-mediated Gαi/o activation in mood disorder patients. This work is ongoing.
G-proteins play a pivotal role in receptor-mediated signal transduction pathways. Enhanced receptor/G-protein coupling has been reported in frontal cortical membranes obtained from postmortem brains of BP patients, compared to age-, sex-, and postmortem interval-matched controls [41]. Enhanced receptor/G-protein coupling in BP subjects has been detected between several receptors and Gα subtypes (i.e., isoproterenol-stimulated Gαs; carbachol-stimulated Gαi, Gαo, and Gαq; 5-HT-stimulated Gαs, Gαi, Gαo, and Gαq). This universal hypersensitivity of receptor-coupled G-protein function in BP patients, along with an epoch-making finding that the anti-bipolar agent lithium inhibits adrenergic and cholinergic increases in GTP binding [42], has led to the so-called “G-protein hypothesis of mood disorders” [43,44,45]. However, such an oversimplified hypothesis appears insufficient, considering controversial results on quantitative and functional status of the heterotrimeric G-proteins and G-protein-mediated signaling in various experimental designs implicated in the pathogenesis and treatment of mood disorders [46, 47]. Our previous efforts aiming to replicate the report of Avissar et al. [42] failed to identify interfering effects of lithium on receptor/G-protein coupling, at least in the case of Gi/o proteins coupled with various neurotransmitter receptors [48]. Further investigation is needed to evaluate the implications of G-protein and G-protein-linked molecular machinery in the pathophysiology and treatment of mood disorders.
In conclusion, we examined mAChR-mediated G-protein activation in postmortem human prefrontal cortical membranes. Although there was no significant correlation in %Emax values or slope factors between M2/M4 mAChR-mediated Gαi/o activation and M1 mAChR-mediated Gαq/11 activation, pEC50 values were significantly correlated with each other, indicating some interactive molecular processes between these two mAChR-mediated signaling pathways. The results of M1 mAChR-mediated Gαq/11 activation in the BP, MDD, schizophrenia, and control subjects, indicated no significant disorder-specific differences in each pharmacological parameters. However, the distribution patterns of the pEC50 values determined for M1 mAChR-mediated Gαq/11 activation showed unequal variances among the groups, and there was a significant inverse correlation between the %Emax values and the pEC50 values restricted to the schizophrenia cohort, but not in the BP, MDD, or controls. The lack of direct evidence indicating heterogeneous subgroups in schizophrenia patients is a major limitation of this study. Furthermore, the densities of M1 mAChRs as well as Gαq/11 proteins were not determined by radioligand binding assay or western blot. Nevertheless, the data may support the notion that schizophrenia patients consist of biologically heterogeneous groups, i.e., a small proportion with M1 mAChR-mediated Gαq/11 signaling deficits, and the majority without such deficit [20, 26].
