
Abstract
Brains of 42 individuals between the ages of 4 and 29 were examined with antibodies (AT8, 4G8) and silver stains for the presence of intraneuronal and extracellular protein aggregates associated with Alzheimer’s disease. Thirty-eight of 42 (38/42) cases displayed abnormally phosphorylated tau protein (pretangle material) in nerve cells or in portions of their cellular processes, and 41/42 individuals showed no extracellular amyloid-β protein deposition or neuritic plaques—an individual with Down syndrome was the only exception. In 16/42 cases abnormal tau was found in the transentorhinal region, and in 3/42 cases this site was Gallyas-positive for isolated NFTs (NFT stage I). Of 26 cases that lacked abnormal tau in the transentorhinal region, 4 did not show pretangle material at subcortical sites. The remaining 22 of these same 26 cases, however, had subcortical lesions confined to non-thalamic nuclei with diffuse projections to the cerebral cortex, and, remarkably, in 19/22 individuals the pretangle material was confined to the noradrenergic coeruleus/subcoeruleus complex. Assuming the pretangle alterations are not transient and do not regress, these findings may indicate that the Alzheimer’s disease-related pathological process leading to neurofibrillary tangle formation does not begin in the cerebral cortex but, rather, in select subcortical nuclei, and it may start quite early, i.e., before puberty or in early young adulthood.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The morphological hallmark lesions and pathological processes underlying sporadic Alzheimer’s disease (AD) are well characterized. Once begun, this tauopathy progresses for decades probably without remission [13, 18]. Within select neuronal types, the AD-related process is marked by abnormal phosphorylation of the microtubule-associated protein tau that appears in both the somata and cellular processes [24, 43]. In its hyperphosphorylated state, the soluble tau protein tends to take on an abnormal configuration and undergoes aggregation into insoluble fibrillary material that appears as neuropil threads (NTs) in cellular processes and neurofibrillary tangles (NFTs) in neuronal somata [2, 7, 12, 20, 41, 53, 57, 62]. The severity of the tau pathology increases gradually during the course of the disorder until it crosses a threshold to induce clinically recognizable dysfunctions. This also implies that incidental AD-associated changes may not be simply innocuous alterations routinely accompanying healthy brain aging. Moreover, such pathological changes can be incorporated into diagnostic protocols to facilitate differential neuropathological assessment of the disease process from other tauopathies, such as progressive supranuclear palsy or fronto-temporal dementia and Pick’s disease, in asymptomatic and symptomatic individuals alike [17, 18, 28–30, 52, 58].
It is widely recognized that the AD-related pathological process in the cerebral cortex occurs at predilection sites and progresses from there in a systematic manner throughout hitherto uninvolved cortical areas. Characteristic changes in the lesional distribution pattern are used to distinguish stages in the course of the process [1, 4, 13, 18, 20, 51, 91]. Nonetheless, studies of late stages are fraught with difficulties because large numbers of vulnerable nerve cells either are no longer visible in the tissue or have undergone structural alterations, and glial cells as well as other non-neuronal tissue components may have reacted upon the neurofibrillary lesions. In the initial disease stages, by contrast, the circumstances are clearer: clinically unremarkable individuals occur very frequently in the normal population [15], and, in the majority of cases, their brains display a single AD-related pathological alteration, i.e., the presence of abnormally phosphorylated tau protein in several nerve cells or even in a single neuron.
To obtain greater insight into the AD-associated pathological process, we studied the intraneuronal tau lesions in a cohort of 42 individuals under the age of 30. The information gained from this group supplements data from an earlier report regarding the frequency of AD-associated lesions in the cerebral cortex in individuals ranging 26–95 years [15]. New considerations drawn from the results are discussed against the background of existing doctrines and hypotheses regarding the pathogenesis of AD.
