
Abstract
X-linked myotubular myopathy (XLMTM), a severe congenital myopathy, is caused by mutations in the MTM1 gene located on the X chromosome. A majority of affected males die in the early postnatal period, whereas female carriers are believed to be usually asymptomatic. Nevertheless, several affected females have been reported. To assess the phenotypic and pathological spectra of carrier females and to delineate diagnostic clues, we characterized 17 new unrelated affected females and performed a detailed comparison with previously reported cases at the clinical, muscle imaging, histological, ultrastructural and molecular levels. Taken together, the analysis of this large cohort of 43 cases highlights a wide spectrum of clinical severity ranging from severe neonatal and generalized weakness, similar to XLMTM male, to milder adult forms. Several females show a decline in respiratory function. Asymmetric weakness is a noteworthy frequent specific feature potentially correlated to an increased prevalence of highly skewed X inactivation. Asymmetry of growth was also noted. Other diagnostic clues include facial weakness, ptosis and ophthalmoplegia, skeletal and joint abnormalities, and histopathological signs that are hallmarks of centronuclear myopathy such as centralized nuclei and necklace fibers. The histopathological findings also demonstrate a general disorganization of muscle structure in addition to these specific hallmarks. Thus, MTM1 mutations in carrier females define a specific myopathy, which may be independent of the presence of an XLMTM male in the family. As several of the reported affected females carry large heterozygous MTM1 deletions not detectable by Sanger sequencing, and as milder phenotypes present as adult-onset limb-girdle myopathy, the prevalence of this myopathy is likely to be greatly underestimated. This report should aid diagnosis and thus the clinical management and genetic counseling of MTM1 carrier females. Furthermore, the clinical and pathological history of this cohort may be useful for therapeutic projects in males with XLMTM, as it illustrates the spectrum of possible evolution of the disease in patients surviving long term.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Myotubular myopathy (XLMTM, MIM #310400) is a severe congenital myopathy linked to the X chromosome. While affected males display severe hypotonia and weakness at birth and have a very short life expectancy even with interventional care and ventilatory support, female carriers were reported to be either asymptomatic or affected through a wide spectrum of severity. The disease in females is much less characterized to date. Moreover, as affected females reach adulthood, they could potentially reveal additional symptoms that are not described in XLMTM in males. In this study, we thus aimed to better define the symptoms, the genotype–phenotype correlation and the disease progression in females with XLMTM through a thorough characterization of a large cohort of novel cases and comparison with the literature.
XLMTM is caused by mutations in the MTM1 gene (MIM 300415) encoding the ubiquitous phosphoinositide phosphatase myotubularin (MTM1) [38]. This is the most severe form of centronuclear myopathies, a group of myopathies caused by different genes and modes of inheritance whose hallmark histological abnormalities are centrally located nuclei [2, 8, 11, 12, 15, 38, 44, 48, 52, 65], without dystrophic features. Around 500 males with XLMTM have been reported and they usually present at birth with severe hypotonia, muscle weakness and respiratory insufficiency [19, 36, 45, 57, 62]. XLMTM is non-progressive and those affected males who survive several years may show additional non-muscle clinical features, the most common being liver peliosis [27, 30, 43, 59]. Atypical forms have been reported in some boys and adult men with a clinical classification based on respiratory and milestone features [30, 36, 42]. Mutations are found in all 15 exons of MTM1 and encompass point mutations (missense, nonsense, splice site mutations), insertions, small and large deletions [10, 19, 31, 36, 37, 57, 62] and duplications [4, 45, 61]. The majority of mutations are clustered in exons 4, 8, 9, 11 and 12. Duplication has been described in some cases and a deletion of exon(s) or of the whole MTM1 gene was reported in 7% of patients. Large deletions including part or the entire neighboring MAMLD1 gene cause a contiguous gene syndrome with hypospadias and myotubular myopathy in males [6, 24, 32, 37].
Female carriers are usually asymptomatic but a total of 26 affected females aged 5–71 years have been reported presenting with severe neonatal form [33, 51] or moderate to mild forms [7, 18, 20, 22, 23, 25, 28, 46, 50, 55, 56]. Truncating mutations as well as missense mutations were observed scattered through the gene. Skewed X inactivation was reported in some as a potential explanation for their affected status compared to non-affected carriers, while others did not show skewing.
In this study, we reviewed the clinical, histological, imaging and molecular data from 43 affected females, including 17 new cases. We have identified consistent asymmetry in muscle involvement and have better defined the spectrum and progression of the disease, the frequency of clinical, investigation and histological findings, and discuss the clinical correlation with the type of mutation and X inactivation.
Patients and methods
Patient cohort
Data from the 43 females affected with XLMTM are summarized in Supplementary Tables 1–3. The 17 new unrelated females originated from Brazil, Finland, France, Israel, The Netherlands, UK and USA. Fifteen were from new families, while F8 is the sister of an affected male previously reported as patient HC19 [10] and patient F1 is the sister of an affected female previously reported as patient 1 by Bevilacqua and colleagues [7]. Clinical data were collected from the clinicians who referred patients for a genetic diagnosis (25 patients) or from the literature (18 patients).
Morphological studies
Muscle biopsies were performed on 14 out of the 17 new patients (Supplementary Table 1). Muscle sections were stained with hematoxylin–eosin (H&E), modified Gomori trichrome, periodic acid Schiff (PAS) and Sudan red and black. Histoenzymology was performed using routine histochemical methods [21]. Ultrastructural study was carried out according to standardized protocols. Briefly, tissue samples were fixed in a 2% glutaraldehyde fixative solution, post-fixed with osmium tetroxide and embedded in resin epoxy. Semi-thin sections were stained with toluidine blue. Ultrathin sections were contrasted with uranyl acetate and lead citrate.
Molecular analysis
DNA from the 17 new patients and relatives was extracted from blood or muscle samples by standard methods.
Sequence analysis of the MTM1 gene was performed by bi-directional sequencing of the 15 exons with flanking intronic sequences (primer sequences and PCR conditions are available on request). A search for copy number variations (deletion or duplication of exon or whole gene) was performed by MLPA analysis (multiplex ligation-dependent probe amplification) using the P309-A1-MTM1 kit (MRC Holland) for patients F2, F3 and F4. Patient F11 was included in MTM1 Sanger analysis due to the detection on Western blot [60] of a decreased level of myotubularin on protein extracts from lymphoblasts and muscle biopsies (manuscript in preparation). Patients F15 and F12 were diagnosed by exome sequencing in the scope of a French consortium research project (MYOCAPTURE). Exome analysis was performed using SureSelect Human all Exon 50 Mb capture library v4 (Agilent, Santa Clara, USA) and paired-end sequenced on Illumina HiSeq 2500 sequencer at the French National Center of Genotypage, Paris, France. Patient F17 was diagnosed by exome sequencing using SureSelectXT Human all Exon 50 Mb capture library (Agilent, Santa Clara, USA) on IlluminaHiSeq at BGI-EUROPE in Denmark. Patient F13 was diagnosed using a multi gene panel, MGZ laboratory, Munich, Germany. Patient F16 was diagnosed using targeted exons sequencing of a custom panel of 210 genes linked to neuromuscular diseases (SureSelect, Agilent, Santa Clara, CA) (MYOdiagHTS panel, Strasbourg, France, manuscript in preparation).