References
Caulfield MP, Birdsall NJM (1998) International union of pharmacology. XVII. Classification of muscarinic acetylcholine receptors. Pharmacol Rev 50:279–290
Eglen RM (2005) Muscarinic receptor subtype pharmacology and physiology. Prog Med Chem 43:105–136. https://doi.org/10.1016/S0079-6468(05)43004-0
Felder CC (1995) Muscarinic acetylcholine receptors: signal transduction through multiple effectors. FASEB J 9:619–625
Scarr E (2012) Muscarinic receptors: their roles in disorders of the central nervous system and potential as therapeutic targets. CNS Neurosci Ther 18:369–379. https://doi.org/10.1111/j.1755-5949.2011.00249.x
Readler TJ, Bymaster FP, Tandon R, Copolov D, Dean B (2007) Towards a muscarinic hypothesis of schizophrenia. Mol Psychiatry 12:232–246. https://doi.org/10.1038/sj.mp.4001924
Bymaster FP, McKinzie DL, Felder CC, Wess J (2003) Use of M1–M5 muscarinic receptor knockout mice as novel tools to delineate the physiological roles of the muscarinic cholinergic system. Neurochem Res 28:437–442
Jeon WJ, Dean B, Scarr E, Gibbons A (2015) The role of muscarinic receptors in the pathophysiology of mood disorders: a potential novel treatment? Curr Neuropharmacol 13:739–749
Scarr E (2009) Muscarinic receptors in psychiatric disorders––can we mimic 'health'? Neurosignals 17:298–310. https://doi.org/10.1159/000231896
Bennett JP Jr, Enna SJ, Bylund DB, Gillin JC, Wyatt RJ, Snyder SH (1979) Neurotransmitter receptors in frontal cortex of schizophrenics. Arch Gen Psychiatry 36:927–934
Toru M, Watanabe S, Shibuya H, Nishikawa T, Noda K, Mitsushio H, Ichikawa H, Kunugi A, Takashima M, Mataga N, Ogawa A (1988) Neurotransmitters, receptors and neuropeptides in post-mortem brains of chronic schizophrenic patients. Acta Psychiatr Scand 78:121–137
Watanabe S, Nishikawa T, Takashima M, Toru M (1983) Increased muscarinic cholinergic receptors in prefrontal cortices of medicated schizophrenics. Life Sci 33:2187–2196
Crook JM, Tomaskovic-Crook E, Copolov DL, Dean B (2000) Decreased muscarinic receptor binding in subjects with schizophrenia: a study of the human hippocampal formation. Biol Psychiatry 48:381–388
Crook JM, Tomaskovic-Crook E, Copolov DL, Dean B (2001) Low muscarinic receptor binding in prefrontal cortex from subjects with schizophrenia: a study of Brodmann’s areas 8, 9, 10, and 46 and the effects of neuroleptic drug treatment. Am J Psychiatry 158:918–925. https://doi.org/10.1176/appi.ajp.158.6.918
Dean B, Crook JM, Opeskin K, Hill C, Keks N, Copolov DL (1996) The density of muscarinic M1 receptors is decreased in the caudate-putamen of subjects with schizophrenia. Mol Psychiatry 1:54–58
Dean B, McLeod M, Keriakous D, McKenzie J, Scarr E (2002) Decreased muscarinic1 receptors in the dorsolateral prefrontal cortex of subjects with schizophrenia. Mol Psychiatry 7:1083–1091
Deng C, Huang XF (2005) Decreased density of muscarinic receptors in the superior temporal gyrus in schizophrenia. J Neurosci Res 15:883–890. https://doi.org/10.1002/jnr.20600
Gibbons AS, Scarr E, McLean C, Sundram S, Dean B (2009) Decreased muscarinic receptor binding in the frontal cortex of bipolar disorder and major depressive disorder subjects. J Affect Disord 116:184–191. https://doi.org/10.1016/j.jad.2008.11.015
Gibbons AS, Scarr E, Boer S, Money T, Jeon WJ, Felder C, Dean B (2013) Widespread decreases in cortical muscarinic receptors in a subset of people with schizophrenia. Int J Neuropsychopharmacol 16:37–46. https://doi.org/10.1017/S1461145712000028