Materials and methods
Study cohort
This retrospective study was performed in compliance with university ethics committee guidelines as well as German federal and state law governing human tissue usage. Forty-two brains had been obtained at autopsy from 14 non-demented females and 28 non-demented males (age range 4–29 years; mean age ± SD, 19.84 ± 7.01 years). One individual had had Down syndrome (case 17), one had suffered from type Duchenne muscular dystrophy (case 8), and five individuals had died as a result of automobile accidents or polytrauma involving acute head injury (cases 18, 20, 23, 25, 36). Others had suffered from or succumbed to congenital heart defects or cardiac disease (cases 6, 12, 15, 19, 27, 32, 39), post-operative or transplantation-related (graft-versus-host) complications (cases 7, 26, 29, 35, 42), non-CNS malignant neoplasias (cases 11, 31, 40, 41), primary CNS tumors (cases 1, 2), leukemia (cases 16, 35, 38), aplastic anemia (case 26), hereditary spherocytosis (case 37), generalized sepsis (cases 3, 4, 22, 29), rubella (case 9), influenza (case 13), spinal cord injury (case 21), strangulation (case 5), renal failure (case 29), or amniotic fluid embolism (case 30). One patient had had a history of chronic asthma (case 37) and two type 1 diabetes (cases 10, 29). Demographics and neuropathological diagnoses for the cohort are shown in Table 1. Neuropathological diagnoses for all cases were made according to published criteria for Alzheimer’s disease (AD) and Parkinson’s disease (PD) [1, 13, 18–20].
Tissue sectioning
The brainstems and a single hemisphere from all individuals were fixed by immersion in a 4% buffered aqueous solution of formaldehyde. A set of six tissue blocks was excised and embedded in polyethylene glycol (PEG 1000, Merck) [18–20, 55]. These cases are designated “bl” (tissue blocks) in Table 1. Multiple 100 μm sections were made from each block on a sliding microtome because this thickness allows for the superimposition of large numbers of biological structures, including nerve cells with their entire dendritic tree.
Four brainstem blocks from each case were cut perpendicular to the brainstem axis of Meynert. An initial block was cut through posterior portions of the medulla oblongata at the latitude of the dorsal motor nucleus of the vagus nerve (dmX), adjoining intermediate reticular zone (irz), and inferior olivary nucleus. A second block consisted of the brainstem at the widest latitude of the fourth ventricle and included the great raphe nucleus (lower raphe). The third block ran through the pontine tegmentum and contained portions of the coeruleus/subcoeruleus complex (coer), and the dorsal raphe nucleus. The fourth block was cut at the level of the inferior colliculus and included posterior portions of the substantia nigra (sn), the tegmental pedunculopontine nucleus, and the supratrochlear portion of the dorsal raphe nucleus (upper raphe) (Table 1).
Two additional tissue blocks were excised perpendicular to the hemisphere axis of Forel: the fifth block contained portions of the magnocellular nuclei of the basal forebrain close to the anterior commissure, usually including the interstitial nucleus of the diagonal band, and/or portions of Meynert’s basal nucleus (magn). The sixth block was cut at mid-uncal level through medial portions of the temporal lobe and encompassed anterior (i.e., uncal) portions of the hippocampal formation and the parahippocampal gyrus (entorhinal region), including the adjoining transentorhinal region (tre/ent) as well as portions of the occipito-temporal gyrus and additional gyri of the basal temporal neocortex [18–20, 90, 91]. A seventh block contained the olfactory bulb and/or anterior olfactory nucleus (olf) (Table 1).
Two cases were processed differently and are designated “ser” (series) in Table 1. Brainstems of these cases were severed between the pontine tegmentum and mesencephalic tegmentum, embedded in PEG, and then microtomed perpendicular to Meynert’s axis into uninterrupted series of 100-μm-thick free-floating sections [14]. Each of these cases was supplemented either by serial sections through one of the hemispheres or by multiple sections from tissue blocks including the motives of the fourth, fifth, and sixth block of the other cases.
Staining and immunocytochemistry
Collections of free-floating sections from all blocks for each case were processed with the following staining techniques or immunocytochemical reactions:
Pigment-Nissl staining was used to show the presence and extent of lipofuscin deposits (aldehyde fuchsin) and basophilic material (Darrow red) [14]. Additional sections were silver-stained with the Campbell–Switzer and Gallyas methods that exploit the physical development of nucleation sites. These techniques were used to visualize neuromelanin granules as well as amyloid-β protein deposition (Campbell–Switzer) and argyrophilic tau-positive inclusions (Gallyas) [9, 14, 76].