Affymetrix Cytogenetics Whole-Genome 2.7M Array was used to delineate deletion breakpoints for patient F3. DNA quantity was insufficient to perform this study for patients F2 and F4.
X-chromosome inactivation (XCI) analysis was performed using HpaII predigestion of DNA followed by PCR of the highly polymorphic CAG repeat of the AR gene (HUMARA) [3] and the CGG repeat within the FMR1 gene [13]. XCI with an allele ratio <80:20 was considered a random pattern, a ratio equal or greater than 80:20 was considered to be skewed and a ratio greater than 90:10 was considered to be highly skewed.
Results
Clinical, imaging, histological, ultrastructural and molecular data are summarized in Supplementary Tables 1–5 for the 43 females including 17 novel cases and 26 previously reported cases, resulting in the largest XLMTM female cohort characterized so far. The main clinical and histological findings representing the new cases are presented (Figs. 4, 5, 6, 7).
Females with XLMTM demonstrate an independently recognizable myopathy
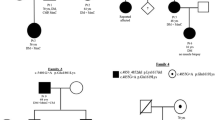
To assess if females with XLMTM signs define a disease entity, we analyzed the familial history for XLMTM or undefined muscle disorder. The female patient was the first diagnosed case of XLMTM in 28 out of the 36 families, with 20 being sporadic cases. The diagnosis of XLMTM was previously established or suspected (based on clinical and histopathological data) in a male relative in only 8 of the 36 families (Fig. 1a; Supplementary Table 2). In four of the seven families including several affected females, there was no affected male (Fig. 1b; Supplementary Table 2). The mutation occurred de novo for the older affected females in 13 of the 19 families in which samples from parents were available for further analysis. Altogether, these findings indicate that females with XLMTM define a specific myopathy cohort, independently of the presence of male XLMTM.

Family history of XLMTM in 36 families. a Families with previously diagnosed XLMTM. b Family history of muscular diseases
Females with XLMTM are usually less severely affected than males, with overlapping clinical hallmarks
Most males with XLMTM are severely affected from birth with weakness and respiratory distress, needing respiratory and feeding support. To assess the distinctive features between male and female XLMTM, we analyzed the age of onset and severity in 43 females with XLMTM to compare with affected males.
The clinical phenotype is highly variable in age of onset and severity, in particular regarding weakness and respiratory muscle involvement (Fig. 2; Supplementary Table 3). The age of onset ranged from birth or fetal life to adulthood. Only eight of 42 females were reported to be hypotonic and have respiratory or feeding problem at birth. The pregnancy for two of these eight patients was associated with reduced fetal movements and/or polyhydramnios and was normal for two others (no data available for four patients). Most of the females affected from birth displayed generalized muscle weakness. For 30 out of 42 female patients, weakness was clinically manifest during childhood (2–14 years) with the first impairment being proximal in the lower limbs in 17 patients (delayed motor milestones, gait difficulties, difficulty with climbing stairs or raising from a squatting position) and in the upper limbs in five.

Initial muscle impairment at the age of onset for 42 females with XLMTM. The initial muscle impairment reported for 42 patients (age of onset unknown for 1 patient) is presented depending on the age of onset: at birth, at childhood (2–14 years) or at adulthood. Facial face, ptosis; upper limb arm weakness; lower limb delayed motor milestones, gait difficulty, difficulty in climbing stairs or in standing from a squatting position; generalized hypotonia at birth ± facial involvement; Other feeding difficulties; NA not available
The most severely affected female in our cohort displayed similar clinical symptoms and course as an XLMTM male (F2). She was affected with severe neonatal form and died aged 18 months old. There was no family history of neuromuscular conditions. She was born eutrophic at 41 weeks of gestation, and Apgar scores were 9 and 10 at 1 and 5 min, respectively. Thirty minutes after birth, she presented with severe hypotonia and respiratory distress. Clinical examination revealed severe left facial weakness with left ptosis, stridor due to laryngomalacia and swallowing difficulties. At the age of 1.5 months, she demonstrated fluctuating and moderate axial hypotonia and a Moebius syndrome diagnosis was initially considered. From the age of 2.5 months, she had recurrent episodes of respiratory distress and a congenital myasthenic syndrome was considered with the findings of axial hypotonia, absent deep tendon reflexes and worsening of the facial weakness. Quadriceps wasting was noted at age 15 months and a muscle biopsy was performed showing multiple anomalies (see below). The child was subsequently treated with anticholinesterase drugs (pyridostigmine) with a very transient improvement. She started walking with support at the age of 18 months, but died from an acute bronchopneumonia shortly after.
In all other cases, there was a slowly progressive course, with limb weakness being the major sign, sometimes predominating in the pelvic girdle and spreading to distal muscles. Several of the 43 patients showed significant difficulties in standing from a seated position and/or in climbing stairs and/or running. Eight patients were never able to walk ([18, 33], F2) or run ([23, 28, 50, 51], F16): seven had lost the ability to walk independently, with three patients needing a cane at ages 40, 50 and 77 years (F11 patient from [46], F10) and four needing a wheelchair at ages 13, 32, 53 and 66 years (F4, F3, F17, the mother in [29]). Pelvic floor weakness with urinary incontinence was reported in seven cases (at the ages of 6, 27, 35, 35, 52, 55 and 79 years, respectively). Neck flexor weakness was reported in 12 cases.
The frequency of additional clinical signs in this cohort of 43 females is summarized in Fig. 3. Bulbar or pseudo bulbar symptoms were reported for 24 patients including facial weakness, limitation of extra-ocular movements, ophthalmoparesis, uni- or bilateral ptosis and dysarthria, whereas no such involvement was noted for ten patients. Respiratory muscle involvement was noted for 14 out of 33 patients, with a decrease in forced vital capacity in nine patients (at 37–83% of age and sex-matched predicted values). Severe restrictive respiratory dysfunctions with a hemidiaphragmatic paresis led to death of the oldest patient [46] at 84 years of age. Serum CK (creatine kinase) levels were normal in 21 patients and slightly elevated in 10 patients, supporting the finding that disease is not dystrophic. Skeletal and joint abnormalities were observed in 17 out of 28 cases, including kyphoscoliosis, scoliosis, joint hyperlaxity, joint contractures of the lower extremities, foot deformities and hand and/or facial contractures.

