
Abstract
Purpose
A growing body of evidence has elucidated that the gut microbiota has a crucial impact on the pathophysiological process of atopic diseases. Eosinophilic chronic rhinosinusitis with nasal polyps (eCRSwNP) is a local atopic disease of the systemic immune response. Alterations in the gut microbiome in eCRSwNP patients remain largely undefined.
Methods
16S rRNA gene sequencing was performed in a cross-sectional study of 17 eCRSwNP patients, 9 noneCRSwNP patients and 13 healthy controls, and gut microbiota DNA sequencing between each pair of groups was compared using bioinformatic methods.
Results
Compared with that of healthy controls, the gut microbiomes of eCRSwNP patients were characterised by a distinct overall microbial composition. However, no significant differences were found in the alpha diversity of the gut microbiota between patients and healthy controls. The distinct differences in microbial composition were significantly correlated with the severity of disease. At the genus level, the abundance of Faecalibacterium positively correlated with Lund-Mackay CT scores. Similarly, the abundance of Turicibacter positively correlated with the percentage of tissue eosinophils.
Conclusions
We found alterations in the gut microbiome in eCRSwNP patients, and the alterations in the gut microbiome were correlated with the severity of disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The interaction between the gut microbiota and the host has a significant impact on the host’s health and has become a research hotspot in recent years [1, 2]. The gut microbiota not only has an important influence on the digestive system but also may affect the immune balance of the whole body by regulating the function of immune cells and inflammatory responses. An imbalance in the gut microbiota may be closely related to the occurrence and development of various diseases. Existing studies have confirmed that an imbalance in the gut microbiota is associated with the occurrence and development of various chronic inflammatory diseases, such as asthma, inflammatory bowel disease, and rheumatoid arthritis [3,4,5].
Chronic sinusitis with polyps (CRSwNP) is a common chronic inflammatory disease of the nasal mucosa, with a prevalence of 4–8% in the population [6, 7]. This disease can be divided into two different intrinsic types based on its clinical manifestations and pathological characteristics: eosinophilic nasal polyps and noneosinophilic nasal polyps. The pathogenesis of CRSwNP is complex, and the body's immune system may be closely related to the occurrence and development of CRSwNP. Its immune response is abnormal and manifests as a Th1/Th2 cell imbalance, abnormal regulatory T-cell function, and eosinophil infiltration [8,9,10,11]. However, no research has been conducted on the relationship between the gut microbiota and chronic sinusitis (CRS).
This study aimed to explore the correlation between the gut microbiota and chronic sinusitis. We used 16S rRNA microbiomic technology to analyse the gut microbiota of patients with different intrinsic types of chronic sinusitis and healthy controls (HCs), compare the differences between them, and explore the relationships between these differences and the pathogenesis, clinical symptoms, and treatment effects of chronic sinusitis. Through this study, we hope to understand the correlation between the gut microbiota and chronic sinusitis and provide new ideas and targets for the prevention and treatment of CRS.
Methods
Patient enrolment
This study recruited 17 eosinophilic CRSwNP patients (eCRSwNPs), 9 non-eCRSwNPs, and 13 HCs between March 2021 and July 2021. This study was performed in accordance with the relevant guidelines and regulations. Written informed consent was obtained from all participants. All subjects were between 24 and 68 years of age. The clinical diagnosis of CRSwNPs was based on the guideline criteria of the European Position Paper on Rhinosinusitis and Nasal Polyps (2020). Subjects with local CRSwNP, pregnancy, fungal sinusitis, antrochoanal polyps, immunodeficiency, cystic fibrosis, and primary ciliary dyskinesia were excluded. Patients who underwent surgery for non-CRS aetiologies, including surgery for oral, thyroid or septum without nasal inflammatory diseases (such as allergic rhinitis), were selected as the HCs.