The presence of intraneuronal lesions associated with neurodegenerative diseases was assessed immunohistochemically using the following antibodies: (1) monoclonal antibody PHF-Tau [1:2,000; Clone AT8; Pierce Biotechnology, Rockford, IL, USA (Thermo Scientific)] for hyperphosphorylated tau protein in pretangle material and in neurofibrillary changes of the Alzheimer type; (2) monoclonal antibody anti-syn-1 (1:2,000; Clone number 42, BD Biosciences, Mountain View, CA, USA) for assessment of Lewy pathology; (3) polyclonal antibody anti-phospho TDP-43 [pS409/410-2] (1:20,000; Cosmo Bio Co., Ltd., Tokyo, Japan) as a marker for phosphorylated TDP-43 protein inclusions in fronto-temporal lobar degeneration and amyotrophic lateral sclerosis; (4) polyclonal antibody anti-ubiquitin (1:1,000; Dako, Glostrup, Denmark); and (5) monoclonal antibody anti-beta-amyloid (1:5,000; Clone 4G8; Covance, Freiburg/Br, Germany) for detection of amyloid-β deposition. Immunoreactions for tyrosine hydroxylase (polyclonal antibody anti-TH; 1:2,000; AB152, Chemicon/Millipore, Schwalbach, Germany) were used to visualize noradrenergic nerve cells of the coeruleus/subcoeruleus complex and dopaminergic neurons in the substantia nigra (Table 1). All immunoreactions can be performed on material that has been stored for long periods of time in formaldehyde [74].
Tissue sections for immunoreactions were first treated in a mixture of 10% methanol plus 10% concentrated (30%) H2O2 and 80% Tris for 30 min. After pretreatment with 100% formic acid for 3 min to facilitate the immunoreactions (anti-syn-1, anti-p62, anti-β-amyloid, anti-TH) [86] or enhancement by microwave pretreatment at 700 W for 18 min in citrate buffer (pH 6.0) solution (anti-phospho TDP-43, anti-ubiquitin), blocking with bovine serum albumin was performed to inhibit endogenous peroxidase and to prevent nonspecific binding. Subsequently, each of the various sets of free-floating sections was incubated for 18 h at 20°C using the primary antibodies. Subsequent to processing with a corresponding secondary biotinylated antibody (anti-mouse IgG, 1:200; Linaris) for 1.5 h, all immunoreactions were visualized with the avidin–biotin complex (ABC, Vectastain, Vector Laboratories, Burlingame, CA, USA) for 2 h and the chromogen 3,3′-diaminobenzidine tetrahydrochloride (DAB, D5637 Sigma, Taufkirchen, Germany). Omission of the primary antiserum resulted in non-staining. Positive as well as negative control sections were routinely included to confirm specificity of the immunostaining. The tissue sections were cleared and mounted in a synthetic resin (Permount, Fisher). All sections were viewed with a Vanox AHB53 Olympus microscope, and digital micrographs were taken using the analysis® Soft Imaging System (Münster, Germany).
Results
The brains of 42/42 individuals staged for AD-related intraneuronal lesions using immunoreactions for abnormally phosphorylated tau protein were immunonegative for α-synuclein, TDP-43 protein, and ubiquitin at levels of the dorsal motor nucleus of the vagal nerve, great raphe nucleus, coeruleus/subcoeruleus complex, substantia nigra, and upper raphe nuclei as well as in forebrain sections through the olfactory stalk and/or bulb, magnocellular nuclei of the basal forebrain and the hippocampal formation at mid-uncal level (Table 1). In addition, with the exception of the individual with Down Syndrome (case 17), none of the cases examined using the 4G8 antibody and/or Campbell–Switzer silver method displayed amyloid-β protein deposition or neuritic plaques in neocortical or allocortical areas of the temporal lobe or within the boundaries of the subcortical nuclei under consideration.
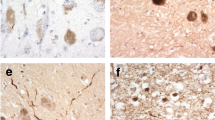
As anticipated in this group of young individuals, overview sections stained for lipofuscin pigment and basophilic material failed to show any obvious pathological alterations. Moreover, nerve cells in catecholaminergic nuclei (coeruleus/subcoeruleus complex, substantia nigra) contained sparse amounts of neuromelanin granules in the youngest members (e.g., Fig. 1d, e from an individual aged 11 years, case 5) and the nuclei themselves were pale macroscopically. Extraneuronal deposits of neuromelanin were not observable in the substantia nigra of any of the cases examined and were seldom present in the coeruleus/subcoeruleus complex.