Frequency of clinical and investigation signs in the cohort of 43 females with XLMTM. The numbers in brackets indicate the number of patients for each item. ↑ Elevated, ↓VC decreased vital capacity, CK creatine kinase, NA not available
Five of the patients were reported to be overweight.
Overall, while a few females presented with similar onset to the male XLMTM (with fetal involvement and/or neonatal generalized weakness) with one case with an identical course (F2), the vast majority of females with XLMTM had later onset and were less severely affected than males with XLMTM. Affected females with XLMTM display similar facial signs as males with XLMTM, namely facial weakness, ophthalmoparesis and ptosis, and these appear to be important clues for diagnosis.
Females with XLMTM frequently show a pattern of asymmetric involvement
At all ages, a consistent and striking feature was asymmetric symptoms, seen in 29 out of 35 patients. These included muscle weakness and wasting, especially of the arm, leg, and calf (Fig. 4). Hemidiaphragm elevation and asymmetric scapular winging were also noted.

Facial and general muscle weakness and asymmetric involvement. a Patient F4 presenting with facial asymmetry (left weaker), deficit in arm elevation and retractions. b Patient F16 presenting with left side ptosis, facial asymmetry, high arched palate, reduced muscle bulk and scapula winging. c Patient F17 presenting with left side ptosis, deficit in arm elevation and needing support for standing. d Patient F5 presenting with mild left ptosis. Right hand smaller than left hand
There was also consistent asymmetry of facial involvement, notably asymmetric ptosis and facial weakness; examples are given for four patients of different ages in Fig. 4a–d. These asymmetries were mainly on a left–right axis.
Asymmetric involvement has been previously reported in several cases (Supplementary Table 1 and 3) and Drouet et al. reported two females with unilateral weakness [20]. In addition to asymmetric weakness, skeletal asymmetry was noted in some patients. For example, patient F5 has a right hand smaller than her left hand (Fig. 4d) and patient F13 has a lower limb length discrepancy. Grogan et al. reported females of two unrelated families with asymmetric weakness, hemidiaphragm elevation, and arms and fingers of different sizes when comparing the right and left sides [25].
Both our large cohort of novel cases and the descriptions of those in the literature highlight the frequency of asymmetric muscle involvement in females with XLMTM and suggest skeletal defects are a primary symptom of the disorder.
Imaging investigations highlight general and asymmetric muscle involvement
We next assessed whether imaging confirms the clinical symptoms, including asymmetry, and whether this yields additional diagnostic clues. Magnetic resonance imaging (MRI) findings for six newly described patients and nine previously reported patients [7, 20, 33, 46, 50, 51] are summarized in Supplementary Table 4 and representative images are depicted in Fig. 5 for several of the novel cases, including whole body MRI.

Representative magnetic resonance imaging (MRI) in four patients. Whole body muscles magnetic resonance imaging (WB MRI). Patient F14: T1 axial sequences of the proximal lower limb (a), thigh (b), lower limb (c), proximal upper leg (d), neck (e) and whole body muscles (f). Patient F17: T1 axial sequences (g, h), hip girdle (i, j), upper (k, l) and lower limb muscles (m, n). Patient F13: T1 axial and STIR sequences of the thigh (o, q) and leg (p, r). Patient F16: T1 axial sequences of the thigh (s, t) and calf (u)
For patient F14, whole body MRI demonstrated asymmetric fatty involvement of the muscles, which predominates in the left side of the upper and lower limbs. In the lower limbs, the fatty involvement involves the anterior and posterior part of the legs: the soleus is near normal in the right side, whereas the left soleus muscle showed a moderate fatty infiltration (Fig. 5a). In the thigh and the pelvis, the asymmetric distribution of the fatty infiltrates is obvious, with gracilis, sartorius, biceps femoris, quadriceps and glutei muscles (minimus, medius and maximus) demonstrating almost complete fatty involvement (Fig. 5b, c). In the upper limbs, the fatty involvement predominates in the deltoid muscles, infra and supraspinatus and subscapularis muscles, with the left side showing the most evident fatty infiltration (Fig. 5e). Asymmetric fatty involvement is also noted in the neck extensors, while facial muscles are spared.
Several of these features are found in other patients (Fig. 5 and Supplementary Table 4). For patient F17, there is severe atrophy and fatty infiltration of paraspinal, gluteal and psoas muscles and the upper leg muscles, while the lower leg muscles are less severely affected (Fig. 5g–n). The more proximal muscles (obturatorius, piriformis and transverse abdominal) are relatively spared. For patient F13, prominent fatty infiltration of the muscles of the lower limbs was noted, both at the thighs and legs (Fig. 5o–q). All the muscles of the thighs were affected, but the right semimembranosus and the left vastus intermedius were slightly less affected. The anterior compartments of the legs were more affected, while the gastrocnemius and soleus were nearly spared. For patient F16, muscle involvement with clear asymmetry was noted mainly for the distal muscles, including involvement of the left tibialis anterior and the right peroneus (Fig. 5s–u). In the thighs, the anterior and posterior muscles were affected, especially visible on the left in the vastus externus and adductor magnus.
Taking both the novel reported cases and previous reports (Supplementary Table 4), these data confirm a general involvement of muscles and often highlight an asymmetric pattern correlating with the asymmetric muscle weakness observed on clinical assessment.
Histological and ultrastructural features are similar in males and females with XLMTM
Biopsies were available for 38 patients, including one that was not interpretable due to massive fatty replacement. Variation in myofiber diameter with atrophic round fibers was reported in 25 cases, type 1 fiber predominance in 17 cases, and endomysial fibrosis and/or fatty replacement in 20 cases (Fig. 6; Supplementary Table 1 and 5). There was no fiber necrosis or inflammatory change. Abnormally positioned nuclei varied from a few scattered to numerous non-peripheral nuclei in all interpretable biopsies. Six biopsies were described with internalized nuclei, 19 biopsies with central nuclei, and 12 biopsies had both. Necklace fibers and radial sarcoplasmic strands (RSS), as previously reported in adult XLMTM patients and also in patients with DNM2-related dominant centronuclear myopathy, were described in 11 and 3 out of the biopsies, respectively. Necklace fibers display a basophilic ring underneath the sarcolemma that is strongly reactive with PAS and oxidative reactions.

Histological features in females with XLMTM. Muscle biopsies from patient F2 (a–d) and F4 (e, f). a A necklace fiber displaying a basophilic ring located underneath the sarcolemma (arrow) (hematoxylin and eosin staining). b A necklace fiber strongly reacting with PAS (arrow). c Type I predominance with small type I (light) and II (dark) fibers (myosin adenosine triphosphatase preincubated at pH 9.4 technique). d A necklace fiber strongly reactive for oxidative reactions (arrow) (nicotinamide adenosine dinucleotide-tetrazolium reductase technique). e Numerous centralized nuclei and fiber size heterogeneity (Gomori trichrome stain). f Necklace fibers strongly reactive for oxidative reactions (arrow) (nicotinamide adenosine dinucleotide-tetrazolium reductase technique)
At the ultrastructural level, internal nuclei may display an altered shape (Fig. 7). There were a range of signs of myofibrillar disruption from focal loss of myofibrils to fibers with complete disorganization of the myofibrillar network. Z disk streaming was also common. Necklaces form a clear ring devoid of organelles under the sarcolemma and are associated with an oblique orientation of the myofibrils.