The demographic and clinical variables recorded for each subject were age, gender, body mass index (BMI), smoking history, allergy and asthma history, total serum immunoglobulin E (IgE) levels, exhaled nitric oxide (NO) levels, history of topical steroid use 4 weeks prior to enrolment, preoperative Lund-Mackay CT (LM) scores and sinonasal outcome test (SNOT-20) scores. ImmunoCAP™ tests were performed to detect specific IgE antibodies. Patients with the use of antibiotics, systemic corticosteroids and a history of acute respiratory infection for 4 weeks prior to enrolment were excluded.
Histological evaluation of polyp tissue
Haematoxylin and eosin were used to stain paraffin-embedded sections. The numbers of different kinds of immune cells were observed at 400 × magnification. The proportion of eosinophils > 27% of the total inflammatory cells or an absolute tissue eosinophil count of 55 eosinophils per high power field (hpf) in the nasal polyp tissue was defined as eCRSwNP. The proportion of eosinophils ≤ 27% of the total inflammatory cells or an absolute tissue eosinophil count of 55 eosinophils per high power field (hpf) in the nasal polyp tissue was defined as noneCRSwNP [12].
Sample collection
All the enrolled patients provided faecal samples before standard operation and drug treatment. After collection, the faecal samples were immediately transported to the biobank within 2 h, where they were stored at –80 °C until DNA extraction.
DNA extraction and polymerase chain reaction amplification
Total DNA was extracted from faecal samples as described previously [3, 4]. The V3-V4 hypervariable regions of the 16S rRNA gene of bacterial DNA were amplified with primers 338F (5′-ACTCCTACGGGAGGCAGCAG-3′) and 806R (5′-GGACTA CHVGGGTWTCTAAT-3′) via PCR. PCRs were performed in triplicate in a 20 μL mixture containing 2 μL of 2.5 mM dNTPs, 0.8 μL of each primer (5 μM), 0.4 μL of FastPfu Polymerase, 4 μL of 5 × FastPfu Buffer and 10 ng of template DNA.
Illumina 16S rRNA sequence processing
The PCR products were extracted from a 2.0% agarose gel, purified using Agencourt AMPure Beads (Beckman Coulter, Indianapolis, IN) and quantified using the PicoGreen dsDNA Assay Kit (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. The purified amplicons were pooled in equimolar amounts and paired-end sequenced (2 × 250 bp) on an Illumina MiSeq platform (Illumina, San Diego, USA) according to the standard protocols by Majorbio Bio-Pharm Technology Co. Ltd. (Shanghai, China).
Data analysis
Raw fastq files were filtered by Trimmomatic and merged by FLASH with the criteria as follows: (1) If the average quality score of the reads at any site was less than 20 over a 50 bp sliding window, they were truncated. (2) Sequences were spliced according to an overlap longer than 10 bp and an overlap with a mismatch of no more than 2 bp. (3) Sequences for each sample were separated according to barcodes and primers. The barcodes needed to be matched accurately and the primers allowed a mismatch of 2 nucleotides; reads containing ambiguous bases have been deleted.
According to FASTQ format, the amplicon sequence variant (ASV) was obtained after quality control, noise elimination and chimerism removal was performed with DADA2 method recommended by QIIME2.
Compare and annotate ASV with GREENGENES database (align the database with V3-V4 region according to 338F/806R primer), the ASV with 99% similarity was classified as an operational taxonomic unit (OTU) to obtain the OTU classification information table. Utilizing the RDP classifier, the OTUs were classified to obtain their numbers at different taxonomic levels.
Statistical analysis
Demographics and clinical characteristics were analysed with analysis of variance, chi-square test, Fisher probabilities in 2 × 2 table data, and independent-samples T test.
The number of observed species as well as Shannon and Faith’s phylogenetic diversity index were calculated using rarefied data (depth = 50,000 sequences/sample) and used to characterise species diversity in a community. Principal component analyses of the unweighted UniFrac distance were used to assess the variation among the eCRSwNPs, non-CRSwNPs and HC groups (beta diversity). Differential analysis was performed using Kruskal–Wallis (KW), LEfSe and DEseq2 methods. Partial least squares discriminant analysis (PLSDA) was performed to predict the sample types corresponding to microbial communities. Permutational multivariate analysis of variance analysis and multivariate logistic regression analysis were used to investigate the correlation between clinical variables and the gut microbiome after adjusting for covariates.
