
Abstract
The EYA4 gene encodes a 640-amino-acid protein that serves as a transcription factor. This protein contains a highly conserved Eya domain (eya-HR) and a variable domain (eya-VR). Mutations of this gene are known to cause postlingual and progressive sensorineural hearing loss, either as non-syndromic (DFNA10) or syndromic hearing loss, depending on the location of truncation of the mutant protein. Since our previous report, we have recruited 14 families segregating autosomal dominant moderate SNHL. A thorough medical history and physical examination including evaluation of heart problems ruled out any syndromic features in these families. Screening of EYA4 was performed by targeted exome sequencing of 134 known deafness genes (TES-134) from the probands. After basic filtering of the variants, we identified one proband who carried a novel truncation mutation, c.1194delT (p.Met401TrpfsX3) of EYA4, making the frequency of DFNA10 to be 7.14 % (1/14) in Koreans. The variant co-segregated perfectly with a slightly down-sloping, moderate degree of SNHL in the family (SH117), and was not detected in any of the 592 normal control chromosomes. This variant is likely to generate protein products that are truncated just downstream of the eya-VR domain. None of the three affected family members showed any syndromic features, including cardiac problems, which was compatible with a previous genotype–phenotype correlation. The identification of a novel EYA4 truncation mutation associated with DFNA10, rather than syndromic hearing loss, supports a previously reported genotype–phenotype correlation in this gene. Considering its detection rate, EYA4 mutations should be suspected in hereditary moderate hearing loss with a corresponding audiologic configuration, and a cardiac examination should be included in the initial evaluation.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The global prevalence of hearing loss is ~70 million people, with 50–60 % having a genetic predisposition for deafness [1]. Non-syndromic hearing loss (NSHL) with severe sensorineural deafness occurs in 1 in 1000 children and at least 60 % of cases are inherited [2]. Numerous cases of hereditary deafness are heterogeneously inherited according to a Mendelian inheritance pattern [2]. The autosomal-recessive form is estimated to account for ~80 % of cases of deafness, autosomal dominant for ~20 %, and mitochondrial and X-linked for only ~1 % of hereditary non-syndromic deafness [2]. In general, autosomal dominant forms induce progressive and post-lingual deafness, while autosomal-recessive forms usually cause severe sensorineural hearing loss [3]. To date, >30 genes and over 50 dominant loci have been reported for NSHL, segregating in an autosomal dominant fashion [4]. An audioprofile is a graph of recorded audiograms from patients of similar age group with mutations of the same autosomal dominant gene. These data have led to speculation regarding the mutant gene or locus responsible for autosomal dominant NSHL [5]. In addition to this audioprofile-driven approach, recent application of next-generation sequencing has enabled accurate and rapid genetic testing and more effective molecular diagnoses. The ability of this technology to detect the causative variant responsible for autosomal dominant hearing loss in the Korean population has been demonstrated [6].
The EYA4 gene, an ortholog of eya (“eyes absent”) of Drosophila, is an autosomal dominant deafness gene (NM_004100). It is engaged in the formation of compound eyes [7]. The EYA4 gene encodes a 640-amino-acid protein, containing a highly conserved C-terminal domain of 271 amino acids (eya-HR) and a transactivation domain, known as the eya-variable region (eya-VR) [8]. EYA4 is a member of the vertebrate EYA family and is a transcription factor that plays a role in the continued function of the mature organ of Corti [9]. Two different mutations in this gene at the DFNA10 locus were detected in an American and a Belgian family with autosomal dominant non-syndromic postlingual progressive hearing loss [9]. To date, seven truncation mutations and one missense mutation in this gene have been described as responsible for autosomal dominant NSHL [6, 9–15]. Interestingly, dilated cardiomyopathy (DCM), also present in addition to NSHL in a family carrying the truncation mutation, led to the shortest EYA4 transcript ever reported [12], suggesting a genotype–phenotype correlation [12, 13].