Ultrastructural defects in females with XLMTM. Electron microscopy for muscles from patient F2 (a–d) and F11 (e–g). a Focal loss of myofibrils in a small muscle fiber (arrow) (OM ×5000). b Z disk streaming running over two sarcomeres (arrow) (OM ×6000). c Internalized nuclei in an atrophic fiber with a complete disorganization of the myofibrillar network (arrow) (OM ×4000). d Ultrastructural pattern of a necklace, forming a clear ring devoid of organelles under the sarcolemma, with an oblique arrangement of the myofibrils (arrow) (OM ×4000), e, f Muscle fibers showing internalized nuclei, cisternae (blue arrow), vacuoles (red arrow) and material accumulation (green arrow)
Overall, the histopathology and ultrastructural defects in females with XLMTM appear very similar to those seen in males with XLMTM, albeit necklace fibers are mainly found in adult patients and are less observed in neonatal affected males.
Most MTM1 mutations in females were associated with a severe phenotype in males
Females with XLMTM are usually more mildly affected than males. We explored if there was a correlation between the type of mutation and the phenotypic severity. First, we analyzed the MTM1 gene through approaches including Sanger sequencing, targeted and exome sequencing, MLPA and DNA microarray to delineate the genetic defects. Analysis of MTM1 revealed four new heterozygous mutations in the 17 new female patients reported: an in-frame deletion encompassing exons 9 and 10, the intronic mutation c.1644+1G>A predicted to disrupt the donor splice site of exon 14 and two stop gain c.548G>A (p.Trp183*) and c.1601G>A (p.Trp534*) (Supplementary Table 1). Patients F3, F4 and F17 were heterozygous carriers of a large deletion of the whole MTM1 gene and the neighboring MTMR1 gene. For patient F3, comparative genomic hybridization (CGH) study delineated the deletion with a minimum size of 595,302 bp (from genomic position chrX: 150,443,004–151,038,095; hg38) and a maximum size of 597,756 bp (from genomic position chrX: 150,442,220–151,039,765;hg38) encompassing MAMLD1, MTM1, MTMR1, CD99L2 and HMGB3 genes. For patient F17, exome analysis delineated a 474,391 bp deletion (from genomic position chrX: 150,512,003–150,986,191; hg38) encompassing MTMR1 and CD99L2, as well as part of MAMLD1 and HMGB3.
Genotype–phenotype correlation in males has established that some mutations are more likely to be associated with a mild or moderate phenotype, probably being compatible with a residual normal function of the mutated myotubularin. Conversely, severely affected males tend to have an undetectable or a marked reduction in the protein level [39]. Twenty of the 29 mutations present in this cohort of 43 affected females have previously been found in male patients, with 16 being reported in association with a severe form of the disease, 3 predominantly with a severe form but occasionally with a mild/moderate form (c.109C>T, c.614C>T, c.1262G>A), and 1 (c.1354-1G>A) described once in a moderate case [30] and also found once in a severe case (Biancalana, unpublished observation). Among the ten mutations described only in females, nine are in-frame or out-of-frame deletions, truncations or splicing mutations predicted to be associated with a severe phenotype in males, and one is a missense mutation (c.1115T>A) which has been shown to lead to a reduced protein level. Thus, females with XLMTM in this cohort do not have specific MTM1 mutations compared with males, but rather harbor mutations usually associated with a severe phenotype in males with XLMTM, while these females display a milder phenotype.
Enhancement of skewed X chromosome inactivation in females with XLMTM
The difference in severity between males with XLMTM and females is not explained by the type of mutation. In a female heterozygous for an X-linked recessive mutation, half of the cells on average should have a normal level of the gene product, while the other half would express the mutated allele. However, skeletal muscle fibers are syncytia formed from the fusion of different myoblasts. It was shown at least in mice that different nuclei from single myofibers have random X-chromosome inactivation (XCI), predicting that each fiber would have about 50% expression level [66]. A skewed X-inactivation pattern favoring the mutated X chromosome has been hypothesized to be a determinant of the variability of the phenotype observed in females with XLMTM, but several reports did not confirm a skewed XCI in affected females (Supplementary Table 1). To better evaluate XCI as a determinant of the phenotype severity in females, we compared the disease onset versus the XCI pattern (random, skewed and highly skewed; see “Patients and methods”). Unfortunately, the test could not determine which X chromosome was preferentially active. XCI studies were informative in 32 out of 43 patients. Evidence of skewed XCI (80:20–90:10) or highly skewed XCI (≥90:10) was identified in 15.6 and 18.75% of patients aged <55 years, respectively, whereas in the general adult population the ratio was around 14 and 4%, respectively [5]. This enhancement in XCI among females with XLMTM is significant for the highly skewed group (p < 0.025) and is even more significant when considering that for the six patients with highly skewed XCI, the ratio was in fact >95:5 (found in only 1.7% of the normal population). In conclusion, there is an increased prevalence of highly skewed X inactivation in the cohort of females affected by XLMTM.
Discussion
We have described or reviewed the clinical, morphological and molecular data obtained from 43 X-linked myotubular myopathy female carriers from 36 unrelated families. To date, only 26 manifesting female carrier cases have been reported in the literature. Analyzing our data on 17 new affected females and those previously reported (see Supplementary Table 1), we were able to establish several specific observations. Females with XLMTM often present with a myopathy, independently of the presence of affected males in the family. This myopathy can be highly heterogeneous in terms of onset and severity and shares several clinical features with male XLMTM including similar facial involvement. However, females with XLMTM frequently display asymmetric involvement and specific histological features develop with age, as necklace fibers. While they usually have a milder phenotype than males with XLMTM, females share MTM1 mutations associated with severe cases in males, potentially due to the fact that they can express a normal allele from the non-mutated X chromosome.