Results
Subject characteristics
Thirty-nine patients, including 17 eCRSwNPs (11 males, 6 females), 9 non-CRSwNPs (8 males, 1 females) and 13 HCs (5 males, 8 females), were enrolled in the cohort. The demographic and clinical characteristics of the three groups are displayed in Table 1. Except for age and BMI, there was no difference among the three groups.
DNA sequencing results of the gut microbiome
The OTUs of the three groups were used to construct the petal diagram and phylogenetic tree (eFigure 1A, B). The dilution curve was gentle, indicating that the results obtained based on the sequencing depth could reflect the diversity of the gut microbiome in the samples (eFigure 2A, B).
At the phylum level, Firmicutes, Actinobacteria and Proteobacteria were the main phyla (Fig. 1A). Compared with those in the eCRSwNP group, the proportions of Actinobacteria in the non-CRSwNP (P = 0.0437) and HC (P = 0.0389) groups were significantly increased, while the proportions of Proteobacteria in the non-CRSwNP (P = 0.2584) and HC (P = 0.1837) groups were decreased. At the genus and species levels, the composition of the gut microbiome in the three groups is displayed (Figs. 1B, C). In summary, at different taxonomic levels, our results suggested the distinct distribution of the gut microbiome in the three groups.

Comparison of relative taxa abundance among the HCs, eCRSwNP patients and noneCRSwNP patients groups at phylum, genus and species levels. A The bar chart of relative taxa abundance among the three groups at phylum levels. B The bar chart of relative taxa abundance among the three groups at genus levels. C The bar chart of relative taxa abundance among the three groups at species levels. n = 39, eCRSwNP patients = 11, noneCRSwNP patients = 9, and HCs = 13. eCRSwNP eosinophilic chronic rhinosinusitis with nasal polyps, HCs healthy controls
The diversity in the gut microbiome among groups
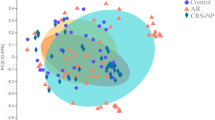
In this cohort, there was no difference in intraindividual diversity among the eCRSwNP, noneCRSwNP and HC groups, as measured by the observed OTUs and Shannon and Faith’s phylogenetic diversity indices (eTable 1). Next, PCoA was used to assess the overall diversity of the gut microbiome (Fig. 2A, B). The gut microbiome of the eCRSwNPs, noneCRSwNPs and HC groups indicated partial but significant clustering in the PCoA diagram. Based on the weighted UniFrac distance index among the three groups (eTable 2), the significant distinctions in overall diversity between eCRSwNP and HC (P = 0.020) and between eCRSwNP and noneCRSwNP (P = 0.020) were significant, while those between noneCRSwNP and HC (P = 0.482) were not significant. Therefore, the gut microbial structure of the eCRSwNP group was significantly different from that of the noneCRSwNP and HC groups. Our results indicated that the gut microbiota may play a crucial role in the pathogenesis of eCRSwNP.

Characteristics of gut microbiome composition in the HCs, eCRSwNP patients and noneCRSwNP patients. A Diagram of the LDA scores calculated at genus levels among HCs, eCRSwNP and noneCRSwNP groups. Only the LDA score > 2 are shown in the figure. B Diagram of the LDA scores calculated at genus level between eCRSwNP and HCs groups. C Diagram of the LDA scores calculated at genus level between noneCRSwNP and HCs groups. n = 39, eCRSwNP patients = 11, noneCRSwNP patients = 9, and HCs = 13. eCRSwNP eosinophilic chronic rhinosinusitis with nasal polyps, HCs healthy controls
Next, the gut microbiome composition of the three groups was clustered by PLS-DA (Fig. 2C). Our results indicated that the gut microbiome composition of the three groups was significantly distinct. The prediction models were established based on the nasal detected distinct genera of the three groups by using PLS-DA. The performance of the models achieved an AUC value of almost 1 (Fig. 2D). Our findings indicated the great potential of the gut microbiome as a noninvasive classifier for eCRSwNP diagnosis and may be recognised as a risk factor in the pathogenesis of eCRSwNP.