In this paper, we tried to identify a frequency of sensorineural hearing loss (SNHL) due to alteration of EYA4 among autosomal dominant moderate SNHL in a Korean population by screening the EYA4 gene based on targeted exome sequencing of a deafness panel. Additionally, we report a novel truncation mutation of EYA4 from a Korean family segregating with NSHL without any heart problems, and confirm a previously proposed genotype–phenotype correlation related to this gene.
Materials and methods
Subjects and phenotyping
Previously, we reported the results of targeted exome sequencing of 80 known deafness genes from 13 autosomal dominant moderate SNHL [6]. Since then, we additionally recruited 14 families segregating moderate SNHL in an autosomal dominant fashion. All of the probands from the 14 families and, whenever available, the family members participated in this study after providing informed consent to the Ethics Committee of either Seoul National University Bundang Hospital (IRB-B-1007-105-402) or Seoul National University Hospital (IRBY-H-0905-041-281). All 14 probands were evaluated by 12-lead ECG and chest X-rays.
All of the probands underwent a medical evaluation including a physical examination and a medical history interview. The medical history included age at onset of hearing loss, degree and progression of hearing impairment, and other relevant clinical manifestations, such as any history of other disease and environmental factors, including infection, ototoxicity and noise. The physical examinations ruled out the probability of syndromic hearing loss and heart problems.
Pure-tone audiometry (PTA) and speech audiometry were obtained for three individuals (SH117-241, SH117-325, and SH117-326), as well as the proband (SH117-240) of SH117. PTA was calculated as an average of the thresholds measured at 0.5, 1.0, 2.0 and 4.0 kHz, and air-conduction threshold measurements were performed at 125–8000 Hz. The level of hearing loss was described depending on the PTA results. Bone conduction thresholds were measured at 250–4000 Hz to assess conductive hearing loss in affected individuals.
The probands also underwent evaluations including computed tomography, magnetic resonance imaging of the inner ears and temporal bone.
Targeted exome sequencing, basic filtering and variant detection
The 14 probands underwent targeted exome sequencing (Otogenetics, Norcross, GA, USA), which was captured by the NimbleGen Sequence Catcher (Roche NimbleGen Inc., Madison, WI, USA), and tested against 134 known deafness genes. Reads were compared to the UCSC hg19 reference genome and non-synonymous SNPs filtered with a depth = 40; dbSNP138 was filtered out, except for the flagged SNP (Suppl. Table 1). Candidate novel or known deafness-causing variants were selected according to an autosomal dominant inheritance pattern and validated by Sanger sequencing. Co-segregation of strong candidates was confirmed among SNUH117 family members and detection of the candidate variants were also checked among 592 unrelated Korean control chromosomes.
Results
Targeted exome sequencing data
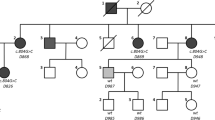
Targeted resequencing of the 134 known deafness genes from the probands was performed and the reads were compared with a human reference genome. Among the 14 probands, SH117-240 carried a potentially pathogenic variant of EYA4. In this family, four members (SH117-240, 241, 325 and 326) were recruited for this study (Fig. 1a). In detail, through basic and further bioinformatic filtering analyses, 13 SNPs remained as candidate variants for SNHL in the family SH117. Considering the autosomal dominant inheritance pattern in this family, three SNPs in known recessive genes were excluded. Sanger sequencing and a segregation study among the 10 inheritance pattern matched candidates were performed (Fig. 2). One candidate variant of EYA4, c.1194delT (p.Met401Trpfs*3) perfectly co-segregated with hearing loss; there were three affected individuals and one unaffected: EYA4 (Fig. 3). This variant was not detected in 426 unrelated Korean control chromosomes.

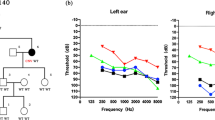
a Family pedigree showing the five affected patients over the three generations indicating the autosomal dominant inheritance of SNHL. b Audiogram of the affected patients (SH 117-240, 117-325, 117-326) and an unaffected member (SH 117-241). The proband (SH117-240) and the affected members (SH117-325 and 326) manifest a slightly down-sloping moderate degree SNHL with a progressive nature

Schematic flow chart showing the filtering of causative variants

Sequence chromatogram of the EYA4 gene of all four family members. The mutation at nucleotide position c.1194 is indicated by an arrow
Frequency of EYA4 mutations in Korean autosomal dominant SNHL
We identified one proband [1/14 (7.14 %)] whose SNHL was due to alteration of EYA4 (DFNA10) among 14 Korean probands segregating autosomal dominant SNHL. Considering the figure, 1/13, that our group previously reported [6], overall frequency of DFNA10 in this population recruited in our center was 2/27 (7.4 %).