Assessing the full clinical spectrum and frequency of females with XLMTM
XLMTM female carriers were previously considered to be usually non-affected or mildly affected. However, including the novel cases characterized in this study, 43 females from 36 unrelated families were reported to show signs of muscle involvement, with several presenting with muscle, respiratory or skeletal involvement significantly impacting on their daily life. Nevertheless, this cohort is not likely to represent accurately the spectrum of involvement, as several recruitment biases may exist. Females with XLMTM may be recognized either because they are severely affected (they are the proband) or are mildly affected and evaluated following the identification of males with XLMTM in the family. Those females from the 12 “female families” include some who are mildly affected, suggesting that an unknown proportion of females with XLMTM may have never come to clinical attention without a prior XLMTM diagnosis in their relatives (i.e., patient F1). It is also likely that some XLMTM manifesting females are not diagnosed because the MTM1 gene is not suspected as the cause of their symptoms, particularly in the absence of suggestive histology. The proportion of reported manifestation of females with XLMTM compared with all diagnosed patients is around 6%, but this figure is not homogeneous across countries. For example, this proportion is increased in France with around 13% of XLMTM French patients being female (19 females of the present cohort). This variation suggests that there is likely to be a lack of consideration of the diagnosis in females and strongly suggests that the frequency of females with XLMTM is underestimated. Thorough characterization and comparison of females with XLMTM in large cohorts, as attempted in this report, should increase awareness and provide diagnostic clues to better recognize this myopathy.
Clinical spectrum and asymmetric involvement in females with XLMTM
Females with XLMTM present usually with a progressive pattern of limb-girdle myopathy, possibly associated with respiratory muscle and/or skeletal involvement. The impairment may be as severe as in a severe male neonatal form which may be responsible for death within the first months, as observed in patient F2, ranging to a milder form occurring mostly during childhood but impacting daily life significantly. Many carriers were considered as non-affected in childhood, but were never athletic at school. Females with XLMTM display similar facial features as affected males, with facial weakness, ophthalmoplegia and ptosis being important diagnostic clues. The predominant proximal and lower leg involvement pattern observed on MRI or CT scan for most of the 15 female patients reviewed in this study is similar to the only two observations reported for affected males [7, 47].
In this analysis of a large patient cohort, we found recurrent asymmetric involvement that is also underlined in MRI analyses. This asymmetry may involve limb and facial muscle weakness and also skeletal development in several cases (this study, [20, 25]). We and others found a strong bias in the XCI in affected females, but this was not always consistent with the severity of the symptoms. Such an asymmetric weakness in manifesting carrier females is indeed described in other X-linked diseases such as Duchenne muscular dystrophy [54, 58], suggesting that a distinct X-chromosome inactivation ratio in different muscles could be responsible for this asymmetry, leading to greater expression of the mutated gene on one side of the body. It was not possible to directly investigate several muscles from the same patient for XCI, but such a study would help delineate a correlation with asymmetric features. Similarly, the skeletal asymmetry may be caused by differential expression of normal MTM1 in the body, as MTM1 is suspected to be linked to skeletal growth, given that males with XLMTM deficient in myotubularin often show features of overgrowth at birth. However, a mildly affected XLMTM male presenting with muscle asymmetry has been described [7] and this clearly could not be linked to XCI. Further observations are needed to establish the prevalence of asymmetry in males.
In females, asymmetric involvement detected through clinical and/or imaging investigations is another diagnostic clue to suggest undertaking MTM1 genetic testing.
Muscle biopsy features which may be an indication for MTM1 sequencing and XLMTM diagnosis
Muscle biopsy is routinely used to direct the molecular diagnosis of congenital myopathies. In females with XLMTM, abnormal nuclei internalization and centralization is similar to that observed in males with XLMTM. Necklace fibers were observed in nearly one-third (11 out of 37) of females with XLMTM for whom muscle biopsies were interpretable, confirming this is a prevalent histological marker in milder XLMTM, particularly in manifesting carriers [7], although they can also be present in severe cases as in patient F2 [1, 26]. However, this marker is not exclusive for XLMTM, being described in other forms of centronuclear myopathy due to mutations in DNM2 [14, 41].
Fatty replacement and/or fibrosis, as described in males with XLMTM, was also observed in 20 out of 38 biopsies. Such a gradual replacement of contractile tissue with non-contractile connective tissue or fat is described in congenital myopathies, in particular in severe, advanced or chronic cases. The fibrosis in XLMTM may be the result of a replacement of fibers which disappear and are not regenerated due to a defect in the number of satellite cells in the muscle of the patients [52].
Overall, the presence of centralized nuclei and/or necklace fibers is a strong diagnostic clue for XLMTM in females. Nevertheless, several females with XLMTM have non-informative muscle biopsies that show dystrophic features.
The genotype is a suggestive, but not a reliable predictor of clinical prognosis for females with XLMTM
Nearly all MTM1 mutations detected in females with XLMTM are described in or predicted to be associated with a severe phenotype in males. It is possible that mutations associated with a mild or moderate phenotype in males are not associated with a phenotype in females due to the MTM1 expression from the non-mutated X chromosome, but a clinical evaluation of females carrying such mutations is needed to assert this hypothesis.
A non-random XCI pattern has been proposed as an explanation for the development of symptoms in manifesting carriers without chromosomal translocations [63]. The skewed X inactivation observed in muscle for patient F2 correlated with previous reports showing a similar bias in females with a severe phenotype [33, 34]. We found here an increased prevalence of highly skewed XCI in the females with XLMTM, but there is no clear correlation between the XCI ratio and the phenotype. Thus, XCI cannot be a reliable molecular diagnostic test for clinical prognosis. Several factors prevent us from drawing correlations from XCI studies including the small number of described manifesting females, the difficulty in defining a classification of the phenotypic severity, the difficulty of defining which chromosome is predominantly active, the fact that XCI varies with age and that the XCI ratio can differ between tissues and hence the results from lymphocytes may not reflect the status in muscle. Moreover, XCI studies have not been performed in asymptomatic MTM1 carrier females to assess potential correlation with disease severity. Thus, while skewed XCI is noted in several females with XLMTM, it does not correlate with disease severity but is potentially the molecular basis of the asymmetric muscle involvement.
Benefits of XLMTM diagnosis in affected females
Identifying the genetic cause of the disease allows appropriate care and management. A female affected with XLMTM benefits from specific care, in particular respiratory monitoring and close follow-up during and after pregnancy (F11 and F14 were reported to worsen after a pregnancy). A study of respiratory muscle function in a cohort of ten surviving males with XLMTM has shown that the respiratory muscle function declines over time [53], suggesting that females are at risk of a gradual deterioration of respiratory function.
Genetic counseling would inform affected women of the risk of occurrence of a severely affected male and the possibility of prenatal diagnosis. When a de novo mutation arises in a female child, with no evidence of parental origin, both parents should be made aware of the risk of recurrence due to possible germinal mosaicism [64].
Females with XLMTM survive longer than most male patients. A study of affected females may thus yield crucial information on the minimal dosage of myotubularin needed to minimize symptoms and also guide future therapeutic development by documenting disease evolution and complications as targets for management. Moreover, several therapeutic proof of concepts recently reported in animal models may represent potential therapies for females with XLMTM [16, 17, 35, 40, 49].