Differences in the gut microbiome structure among groups
The DESeq2 method was performed to identify differences in the gut microbiome structure among groups (eTable 3). The relative abundances of 2 genera in the noneCRSwNP and HC groups were significantly different, and the relative abundances of 7 genera in the eCRSwNP and noneCRSwNP groups were significantly different. Compared with HCs, the abundances of Escherichia and Enterococcus were significantly reduced in the group at the genus level. Compared with those in noneCRSwNPs, the increased abundances in gut microbiota such as Enterobacter, Escherichia, Megamonas and SMB53 were observed in eCRSwNPs, and the abundances of Bifidobacterium, Streptococcus and Collinsella were significantly increased in noneCRSwNPs. These differential genera can be used to build a noninvasive classifier for the distinct abundant taxa between eCRSwNPs or non-CRSwNPs and HCs.
To identify the distinct abundant taxa among the eCRSwNP, noneCRSwNPs and HC groups, LEfSe analysis was performed on the gut microbiome composition. At the genus level, 14 bacterial taxa showed distinct relative abundances among the three groups (LDA score > 2.0, p < 0.05). Increased abundances of Clostridia, Clostridiales, Firmicutes, and Gemmiger were observed in the non-CRSwNP group, and increased abundances of Bifidobacterium, Actinobacteria, Bifidobacteriales, etc., were observed in the eCRSwNP group (Fig. 3A). Compared with HCs, it was found that the abundances of Turicibacter, Clostridium, Gemmiger, etc., were increased significantly in eCRSwNPs (Fig. 3B); the abundances of Peptostreptococcus, Eubacterium, Clostridium, etc., increased significantly in noneCRSwNPs (Fig. 3C).

PCoA and PLS-DA analysis of the microbiome among the HCs, eCRSwNP patients and noneCRSwNP patients. A Bray–Curtis PCoA based on the relative abundance of OTU (99% similarity level). B Unweighted UniFrac PCoA based on the relative abundance of OTU (99% similarity level). C The PLS-DA analysis on OTUs among the HCs, eCRSwNP patients and noneCRSwNP groups. D ROC analysis for the predictive value of the predictive model constructed based on PLS-DA analysis. The AUCs of the HCs, eCRSwNP patients and noneCRSwNP groups almost are 1. n = 39, eCRSwNP patients = 11, noneCRSwNP patients = 9, and HCs = 13. AUC the area under the curve, eCRSwNP eosinophilic chronic rhinosinusitis with nasal polyps, HCs healthy controls, PCoA principal coordinate analysis, PLS-DA analysis partial least squares Discriminant Analysis
Correlation analysis between the gut microbiome and clinical variables
Partial Spearman’s rank-based correlation test performed on the age, sex, IgE, serum eosinophil count, serum eosinophil percent, BMI, SNOT-20, NO and LM scores was employed to explore the link between clinical variables and the disease-associated abundant taxa in all CRSwNP patients (Fig. 4A). At the genus level, the abundance of Haemophilus and Faecalibacterium positively correlated and Corynebacterium negatively correlated with LM CT scores. Dialister and Enterococcus were positively correlated, and Clostridium, Coprococcus and SMB53 were negatively correlated with SNOT-20. IgE and NO showed similar correlations with the gut microbiome. These results suggested that the faecal microbiota correlates with eCRSwNP disease severity. In addition, permutational multivariate analysis of variance results revealed that the above subject characteristics, such as age, sex and BMI, did not have a significant impact on the gut microbiota of different groups in our cohort (etable 4).