Scarr E, Sundram S, Keriakous D, Dean B (2007) Altered hippocampal muscarinic M4, but not M1, receptor expression from subjects with schizophrenia. Biol Psychiatry 61:1161–1170. https://doi.org/10.1016/j.biopsych.2006.08.050
Scarr E, Cowie TF, Kanellakis S, Sundram S, Pantelis C, Dean B (2009) Decreased cortical muscarinic receptor define a subgroup of subjects with schizophrenia. Mol Psychiatry 14:1017–1023. https://doi.org/10.1038/mp.2008.28
Zavitsanou K, Katsifis A, Mattner F, Huang XF (2004) Investigation of M1/M4 muscarinic receptors in the anterior cingulate cortex in schizophrenia, bipolar disorder, and major depression disorder. Neuropsychopharmacology 29:619–625. https://doi.org/10.1038/sj.npp.1300367
Crook JM, Dean B, Pavey G, Copolov D (1999) The binding of [3H]AF-DX 384 is reduced in the caudate-putamen of subjects with schizophrenia. Life Sci. 64:1761–1771
Gibbons AS, Jeon WJ, Scarr E, Dean B (2016) Changes in muscarinic M2 receptor levels in the cortex of subjects with bipolar disorder and major depressive disorder and in rats after treatment with mood stabilizers and antidepressants. Int J Neuropsychopharmacol 19:118. https://doi.org/10.1093/ijnp/pyv118
Zavitsanou K, Katsifis A, Yu Y, Huang XF (2005) M2/M4 muscarinic receptor binding in the anterior cingulate cortex in schizophrenia and mood disorders. Brain Res Bull 65:397–403. https://doi.org/10.1016/j.brainresbull.2005.02.007
Jeon WJ, Gibbons AS, Dean B (2013) The use of a modified [3H]4-DAMP radioligand binding assay with increased selectivity for muscarinic M3 receptor shows that cortical CHRM3 levels are not altered in mood disorders. Prog Neuropsychopharmacol Biol Psychiatry 47:7–12. https://doi.org/10.1016/j.pnpbp.2013.08.001
Salah-Uddin H, Scarr E, Pavey G, Harris K, Hagan JJ, Dean B, Challiss RA, Watson JM (2009) Altered M1 muscarinic acetylcholine receptor (CHRM1)-Gαq/11 coupling in a schizophrenia endophenotype. Neuropsychopharmacology 34:2156–2166. https://doi.org/10.1038/npp.2009.41
Salah-Uddin H, Thomas DR, Davies CH, Hagan JJ, Wood MD, Watson JM, Challiss RA (2008) Pharmacological assessment of M1 muscarinic acetylcholine receptor-Gq/11 protein coupling in membranes prepared from postmortem human brain tissue. J Pharmacol Exp Ther 325:869–874. https://doi.org/10.1124/jpet.108.137968
Odagaki Y, Kinoshita M, Ota T, Meana JJ, Callado LF, García-Sevilla JA (2017) Functional activation of Gαq coupled to 5-HT2A receptor and M1 muscarinic acetylcholine receptor in postmortem human cortical membranes. J Neural Transm 124:1123–1133. https://doi.org/10.1007/s00702-017-1749-0
Odagaki Y (2019) Guanosine-5′-O-(3-[35S]thio)triphosphate ([35S]GTPγS) binding/immunoprecipitation assay using magnetic beads coated with anti-Gα antibody in mammalian brain membranes. In: Odagaki Y, Borroto-Escuela DO (eds) Co-immunoprecipitation methods for brain tissue neuromethods, vol 144. Springer Nature, New York, pp 97–108
Odagaki Y, Kinoshita M, Ota T, Meana JJ, Callado LF, García-Sevilla JA (2015) Adenosine A1 receptors are selectively coupled to Gαi-3 in postmortem human brain cortex: guanosine-5′-O-(3-[35S]thio)triphosphate ([35S]GTPγS) binding/immunoprecipitation study. Eur J Pharmacol 764:592–598. https://doi.org/10.1016/j.ejphar.2015.07.049
Odagaki Y, Kinoshita M, Ota T, Meana JJ, Callado LF, García-Sevilla JA (2019) Optimization and pharmacological characterization of receptor-mediated Gi/o activation in postmortem human prefrontal cortex. Basic Clin Pharmacol Toxicol 124:649–659. https://doi.org/10.1111/bcpt.13183
Bowie CR, Harvey PD (2006) Cognitive deficits and functional outcome in schizophrenia. Neuropsychiatr Dis Treat 2:531–536