Conclusion
The diagnosis of XLMTM should be suspected in a female presenting with a myopathy, despite the absence of a family history and/or the presence in a muscle biopsy of some dystrophic features, particularly if facial weakness and asymmetric involvement are noted. The diagnosis of XLMTM affected females is likely to increase with the increasing use of massively parallel sequencing in a diagnostic setting ([50], this study). The occurrence of large heterozygous deletions in 4 of the 17 newly described patients underlines the importance of searching for copy number variations in the MTM1 gene [9]. Systematic long-term clinical assessment of heterozygous female relatives of any XLMTM male would allow a better ascertainment of the clinical spectrum and frequency of involvement in XLMTM carrier females. As more than 500 males have been reported to date in the literature, around 425 of their mothers were heterozygous carriers (85% of mother of affected males are carriers) as well as their other female relatives.
References
Abath Neto O, Silva MR, Martins CA, Oliveira AS, Reed UC, Biancalana V, Pesquero JB, Laporte J, Zanoteli E (2016) A study of a cohort of X-linked myotubular myopathy at the clinical, histologic, and genetic levels. Pediatr Neurol 58:107–112
Agrawal PB, Pierson CR, Joshi M, Liu X, Ravenscroft G, Moghadaszadeh B, Talabere T, Viola M, Swanson LC, Haliloglu G et al (2014) SPEG interacts with myotubularin, and its deficiency causes centronuclear myopathy with dilated cardiomyopathy. Am J Hum Genet 95:218–226
Allen RC, Zoghbi HY, Moseley AB, Rosenblatt HM, Belmont JW (1992) Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am J Hum Genet 51:1229–1239
Amburgey K, Lawlor MW, Del Gaudio D, Cheng YW, Fitzpatrick C, Minor A, Li X, Aughton D, Das S, Beggs AH et al (2013) Large duplication in MTM1 associated with myotubular myopathy. Neuromuscul Disord 23:214–218
Amos-Landgraf JM, Cottle A, Plenge RM, Friez M, Schwartz CE, Longshore J, Willard HF (2006) X chromosome-inactivation patterns of 1,005 phenotypically unaffected females. Am J Hum Genet 79:493–499
Bartsch O, Kress W, Wagner A, Seemanova E (1999) The novel contiguous gene syndrome of myotubular myopathy (MTM1), male hypogenitalism and deletion in Xq28: report of the first familial case. Cytogenet Cell Genet 85:310–314
Bevilacqua JA, Bitoun M, Biancalana V, Oldfors A, Stoltenburg G, Claeys KG, Lacene E, Brochier G, Manere L, Laforet P et al (2009) “Necklace” fibers, a new histological marker of late-onset MTM1-related centronuclear myopathy. Acta Neuropathol 117:283–291
Bevilacqua JA, Monnier N, Bitoun M, Eymard B, Ferreiro A, Monges S, Lubieniecki F, Taratuto AL, Laquerriere A, Claeys KG et al (2011) Recessive RYR1 mutations cause unusual congenital myopathy with prominent nuclear internalization and large areas of myofibrillar disorganization. Neuropathol Appl Neurobiol 37:271–284
Biancalana V, Beggs AH, Das S, Jungbluth H, Kress W, Nishino I, North K, Romero NB, Laporte J (2012) Clinical utility gene card for: centronuclear and myotubular myopathies. Eur J Hum Genet. doi:10.1038/ejhg.2012.91
Biancalana V, Caron O, Gallati S, Baas F, Kress W, Novelli G, D’Apice MR, Lagier-Tourenne C, Buj-Bello A, Romero NB et al (2003) Characterisation of mutations in 77 patients with X-linked myotubular myopathy, including a family with a very mild phenotype. Hum Genet 112:135–142
Bitoun M, Maugenre S, Jeannet PY, Lacene E, Ferrer X, Laforet P, Martin JJ, Laporte J, Lochmuller H, Beggs AH et al (2005) Mutations in dynamin 2 cause dominant centronuclear myopathy. Nat Genet 37:1207–1209
Bohm J, Biancalana V, Malfatti E, Dondaine N, Koch C, Vasli N, Kress W, Strittmatter M, Taratuto AL, Gonorazky H et al (2014) Adult-onset autosomal dominant centronuclear myopathy due to BIN1 mutations. Brain 137(12):3160–3170
Carrel L, Willard HF (1996) An assay for X inactivation based on differential methylation at the fragile X locus, FMR1. Am J Med Genet 64:27–30
Casar-Borota O, Jacobsson J, Libelius R, Oldfors CH, Malfatti E, Romero NB, Oldfors A (2015) A novel dynamin-2 gene mutation associated with a late-onset centronuclear myopathy with necklace fibres. Neuromuscul Disord 25:345–348
Ceyhan-Birsoy O, Agrawal PB, Hidalgo C, Schmitz-Abe K, DeChene ET, Swanson LC, Soemedi R, Vasli N, Iannaccone ST, Shieh PB et al (2013) Recessive truncating titin gene, TTN, mutations presenting as centronuclear myopathy. Neurology 81:1205–1214
Childers MK, Joubert R, Poulard K, Moal C, Grange RW, Doering JA, Lawlor MW, Rider BE, Jamet T, Daniele N et al (2014) Gene therapy prolongs survival and restores function in murine and canine models of myotubular myopathy. Sci Transl Med. doi:10.1126/scitranslmed.3007523
Cowling BS, Chevremont T, Prokic I, Kretz C, Ferry A, Coirault C, Koutsopoulos O, Laugel V, Romero NB, Laporte J (2014) Reducing dynamin 2 expression rescues X-linked centronuclear myopathy. J Clin Invest 124:1350–1363
Dahl N, Hu LJ, Chery M, Fardeau M, Gilgenkrantz S, Nivelon-Chevallier A, Sidaner-Noisette I, Mugneret F, Gouyon JB, Gal A et al (1995) Myotubular myopathy in a girl with a deletion at Xq27–q28 and unbalanced X inactivation assigns the MTM1 gene to a 600-kb region. Am J Hum Genet 56:1108–1115
de Gouyon BM, Zhao W, Laporte J, Mandel JL, Metzenberg A, Herman GE (1997) Characterization of mutations in the myotubularin gene in twenty six patients with X-linked myotubular myopathy. Hum Mol Genet 6:1499–1504
Drouet A, Ollagnon-Roman E, Streichenberger N, Biancalana V, Cossee M, Guilloton L, Petiot P (2008) Unilateral presentation of X-linked myotubular myopathy (XLMTM) in two out of three female carriers in a family with no affected male. Rev Neurol (Paris) 164:169–176
Dubowitz V, Sewry CA, Oldfors A (2013) Muscle biopsy: a practical approach, 4th edn. Saunders Elsevier, London
Fattori F, Maggi L, Bruno C, Cassandrini D, Codemo V, Catteruccia M, Tasca G, Berardinelli A, Magri F, Pane M et al (2015) Centronuclear myopathies: genotype-phenotype correlation and frequency of defined genetic forms in an Italian cohort. J Neurol 262:1728–1740