The relationship between gut microbiome and clinical variables at the genus level. A Heat map for Spearman correlation analysis between gut microbiome and clinical variables at the genus level. B The patients with high abundance of Turicibacter (n = 13) had higher level of percentage tissue eosinophil compared to those with low abundance of the species (n = 13). C The patients with high abundance of Faecalibacterium (n = 13) had higher LM CT scores compared to those with low abundance of the species (n = 13). P < 0.05 is showed in the figure. *P < 0.05, **P < 0.01, ***P < 0.001. BMI body mass index, CRSwNP chronic rhinosinusitis with nasal polyps, eCRSwNP eosinophilic chronic rhinosinusitis with nasal polyps, EOS_NUMB absolute tissue eosinophil count, EOS_PERCENT percentage tissue eosinophil, HCs healthy controls, IgE immunoglobulin E, L_M_SCORE Lund-Mackay CT scores, sEOS_NUMB serum eosinophil count, sEOS_PERCENT serum eosinophil percent, SNOT20 sinonasal outcome test scores
In patients with CRSwNP, the degree of eosinophil infiltration and the intensity of the nasal mucosal inflammatory response played a crucial role in the prognosis and disease severity. At the genus level, the abundances of Escherichia and Turicibacter positively correlated with absolute tissue eosinophil count. Gemmiger and Turicibacter were positively correlated, and Lachnospiraceae Clostridium was negatively correlated with the percentage of tissue eosinophils. Similarly, Turicibacter was also found to be positively correlated with serum eosinophil count and serum eosinophil percentage. In addition, Parabacteroides negatively correlated with serum eosinophil percentage, while Parabacteroides and Oscillospira negatively correlated with serum eosinophil count.
To further clarify the relationship between the microbiome and clinical variables, the CRSwNP patients were divided into two groups based on the median levels of the gut microbiome. After adjusting for age, sex and BMI, multivariate logistic regression results revealed that CRSwNPs with a high abundance of Turicibacter were relevant to a higher percentage of tissue eosinophils (P = 0.032, OR = 1.052, 95% CI 1.004–1.102, Fig. 4B). A high abundance of Faecalibacterium was associated with higher LM CT scores (P = 0.047, OR = 1.272, 95% CI 1.003–1.613, Fig. 4C).
Discussion
The human microbiome covaries with host health status and plays a critical role in host immune responses [13]. There is growing evidence that altered gut microbiome dysbiosis has become a topic of great concern to chronic inflammation, even in distant sites, such as the nose [2,3,4,5]. However, alterations in the gut microbiome in eCRSwNP patients remain largely undefined. Herein, our results showed the faecal microbiome community composition in a newly diagnosed eCRSwNP cohort. We found that the faecal microbiome of eCRSwNPs was significantly different from that of HCs, and faecal microbes were associated with eCRSwNP disease severity. Our findings provide a novel perspective into the heterogeneous pathophysiology of eCRSwNP. Moreover, several clinical studies aimed at evaluating the therapeutic effect of probiotics in allergic rhinitis patients have been reported for the last few years [14, 15]. Our findings may suggest a new therapeutic approach for eCRSwNP.
In our cohort, our findings suggested that eCRSwNPs had distinct gut microbiota compositions from noneCRSwNPs and HCs. At the phylum level, compared to HCs, the significantly increased abundance was Proteobacteria in eCRSwNPs. The evidence from previous studies suggested that Proteobacteria was significantly increased in chronic inflammatory diseases, such as nonalcoholic fatty liver disease, IBD, asthma and COPD [16,17,18,19]. In contrast, Actinobacteria were significantly decreased in eCRSwNPs. Several studies have shown that the decreased abundance of Actinobacteria was positively linked to better prognostic outcomes in Crohn's disease patients who received faecal microbiota transplantation and in the treatment of inflammatory diseases, such as those in acne patients, and worse outcomes after CRS surgery [20,21,22].
Our findings indicate that the gut microbiota can reflect disease severity to a certain extent. In the CRSwNPs group, the abundance of Haemophilus and Faecalibacterium positively correlated with LM CT scores, while Streptococcus and Ruminococcus positively correlated with NO. Several studies have found that Haemophilus, Streptococcus and Faecalibacterium had a relatively high prevalence and were relatively abundant in asthma patients [23,24,25]. Haemophilus and Streptococcus are pathogenic factors that significantly increase the risk of acute and chronic airway inflammation [24, 25]. Faecalibacterium was also shown to be significantly correlated with atopic diseases and represents only a single known species, Faecalibacterium prausnitzii. A previous study found that F. prausnitzii played an important role in the dysbiosis of TH2 immune responses in the skin [26]. In addition, in allergic 8-year-olds, the abundance of Faecalibacterium was consistently overrepresented, and Ruminococcus increased gradually over time from 6 months to 8 years old [27].