Abnormal tau protein (pretangle material) in the coeruleus/subcoeruleus complex of young individuals (AT8-immunoreactions for hyperphosphorylated tau protein, 100 μm sections). a–c A single AT8-immunoreactive (ir) neuronal process confined to the locus coeruleus (coer) in a 14-year-old male (case 9). Such individuals are designated as “a” in Table 1. Characteristics of the ir-process resemble those of an axon. Framed area in a represents detail in c. d, e An 11-year-old male (case 5) shows presence of both ir neurites and a few nerve cells with ir-axonal and somatodendritic compartments. Framed area in d represents detail in e. Note the pallor of the locus coeruleus here, which is typical for newborns and children. f Neuromelanin-containing projection neurons of the locus coeruleus normally generate only a few elongated dendrites from a smoothly contoured cell body. Affected nerve cells frequently have quill-like or studded structures (arrow) along the surface or rim of the soma (here, in a 25-year-old male, case 29). g A single ir-neurite showing small varicosities (same case as in d, e above). h, k AT8-ir coeruleus cells (h female, 14 years of age, case 7; k male, 26 years of age, case 33). Such cases are designated as “b” in Table 1. l One of the few AT8-ir nerve cells from Meynert’s basal nucleus as seen in a 29-year-old female (case 41)
All 42 cases were free of AT8-immunoreactive (ir) nerve cells or neuronal processes in anterior olfactory structures (Table 1). Twenty-six of 42 (26/42) individuals failed to show AT8-ir cortical projection neurons, or portions thereof, in the temporal lobe; particularly in the transentorhinal region (Table 1) [13]. The transentorhinal region of 3/42 cases was Gallyas-positive for isolated NFTs (cases 21, 23, 36, i.e., NFT stage I) (Table 1), whereas the selected brainstem areas of all 42 cases were Gallyas-negative (data not shown). Abnormal tau material did not occur in non-neuronal cells [32]. Notably, 22/42 individuals revealed the presence of AT8-ir pretangle material in select subcortical sites in the absence of cortical lesions. This AT8-ir material occurred only in nuclei that send diffuse projections to the cerebral cortex (Table 1). Cases with the most subtle pretangle material (19/42) showed AT8-ir predominantly in the coeruleus/subcoeruleus complex (“a” and “b” in Table 1). Finally, none of the cases displayed AT8-ir inclusions in the inferior olivary nucleus, tegmental pedunculopontine nucleus, or pallidum [32].
Remarkably, 12/42 cases displayed immunoreactivity only in portions of nerve cell processes (Figs. 1a–c, 2d, cases indicated by “a” in Table 1). Many of these nerve cell-adjacent, elongated thready structures very closely resembled axons. In some instances, their diameters varied somewhat at short intervals (e.g., weak varicosities) (Fig. 1g). The AT8-ir material was evenly distributed therein. In 7/19 cases with involvement confined to the coeruleus/subcoeruleus complex, numerous AT8-ir neurites were present and, in addition, isolated nerve cell somata together with all of their neuronal processes filled with the abnormal material (cases indicated by “b” in Table 1) (Figs. 1d–f, h, k, 2d). Normally, the somata of coeruleus projection cells are smoothly contoured. Here, by contrast, the outer somatic rim of AT8-ir neurons often was studded with spiked protrusions (Fig. 1f). These “b” cases also tended to display AT8-ir pretangle material in axons along ascending portions of the anterior catecholaminergic tract [73] and/or in other non-thalamic nuclei with diffuse cortical projections (Table 1). AT8-ir axons were much less frequently observed at levels inferior to the locus coeruleus, e.g., in catecholaminergic fiber bundles of the medial longitudinal catecholaminergic tract that descend to the spinal cord [73].