Flex E, De Luca A, D’Apice MR, Buccino A, Dallapiccola B, Novelli G (2002) Rapid scanning of myotubularin (MTM1) gene by denaturing high-performance liquid chromatography (DHPLC). Neuromuscul Disord 12:501–505
Fukami M, Wada Y, Miyabayashi K, Nishino I, Hasegawa T, Nordenskjold A, Camerino G, Kretz C, Buj-Bello A, Laporte J et al (2006) CXorf6 is a causative gene for hypospadias. Nat Genet 38:1369–1371
Grogan PM, Tanner SM, Orstavik KH, Knudsen GP, Saperstein DS, Vogel H, Barohn RJ, Herbelin LL, McVey AL, Katz JS (2005) Myopathy with skeletal asymmetry and hemidiaphragm elevation is caused by myotubularin mutations. Neurology 64:1638–1640
Gurgel-Giannetti J, Zanoteli E, de Castro Concentino EL, Abath Neto O, Pesquero JB, Reed UC, Vainzof M (2012) Necklace fibers as histopathological marker in a patient with severe form of X-linked myotubular myopathy. Neuromuscul Disord 22:541–545
Hagiwara SI, Kubota M, Sakaguchi K, Hiwatari E, Kishimoto H, Kagimoto S (2013) Fatal hepatic hemorrhage from peliosis hepatis with X-linked myotubular myopathy: a case report. J Pediatr Gastroenterol Nutr 60(5):e45–e46
Hammans SR, Robinson DO, Moutou C, Kennedy CR, Dennis NR, Hughes PJ, Ellison DW (2000) A clinical and genetic study of a manifesting heterozygote with X-linked myotubular myopathy. Neuromuscul Disord 10:133–137
Hedberg C, Lindberg C, Mathe G, Moslemi AR, Oldfors A (2011) Myopathy in a woman and her daughter associated with a novel splice site MTM1 mutation. Neuromuscul Disord 22:244–251
Herman GE, Finegold M, Zhao W, de Gouyon B, Metzenberg A (1999) Medical complications in long-term survivors with X-linked myotubular myopathy. J Pediatr 134:206–214
Herman GE, Kopacz K, Zhao W, Mills PL, Metzenberg A, Das S (2002) Characterization of mutations in fifty North American patients with X-linked myotubular myopathy. Hum Mutat 19:114–121
Hu LJ, Laporte J, Kress W, Kioschis P, Siebenhaar R, Poustka A, Fardeau M, Metzenberg A, Janssen EA, Thomas N et al (1996) Deletions in Xq28 in two boys with myotubular myopathy and abnormal genital development define a new contiguous gene syndrome in a 430 kb region. Hum Mol Genet 5:139–143
Jungbluth H, Sewry CA, Buj-Bello A, Kristiansen M, Orstavik KH, Kelsey A, Manzur AY, Mercuri E, Wallgren-Pettersson C, Muntoni F (2003) Early and severe presentation of X-linked myotubular myopathy in a girl with skewed X-inactivation. Neuromuscul Disord 13:55–59
Kristiansen M, Knudsen GP, Tanner SM, McEntagart M, Jungbluth H, Muntoni F, Sewry C, Gallati S, Orstavik KH, Wallgren-Pettersson C (2003) X-inactivation patterns in carriers of X-linked myotubular myopathy. Neuromuscul Disord 13:468–471
Kutchukian C, Lo Scrudato M, Tourneur Y, Poulard K, Vignaud A, Berthier C, Allard B, Lawlor MW, Buj-Bello A, Jacquemond V (2016) Phosphatidylinositol 3-kinase inhibition restores Ca2+ release defects and prolongs survival in myotubularin-deficient mice. Proc Natl Acad Sci USA 113:14432–14437
Laporte J, Biancalana V, Tanner SM, Kress W, Schneider V, Wallgren-Pettersson C, Herger F, Buj-Bello A, Blondeau F, Liechti-Gallati S et al (2000) MTM1 mutations in X-linked myotubular myopathy. Hum Mutat 15:393–409
Laporte J, Guiraud-Chaumeil C, Vincent MC, Mandel JL, Tanner SM, Liechti-Gallati S, Wallgren-Pettersson C, Dahl N, Kress W, Bolhuis PA et al (1997) Mutations in the MTM1 gene implicated in X-linked myotubular myopathy. ENMC International Consortium on Myotubular Myopathy. European Neuro-Muscular Center. Hum Mol Genet 6:1505–1511
Laporte J, Hu LJ, Kretz C, Mandel JL, Kioschis P, Coy JF, Klauck SM, Poustka A, Dahl N (1996) A gene mutated in X-linked myotubular myopathy defines a new putative tyrosine phosphatase family conserved in yeast. Nat Genet 13:175–182
Laporte J, Kress W, Mandel JL (2001) Diagnosis of X-linked myotubular myopathy by detection of myotubularin. Ann Neurol 50:42–46
Lawlor MW, Armstrong D, Viola MG, Widrick JJ, Meng H, Grange RW, Childers MK, Hsu CP, O’Callaghan M, Pierson CR et al (2013) Enzyme replacement therapy rescues weakness and improves muscle pathology in mice with X-linked myotubular myopathy. Hum Mol Genet 22:1525–1538
Liewluck T, Lovell TL, Bite AV, Engel AG (2010) Sporadic centronuclear myopathy with muscle pseudohypertrophy, neutropenia, and necklace fibers due to a DNM2 mutation. Neuromuscul Disord 20:801–804
McEntagart M, Parsons G, Buj-Bello A, Biancalana V, Fenton I, Little M, Krawczak M, Thomas N, Herman G, Clarke A et al (2002) Genotype-phenotype correlations in X-linked myotubular myopathy. Neuromuscul Disord 12:939–946
Motoki T, Fukuda M, Nakano T, Matsukage S, Fukui A, Akiyoshi S, Hayashi YK, Ishii E, Nishino I (2013) Fatal hepatic hemorrhage by peliosis hepatis in X-linked myotubular myopathy: a case report. Neuromuscul Disord 23(11):917–921
Nicot AS, Toussaint A, Tosch V, Kretz C, Wallgren-Pettersson C, Iwarsson E, Kingston H, Garnier JM, Biancalana V, Oldfors A et al (2007) Mutations in amphiphysin 2 (BIN1) disrupt interaction with dynamin 2 and cause autosomal recessive centronuclear myopathy. Nat Genet 39:1134–1139
Oliveira J, Oliveira ME, Kress W, Taipa R, Pires MM, Hilbert P, Baxter P, Santos M, Buermans H, den Dunnen JT et al (2012) Expanding the MTM1 mutational spectrum: novel variants including the first multi-exonic duplication and development of a locus-specific database. Eur J Hum Genet 21:540–549
Penisson-Besnier I, Biancalana V, Reynier P, Cossee M, Dubas F (2007) Diagnosis of myotubular myopathy in the oldest known manifesting female carrier: a clinical and genetic study. Neuromuscul Disord 17:180–185
Quijano-Roy S, Carlier RY, Fischer D (2011) Muscle imaging in congenital myopathies. Semin Pediatr Neurol 18:221–229
Romero NB (2010) Centronuclear myopathies: a widening concept. Neuromuscul 20:223–228