Our findings suggest that Turicibacter was more abundant in eCRSwNPs than in HCs. It has been reported that Turicibacter may have a significant impact on the occurrence and development of chronic spontaneous urticaria [28]. The survival of gastrointestinal Turicibacter is closely related to the host immune system and bacterial sensors. Several previous studies have revealed that Turicibacter does not survive in the gastrointestinal tract of immunodeficient mouse models and Toll-like receptor 2 knockout mice. These results suggest an interaction between the bacteria and host immune regulation [29, 30]. In addition, the protein that Turicibacter sanguinis expresses is partly responsible for the acute response in atopic conditions, such as asthma, rheumatoid arthritis, urticaria, and anaphylaxis [31, 32]. In our study, a correlation with high levels of absolute tissue eosinophil count and percentage tissue eosinophil was seen in the genus of Turicibacter. Thus, the biological interaction between Turicibacter and eCRSwNP, which remains largely undefined, warrants further investigation.
Through the collection of faecal samples from detailed phenotyping of pathology-proven noneCRSwNPs and eCRSwNPs, our findings uncovered a disordered gut microbial constitution, which provides a novel perspective into the heterogeneous pathophysiology of eCRSwNP. Our work provides a new theoretical framework for exploring the link between the gut microbiome and host immune system in eCRSwNP in the future. This progress may also provide novel potential targets for intervention of this sophisticated disease. Nevertheless, there are some limitations that need to be mentioned. First, the fact that we did not use metagenomes in our study is a disadvantage, which limits us to further analysis of the species level and function. Second, as a single-centre study with a limited sample size, the generalisability of our findings should be cautiously appraised. Finally, since this was a cross-sectional study, it was challenging to conclude the role of the identified bacteria in the progression and pathogenesis of eCRSwNP. Further studies are warranted to assess whether and how disease-associated bacteria play a role in immune dysfunction and inflammation in eCRS pathogenesis.
In conclusion, we defined the characteristics of the gut microbiota in eCRSwNP patients, and increased abundances of Bifidobacterium, Actinobacteria, Bifidobacteriales, etc., were observed in the eCRSwNP group. Our study demonstrated that faecal microbiota correlates with eCRSwNP disease severity. At the genus level, the abundance of Faecalibacterium positively correlated with LM CT scores. Similarly, the abundance of Turicibacter positively correlated with the percentage of tissue eosinophils. Our work may provide a novel perspective on the heterogeneous pathophysiology of eCRSwNP. Further studies are warranted to assess whether and how disease-associated bacteria play a role in immune dysfunction and nasal inflammation in eCRSwNP pathogenesis. The findings in this study may provide a knowledge framework for the clinical application of probiotics in eCRSwNP.
Availability of data and materials
The clean reads were deposited in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) database (Accession no. PRJNA803810).