Clinical features
SH117-240 (F/33YO) manifested a bilateral, slightly down-sloping, moderate degree of SNHL, involving all frequencies (Fig. 1b). Her hearing loss had progressed significantly over the past two decades. SH117-325 (F/58YO) and SH117-326 (F/50YO) showed an advanced degree of SNHL, preserving the configuration of audiograms (Fig. 1b). None of the affected subjects displayed symptoms of tinnitus, vestibular dysfunction or other clinical abnormalities, including heart problems indicating syndromic hearing loss. Chest X-ray and 12-lead ECG of four family members showed no evidence of DCM. Computed tomography and magnetic resonance imaging of the temporal bone of the proband revealed no structural malformations.
Prediction of candidate genes based on audiogram configurations
It was not possible to extract accurate pure-tone threshold averaged data for some published families segregating with EYA4 mutations. The pure-tone audiogram of SH117-240 showed a moderate and sensorineural type of hearing loss involving all frequencies. AudioGene 4.0 (http://audiogene.eng.uiowa.edu/) was used to identify the three genes most likely to be mutated in this patient: DFNA2A, DFNA13 and DFNA10.
Discussion
Through this and our previous study [6], we identified the frequency of EYA4 mutations which might cause heart problems depending on the location of the mutation to be 7.4 % among autosomal dominant SNHL. Another study focusing on Korean autosomal dominant SNHL by other group also reported a detection rate of DFNA10 to be 12.5 % (1/8) [15]. Collectively, the overall detection rate of DFNA10 among Korean autosomal dominant SNHL is calculated to be 8.57 % [6, 15]. This figure is high considering the extreme heterogeneity of molecular etiology of autosomal dominant SNHL in the Korean population [6].
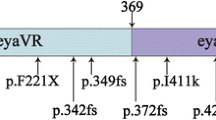
A novel frameshift mutation of EYA4, p.Met401Trpfs*3, was identified in a mid-sized family with non-syndromic, autosomal dominant hearing loss. Our results were compatible with those of previous genotype–phenotype correlation studies. The EYA4 gene encodes a 640-amino-acid protein that serves as a transcription factor. This protein contains a highly conserved C-terminal, known as the Eya domain (eya-HR) and a variable domain (eya-VR). To date, all mutations of the EYA4 gene were reported to affect the eya-HR domain, and all except one led to non-syndromic, postlingual, progressive, autosomal dominant hearing loss (Table 1). In contrast, a 4846-bp genomic deletion, which affected both the eya-VR and eya-HR, and led to the generation of the shortest EYA4 mutant transcript, was associated with DCM as well as NSHL [12]. As an explanation, Schonberger et al. showed that only the shortest of the four EYA4 transcripts generated by injection of four different antisense morpholino nucleotides lacked the ability to form a dimer with a wild-type EYA4 transcript, and to bind to one of the six binding proteins. Only the shortest transcript was associated with markedly compromised cardiac function, suggesting a genotype–phenotype correlation [12]. Makishima et al. documented an association between the fifth-shortest EYA4 transcript reported to date and non-syndromic hearing loss without any heart problems [13], further refining the genotype–phenotype correlation that mutations related to the eya-HR gave rise to sensorineural hearing loss alone, while mutations that truncated a significant portion of the eya-VR domain caused hearing loss with DCM [12, 13]. Previously, NSHL was diagnosed mostly by reference to patient medical histories and recollection data [6, 9, 10, 14, 15] (Fig. 4).