Tripathi A, Kar SK, Shukla R (2018) Cognitive deficits in schizophrenia: understanding the biological correlates and remediation strategies. Clin Psychopharmacol Neurosci 16:7–17. https://doi.org/10.9758/cpn.2018.16.1.7
MacQueen GM, Memedovich KA (2017) Cognitive dysfunction in major depression and bipolar disorder: assessment and treatment options. Psychiatry Clin Neurosci 71:18–27. https://doi.org/10.1111/pcn.12463
Levey AI, Kitt CA, Simonds WF, Price DL, Brann MR (1991) Identification and localization of muscarinic acetylcholine receptor proteins in brain with subtype-specific antibodies. J Neurosci 11:3218–3226
Levey AI, Edmunds SM, Heilman CJ, Desmond TJ, Frey KA (1994) Localization of muscarinic m3 receptor protein and M3 receptor binding in rat brain. Neuroscience 63:207–221
Mysliveček J, Kvetňanský R (2006) The effects of stress on muscarinic receptors. Heterologous receptor regulation: yes or no? Auton Autacoid Pharmacol 26:235–251
van Koppen CJ, Kaiser B (2003) Regulation of muscarinic acetylcholine receptor signaling. Pharmacol Ther 98:197–220
Quitterer U, Lohse MJ (1999) Crosstalk between Gαi- and Gαq-coupled receptors is mediated by Gβγ exchange. Proc Natl Acad Sci USA 96:10626–10631
Cannon DM, Carson RE, Nugent AC, Eckelman WC, Kiesewetter DO, Williams J, Rollis D, Drevets M, Gandhi S, Solorio G, Drevets WC (2006) Reduced muscarinic type 2 receptor binding in subjects with bipolar disorder. Arch Gen Psychiatry 63:741–747
Friedman E, Wang HY (1996) Receptor-mediated activation of G proteins is increased in postmortem brains of bipolar affective disorder subjects. J Neurochem 67:1145–1152
Avissar S, Schreiber G, Danon A, Belmaker RH (1988) Lithium inhibits adrenergic and cholinergic increases in GTP binding in rat cortex. Nature 331:440–442
Avissar S, Schreiber G (1992) Ziskind-Somerfeld research Award. The involvement of guanine nucleotide binding proteins in the pathogenesis and treatment of affective disorders. Biol Psychiatry 31:435–459
Avissar S, Schreiber G (2002) Toward molecular diagnostics of mood disorders in psychiatry. Trends Mol Med 8:294–300
Schreiber G, Avissar S (1991) Lithium sensitive G protein hyperfunction: a dynamic model for the pathogenesis of bipolar affective disorder. Med Hypotheses 35:237–243
Odagaki Y (2005) Transmembrane signal transduction via G proteins implicated in affective disorders. In: Brown MR (ed) Focus on bipolar disorder research. Nova Science Publishers, New York, pp 75–112
González-Maeso J, Meana JJ (2006) Heterotrimeric G proteins: insights into the neurobiology of mood disorders. Curr Neuropharmacol 4:127–138
Odagaki Y, Nishi N, Koyama T (1997) Lack of interfering effects of lithium on receptor/G protein coupling in human platelet and rat brain membranes. Biol Psychiatry 42:697–703
Acknowledgements
This work was supported by the Saitama Medical University Internal Grant 18-B-1–03 to Y.O., the Spanish MINECO‐FEDER (SAF 2017‐88126R, SAF 2013‐48586‐R and 2011‐29918 to J.J.M., L.F.C. and J.A.G‐S., respectively) and the Basque Government (IT‐616‐13). The authors thank the collaboration of the staff members of the Basque Institute of Legal Medicine. We would like to thank Editage (www.editage.com) for English language editing.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Odagaki, Y., Kinoshita, M., Meana, J.J. et al. Functional coupling of M1 muscarinic acetylcholine receptor to Gαq/11 in dorsolateral prefrontal cortex from patients with psychiatric disorders: a postmortem study. Eur Arch Psychiatry Clin Neurosci 270, 869–880 (2020). https://doi.org/10.1007/s00406-019-01088-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00406-019-01088-9