Sabha N, Volpatti JR, Gonorazky H, Reifler A, Davidson AE, Li X, Eltayeb NM, Dall’Armi C, Di Paolo G, Brooks SV et al (2016) PIK3C2B inhibition improves function and prolongs survival in myotubular myopathy animal models. J Clin Invest 126:3613–3625
Savarese M, Musumeci O, Giugliano T, Rubegni A, Fiorillo C, Fattori F, Torella A, Battini R, Rodolico C, Pugliese A et al (2016) Novel findings associated with MTM1 suggest a higher number of female symptomatic carriers. Neuromuscul Disord 26:292–299
Schara U, Kress W, Tucke J, Mortier W (2003) X-linked myotubular myopathy in a female infant caused by a new MTM1 gene mutation. Neurology 60:1363–1365
Shichiji M, Biancalana V, Fardeau M, Hogrel JY, Osawa M, Laporte J, Romero NB (2013) Extensive morphological and immunohistochemical characterization in myotubular myopathy. Brain Behav 3:476–486
Smith BK, Renno MS, Green MM, Sexton TM, Lawson LA, Martin AD, Corti M, Byrne BJ (2016) Respiratory motor function in individuals with centronuclear myopathies. Muscle Nerve 53:214–221
Soltanzadeh P, Friez MJ, Dunn D, von Niederhausern A, Gurvich OL, Swoboda KJ, Sampson JB, Pestronk A, Connolly AM, Florence JM et al (2010) Clinical and genetic characterization of manifesting carriers of DMD mutations. Neuromuscul Disord 20:499–504
Sutton IJ, Winer JB, Norman AN, Liechti-Gallati S, MacDonald F (2001) Limb girdle and facial weakness in female carriers of X-linked myotubular myopathy mutations. Neurology 57:900–902
Tanner SM, Orstavik KH, Kristiansen M, Lev D, Lerman-Sagie T, Sadeh M, Liechti-Gallati S (1999) Skewed X-inactivation in a manifesting carrier of X-linked myotubular myopathy and in her non-manifesting carrier mother. Hum Genet 104:249–253
Tanner SM, Schneider V, Thomas NS, Clarke A, Lazarou L, Liechti-Gallati S (1999) Characterization of 34 novel and six known MTM1 gene mutations in 47 unrelated X-linked myotubular myopathy patients. Neuromuscul Disord 9:41–49
Tasca G, Monforte M, Iannaccone E, Laschena F, Ottaviani P, Silvestri G, Masciullo M, Mirabella M, Servidei S, Ricci E (2012) Muscle MRI in female carriers of dystrophinopathy. Eur J Neurol 19:1256–1260
Terlizzi JP, Azizi R, Chow MD, Underberg-Davis S, Nosher JL, Stafford PW, Pierre J (2013) Peliosis hepatis in a child with myotubular myopathy: successful treatment using hepatic artery embolization. J Pediatr Surg 48:e9–e12
Tosch V, Vasli N, Kretz C, Nicot AS, Gasnier C, Dondaine N, Oriot D, Barth M, Puissant H, Romero NB et al (2010) Novel molecular diagnostic approaches for X-linked centronuclear (myotubular) myopathy reveal intronic mutations. Neuromuscul Disord 20:375–381
Trump N, Cullup T, Verheij JB, Manzur A, Muntoni F, Abbs S, Jungbluth H (2012) X-linked myotubular myopathy due to a complex rearrangement involving a duplication of MTM1 exon 10. Neuromuscul 22:384–388
Tsai TC, Horinouchi H, Noguchi S, Minami N, Murayama K, Hayashi YK, Nonaka I, Nishino I (2005) Characterization of MTM1 mutations in 31 Japanese families with myotubular myopathy, including a patient carrying 240 kb deletion in Xq28 without male hypogenitalism. Neuromuscul Disord 15:245–252
Viggiano E, Ergoli M, Picillo E, Politano L (2016) Determining the role of skewed X-chromosome inactivation in developing muscle symptoms in carriers of Duchenne muscular dystrophy. Hum Genet 135:685–698
Vincent MC, Guiraud-Chaumeil C, Laporte J, Manouvrier-Hanu S, Mandel JL (1998) Extensive germinal mosaicism in a family with X linked myotubular myopathy simulates genetic heterogeneity. J Med Genet 35:241–243
Wilmshurst JM, Lillis S, Zhou H, Pillay K, Henderson H, Kress W, Muller CR, Ndondo A, Cloke V, Cullup T et al (2010) RYR1 mutations are a common cause of congenital myopathies with central nuclei. Ann Neurol 68:717–726
Wu H, Luo J, Yu H, Rattner A, Mo A, Wang Y, Smallwood PM, Erlanger B, Wheelan SJ, Nathans J (2014) Cellular resolution maps of X chromosome inactivation: implications for neural development, function, and disease. Neuron 81:103–119
Acknowledgements
We thank the patients and their families. Because facial features are part of the clinical picture, patients have kindly consented to the use of their clinical photographs without masking their faces. MRI images of patients F13 and F16 were kindly provided, respectively, by Dr Milja Holstila, Finland, and Dr Florence Caillon, France, and muscle biopsy from patient F3 was managed by Cardiobiotec Biobanck, Lyon, France. We thank Claire FEGER and Nadine KEMPF for technical assistance. This work was supported by the Institut National de la Santé et de la Recherche Médicale (INSERM), Centre National de la Recherche Scientifique (CNRS), University of Strasbourg, the France Génomique National infrastructure, funded as part of the Investissements d’Avenir program managed by the Agence Nationale pour la Recherche (ANR-10-INBS-09), and Fondation Maladies Rares within the frame of the “Myocapture” sequencing project, ANR-10-LABX-0030-INRT under the frame program Investissements d’Avenir ANR-10-IDEX-0002-02, Fondation pour la Recherche Médicale (DBI20131228569) and AFM (AFM-16992) and CREGEMES for the MYOdiagHTS Project.
Author information
Authors and Affiliations
Contributions
VB and JL directed the study; VB, SS and MM performed the study; VB analyzed the data; all the others authors contributed materials; VB and JL wrote the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflicts of interest.
Statement of human rights
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study. Additional informed consent was obtained from patients F4, F5, F16 and F17 for whom identifying photographic information is included in this article.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Biancalana, V., Scheidecker, S., Miguet, M. et al. Affected female carriers of MTM1 mutations display a wide spectrum of clinical and pathological involvement: delineating diagnostic clues. Acta Neuropathol 134, 889–904 (2017). https://doi.org/10.1007/s00401-017-1748-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-017-1748-0