The known EYA4 mutations and their products. The EYA4 mutation associated with DFNA10 shows a genotype–phenotype correlation according to the degree of truncation
In this study, DCM was excluded as an EKG and chest X-ray were performed for all three affected members. Given the length of the protein generated by the mutation, the NSHL phenotype appeared to be compatible with the genotype–phenotype correlation proposed by Makishima et al. [12, 13]. Non-syndromic hearing loss constitutes ~75 % of hereditary hearing loss; however, a certain portion of apparently non-syndromic deafness may later be determined to be syndromic after rigorous clinical evaluation. Sometimes, knowing the genotype inadvertently discloses associated anomalies that would otherwise be missed. Likewise, the genotype–phenotype correlation related to the EYA4 mutations may serve as a useful biomarker to predict the development of DCM in DFNA10 deafness. This is especially useful as symptoms of DCM related to DFNA10 deafness tend to appear after sensorineural hearing loss, and show age-related expressivity [12]. Alternatively, it is possible that previous studies reporting DFNA10 hearing loss caused by diverse EYA4 mutations failed to notice later-onset cardiomyopathy. Given this, it may be necessary to perform regular cardiologic tests in families segregating with DFNA10 deafness, irrespective of the mutations in the EYA4 gene. In this study, the 61-year-old affected subject had a normal EKG and chest X-ray (SH117-325), reducing the likelihood of missing any cardiomyopathy. The audiogram of SH117-240 (F/33YO) showed a moderate degree of NSHL across all frequencies, but slightly pronounced in the mid-frequencies. During the second to fourth decade of life, non-syndromic hearing loss related to EYA4 mutations generally begins in the mid-frequencies and progresses to moderate-to-severe hearing loss affecting all frequencies [14].
A new diagnostic tool, combining Sanger sequencing and targeted resequencing (TES), increased the mutation detection rate to 78.1 % in multiplex families [6]. Use of this strategy enabled the identification of the novel mutation of EYA4 that led to a lack of most of the eya-HR domain in SH117. Initially, a machine-based candidate gene prediction tool, AudioGene 4.0 (http://audiogene.eng.uiowa.edu/) was used, because mutations in certain autosomal dominant deafness genes were associated with characteristic audiogram profiles. The prediction software provided us with the list of the most likely candidate genes (KCNQ4, COL11A2, and EYA4) based upon the audiogram configuration. Candidate gene screening based on an audioprofile can occasionally lead to the discovery of a causative mutation. Therefore, when patients with hereditary moderate hearing loss with such an audiogram configuration were encountered, EYA4 mutations could be suspected, indicating the patient initial evaluations should comprise a complete physical examination—including a cardiologic examination. However, the correlation of genotype and phenotype with the audioprofile alone is difficult in many cases, and clinicians must make an overall judgment after more information, including clinical and genetic examinations, has been gathered [5].
The EYA4 protein regulates the gene function necessary for maintenance of normal hearing, development of the inner ear, and heart function. This novel mutant EYA4 protein would lack the most of the EYA domain, but would retain the intact eya-VR domain, thereby causing only NSHL. Moreover, p.Met401Trpfs*3 co-segregated with a moderate degree of hearing loss in all three affected family members, and was not detected in the ethnicity-matched control chromosome with normal hearing. Our identification of a novel EYA4 truncation mutation associated with DFNA10, and not syndromic hearing loss, supports the previously reported genotype–phenotype correlation for this gene. Therefore, when patients with moderate hearing loss are encountered, the evaluation should comprise a complete physical examination—including cardiologic examination—and EYA4 mutations should be suspected considering the detection rate of 8.57 % in this population.