References
Breban M, Tap J, Leboime A, Said-Nahal R, Langella P, Chiocchia G et al (2017) Faecal microbiota study reveals specific dysbiosis in spondyloarthritis. Ann Rheum Dis 76(9):1614–1622
Bowerman KL, Rehman SF, Vaughan A, Lachner N, Budden KF, Kim RY et al (2020) Disease-associated gut microbiome and metabolome changes in patients with chronic obstructive pulmonary disease. Nat Commun 11(1):5886
Thorburn AN, McKenzie CI, Shen S, Stanley D, Macia L, Mason LJ et al (2015) Evidence that asthma is a developmental origin disease influenced by maternal diet and bacterial metabolites. Nat Commun 23(6):7320
Trompette A, Gollwitzer ES, Yadava K, Sichelstiel AK, Sprenger N, Ngom-Bru C et al (2014) Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat Med 20(2):159–166
Book DT, Smith TL, McNamar JP, Saeian K, Binion DG, Toohill RJ (2003) Chronic sinonasal disease in patients with inflammatory bowel disease. Am J Rhinol 17(2):87–90
Divekar R, Patel N, Jin J, Hagan J, Rank M, Lal D et al (2015) Symptom-based clustering in chronic rhinosinusitis relates to history of aspirin sensitivity and postsurgical outcomes. J Allergy Clin Immunol Pract 3(6):934–040. e3
Fokkens WJ, Lund VJ, Hopkins C, Hellings PW, Kern R, Reitsma S et al (2020) European position paper on rhinosinusitis and nasal polyps 2020. Rhinology 58(Suppl S29):1–464
Cao PP, Li HB, Wang BF, Wang SB, You XJ, Cui YH et al (2009) Distinct immunopathologic characteristics of various types of chronic rhinosinusitis in adult Chinese. J Allergy Clin Immunol 124(3):478–84
Tomassen P, Vandeplas G, Van Zele T, Cardell LO, Arebro J, Olze H et al (2016) Inflammatory endotypes of chronic rhinosinusitis based on cluster analysis of biomarkers. J Allergy Clin Immunol 137:1449-1456.e4
Vlaminck S, Vauterin T, Hellings PW, Jorissen M, Acke F, Van Cauwenberge P et al (2014) The importance of local eosinophilia in the surgical outcome of chronic rhinosinusitis: a 3-year prospective observational study. Am J Rhinol Allergy 28(3):260–264
Zhang N, Crombruggen KV, Gevaert E, Bachert C (2016) Barrier function of the nasal mucosa in health and type-2 biased airway diseases. Allergy 71(3):295–307
Lou H, Meng Y, Piao Y, Wang C, Zhang L, Bachert C (2015) Predictive significance of tissue eosinophilia for nasal polyp recurrence in the Chinese population. Am J Rhinol Allergy 29(5):350–356
Zhong H, Penders J, Shi Z, Ren H, Cai K, Fang C et al (2019) Impact of early events and lifestyle on the gut microbiota and metabolic phenotypes in young school-age children. Microbiome 7(1):2
Anania C, Di Marino VP, Olivero F, De Canditiis D, Brindisi G, Iannilli F et al (2021) Treatment with a probiotic mixture containing bifidobacterium animalis Subsp. Lactis BB12 and Enterococcus faecium L3 for the prevention of allergic rhinitis symptoms in children: a randomized controlled trial. Nutrients 13(4):1315
Kang MG, Han SW, Kang HR, Hong SJ, Kim DH, Choi JH (2020) Probiotic NVP-1703 alleviates allergic rhinitis by inducing IL-10 expression: a four-week clinical trial. Nutrients 12(5):1427
Kapil S, Duseja A, Sharma BK, Singla B, Chakraborti A, Das A et al (2016) Small intestinal bacterial overgrowth and toll-like receptor signaling in patients with non-alcoholic fatty liver disease. J Gastroenterol Hepatol 31(1):213–221
Selvanantham T, Lin Q, Guo CX, Surendra A, Fieve S, Escalante NK et al (2016) NKT cell-deficient mice harbor an altered microbiota that fuels intestinal inflammation during chemically induced colitis. J Immunol 197(11):4464–4472
Marri PR, Stern DA, Wright AL, Billheimer D, Martinez FD (2013) Asthma-associated differences in microbial composition of induced sputum. J Allergy Clin Immunol 131(2):346–52.e1-3
Pragman AA, Kim HB, Reilly CS, Wendt C, Isaacson RE (2012) The lung microbiome in moderate and severe chronic obstructive pulmonary disease. PLoS One 7(10):e47305
Yan HM, Zhao HJ, Guo DY, Zhu PQ, Zhang CL, Jiang W (2018) Gut microbiota alterations in moderate to severe acne vulgaris patients. J Dermatol 45(10):1166–1171
Kong L, Lloyd-Price J, Vatanen T, Seksik P, Beaugerie L, Simon T et al (2020) Linking strain engraftment in fecal microbiota transplantation with maintenance of remission in Crohn’s disease. Gastroenterology 159(6):2193-2202.e5
Ramakrishnan VR, Hauser LJ, Feazel LM, Ir D, Robertson CE, Frank DN (2015) Sinus microbiota varies among chronic rhinosinusitis phenotypes and predicts surgical outcome. J Allergy Clin Immunol 136(2):334–42.e1
Goldman DL, Chen Z, Shankar V, Tyberg M, Vicencio A, Burk R (2018) Lower airway microbiota and mycobiota in children with severe asthma. J Allergy Clin Immunol 141(2):808-811.e7
Teo SM, Mok D, Pham K, Kusel M, Serralha M, Troy N et al (2015) The infant nasopharyngeal microbiome impacts severity of lower respiratory infection and risk of asthma development. Cell Host Microbe 17(5):704–715
Zhang Q, Cox M, Liang Z, Brinkmann F, Cardenas PA, Duff R et al (2016) Airway microbiota in severe asthma and relationship to asthma severity and phenotypes. PLoS One 11(4):e0152724
Song H, Yoo Y, Hwang J, Na YC, Kim HS (2016) Faecalibacterium prausnitzii subspecies-level dysbiosis in the human gut microbiome underlying atopic dermatitis. J Allergy Clin Immunol 137(3):852–860
Simonyté Sjödin K, Hammarström ML, Rydén P, Sjödin A, Hernell O et al (2019) Temporal and long-term gut microbiota variation in allergic disease: a prospective study from infancy to school age. Allergy 74(1):176–185
Wang X, Yi W, He L, Luo S, Wang J, Jiang L et al (2021) Abnormalities in gut microbiota and metabolism in patients with chronic spontaneous urticaria. Front Immunol 15(12):691304
Dimitriu PA, Boyce G, Samarakoon A, Hartmann M, Johnson P, Mohn WW (2013) Temporal stability of the mouse gut microbiota in relation to innate and adaptive immunity. Environ Microbiol Rep 5(2):200–210
Kellermayer R, Dowd SE, Harris RA, Balasa A, Schaible TD, Wolcott RD et al (2011) Colonic mucosal DNA methylation, immune response, and microbiome patterns in Toll-like receptor 2-knockout mice. FASEB J 25(5):1449–1460
Fung TC, Vuong HE, Luna CDG, Pronovost GN, Aleksandrova AA, Riley NG et al (2019) Intestinal serotonin and fluoxetine exposure modulate bacterial colonization in the gut. Nat Microbiol 4(12):2064–2073
Modena BD, Dazy K, White AA (2016) Emerging concepts: mast cell involvement in allergic diseases. Transl Res 174:98–121
Acknowledgements
The authors thank all the subjects who participated in this study. This work was funded by Tianjin Key Medical Discipline Construction Project.
Funding
This work was supported by Tianjin Health Research Project (TJWJ2022XK020); and Tianjin Natural Science Foundation (19JCYBJC27200). This work was funded by Tianjin Key Medical Discipline Construction Project.
Author information
Authors and Affiliations
Contributions
YL, RX and GZ all developed the study concept and design. YL, XX and CZ collected nasal samples. WW and XM guided statistical analysis. ZH performed histologic evaluation of polyp tissue. YL and RX interpreted the microbiome wrote and critically reviewed the manuscript. PS, ZY, WW, and GZ recruited patients and collected specimens, collected clinical metadata, interpreted the microbiome data, and YL, RX and GZ were major contributors in writing the manuscript and reviewing it critically. The authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethical approval and consent to participate
This study was approved by the Institutional Review Board of the Tianjin First Central Hospital, Tianjin, China (Approval number. 2021N037KY). This study was performed in accordance with the relevant guidelines and regulations. Written informed consent was obtained from all participants. all participants agreed to publish their data.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liang, Y., Zhang, C., Xiong, X. et al. Alterations of gut microbiome in eosinophilic chronic rhinosinusitis. Eur Arch Otorhinolaryngol (2024). https://doi.org/10.1007/s00405-024-08931-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00405-024-08931-3