References
Tekin M, Arnos KS, Pandya A (2001) Advances in hereditary deafness. Lancet 358(9287):1082–1090. doi:10.1016/S0140-6736(01)06186-4
Morton NE (1991) Genetic epidemiology of hearing impairment. Ann N Y Acad Sci 630:16–31
Petit C (1996) Genes responsible for human hereditary deafness: symphony of a thousand. Nat Genet 14(4):385–391. doi:10.1038/ng1296-385
Van Camp G, Smith RJ. Hereditary Hearing Loss Homepage. http://hereditaryhearingloss.org/. Accessed 1 May 2015
Hildebrand MS, DeLuca AP, Taylor KR, Hoskinson DP, Hur IA, Tack D, McMordie SJ, Huygen PL, Casavant TL, Smith RJ (2009) A contemporary review of AudioGene audioprofiling: a machine-based candidate gene prediction tool for autosomal dominant nonsyndromic hearing loss. Laryngoscope 119(11):2211–2215. doi:10.1002/lary.20664
Choi BY, Park G, Gim J, Kim AR, Kim BJ, Kim HS, Park JH, Park T, Oh SH, Han KH, Park WY (2013) Diagnostic application of targeted resequencing for familial nonsyndromic hearing loss. PLoS One 8(8):e68692. doi:10.1371/journal.pone.0068692
Hentze MW, Kulozik AE (1999) A perfect message: RNA surveillance and nonsense-mediated decay. Cell 96(3):307–310
Zimmerman JE, Bui QT, Steingrimsson E, Nagle DL, Fu W, Genin A, Spinner NB, Copeland NG, Jenkins NA, Bucan M, Bonini NM (1997) Cloning and characterization of two vertebrate homologs of the Drosophila eyes absent gene. Genome Res 7(2):128–141
Wayne S, Robertson NG, DeClau F, Chen N, Verhoeven K, Prasad S, Tranebjarg L, Morton CC, Ryan AF, Van Camp G, Smith RJ (2001) Mutations in the transcriptional activator EYA4 cause late-onset deafness at the DFNA10 locus. Hum Mol Genet 10(3):195–200
Pfister M, Toth T, Thiele H, Haack B, Blin N, Zenner HP, Sziklai I, Nurnberg P, Kupka S (2002) A 4-bp insertion in the eya-homologous region (eyaHR) of EYA4 causes hearing impairment in a Hungarian family linked to DFNA10. Mol Med 8(10):607–611
Schonberger J, Levy H, Grunig E, Sangwatanaroj S, Fatkin D, MacRae C, Stacker H, Halpin C, Eavey R, Philbin EF, Katus H, Seidman JG, Seidman CE (2000) Dilated cardiomyopathy and sensorineural hearing loss: a heritable syndrome that maps to 6q23-24. Circulation 101(15):1812–1818
Schonberger J, Wang L, Shin JT, Kim SD, Depreux FF, Zhu H, Zon L, Pizard A, Kim JB, Macrae CA, Mungall AJ, Seidman JG, Seidman CE (2005) Mutation in the transcriptional coactivator EYA4 causes dilated cardiomyopathy and sensorineural hearing loss. Nat Genet 37(4):418–422. doi:10.1038/ng1527
Makishima T, Madeo AC, Brewer CC, Zalewski CK, Butman JA, Sachdev V, Arai AE, Holbrook BM, Rosing DR, Griffith AJ (2007) Nonsyndromic hearing loss DFNA10 and a novel mutation of EYA4: evidence for correlation of normal cardiac phenotype with truncating mutations of the Eya domain. Am J Med Gen 143A(14):1592–1598. doi:10.1002/ajmg.a.31793
Hildebrand MS, Coman D, Yang T, Gardner RJ, Rose E, Smith RJ, Bahlo M, Dahl HH (2007) A novel splice site mutation in EYA4 causes DFNA10 hearing loss. Am J Med Gen 143A(14):1599–1604. doi:10.1002/ajmg.a.31860
Baek JI, Oh SK, Kim DB, Choi SY, Kim UK, Lee KY, Lee SH (2012) Targeted massive parallel sequencing: the effective detection of novel causative mutations associated with hearing loss in small families. Orphanet J Rare Dis 7:60. doi:10.1186/1750-1172-7-60
Conflict of interest
None of the authors has any conflict of interest, financial or otherwise.
Author information
Authors and Affiliations
Corresponding author
Additional information
H. S. Choi and A. R. Kim contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Choi, H.S., Kim, A.R., Kim, S.H. et al. Identification of a novel truncation mutation of EYA4 in moderate degree hearing loss by targeted exome sequencing. Eur Arch Otorhinolaryngol 273, 1123–1129 (2016). https://doi.org/10.1007/s00405-015-3661-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00405-015-3661-2