AT8-ir pretangle material in the transentorhinal region and summary diagram. a Processes of nerve cells are the first structures displaying pretangle material in the transentorhinal (tre) region. Such loosely distributed and variably shaped portions of neuronal processes possibly correspond to altered terminal axons of non-thalamic nuclei with cortical projections (26-year-old male, case 31). b AT8-ir pyramidal cell in outer layers of the transentorhinal region (29-year-old male, case 39). Note that abnormal tau protein is visible initially at branching points of dendrites. c Thereafter, the pretangle material fills the entire nerve cell (29-year-old female, case 42). d Schema summarizing the postulated steps in the development of early AD-associated tau pathology. These are indicated as “a,” “b,” “c,” “Ia,” or “Ib” in Table 1. The presence of AT8-ir material (shown in red) in neurites of noradrenergic nerve cells in the coeruleus/subcoeruleus complex (stage a) is followed by lesions filling some of these neuromelanin-containing neurons (stage b). In a further step, AT8-ir neurites and/or nerve cells occur in other non-thalamic brainstem nuclei with diffuse cortical projections (stage c). Immunopositive portions of neuronal processes appear for the first time in predisposed areas of the cerebral cortex, e.g., the transentorhinal region (stage Ia) [13, 20]. It remains to be seen whether these subtle lesions actually represent pathologically altered terminals of coeruleus axons. Finally, isolated pyramidal cells of the transentorhinal region together with their cellular processes fill up with AT8-ir pretangle material, and the immunolabeled pyramidal cells increase in number (stage Ib)
Three of the 42 cases had more widely distributed subcortical lesions: AT8-ir nerve cells were found not only in the coeruleus/subcoeruleus complex but also in other non-thalamic nuclei with cortical projections (cases indicated by “c” in Table 1; Fig. 2d). In addition to subcortical pathology, 7/38 cases had mild-cortical lesions. In medial portions of the temporal lobe and, in particular, in the transentorhinal region (tre), structures were seen that could correspond to terminal portions of ascending coeruleus axons (Fig. 2a). Such cases with subtle cortical AT8-immunoreactivity limited solely to neuronal processes are indicated by “Ia” in Table 1 (see also Fig. 2d). Comparable entities were seen in proximity to isolated AT8-ir pyramidal cells that appeared in the transentorhinal cortex. The somata and all of their cellular processes were filled with AT8-ir pretangle material. Such cases (i.e., six with AT8-ir cortical projection cells) are designated “Ib” in Table 1 (Fig. 2b–d).
Discussion
Alzheimer’s disease (AD) is traditionally viewed as a disorder associated with aging, and more specifically, with old age. Some, on the other hand, regard it as “age-dependent” (i.e., vs. age-related or age-associated), under the assumption that the pathological lesions developing during the course of AD are directly caused by the aging process itself or, at the very least, are somehow indirectly attributable to aging. Thereby, a central position in the pathogenesis of AD has been variously assigned to all factors that are known to be capable of damaging postmitotic cells—a risk that increases during aging and old age—such as greater oxidative stress, mitochondrial dysfunction, chronic inflammation, and/or failure of the ubiquitin–proteasome system [34, 61, 63]. Ultimately, however, these findings and viewpoints do not adequately reflect the fact that the pathological process underlying AD is a markedly selective one from beginning to end: only a few types of nerve cells become involved and, notably, not all postmitotic cells throughout the human nervous system [51].
A coherent explanation for the selectivity of the AD process is still elusive. The selective involvement or vulnerability of only a few neuronal types has implications for the development and appearance of clinical symptoms. Moderate and random dysfunction or loss of nerve cells in the brain most probably remains unrecognizable, whereas a non-random loss of similar magnitude and, at the same time, confined to a single nerve cell type can be anticipated to reach the threshold of clinically detectable symptoms. Moreover, the pathological process requires an unusually long prodromal period in excess of three to four decades [3, 15]. In many instances, as the unexpected incidental findings reported here would seem to indicate, it can manifest itself in very young individuals of both genders (Table 1). Yet, it is also known that persons whose brains are involved in the process underlying AD are capable of living to a very old age. As such, it seems more than fair to say that aging or advanced age are not preconditions for the formation of AD-associated intraneuronal lesions. AD, therefore, is almost certainly not an “age-dependent” disorder; rather, it is an uncommonly slowly progressing disease that extends into old age [15].
Pathological changes accompanying AD include intraneuronal lesions (pretangle material, NFTs, NTs) and extracellular amyloid-β protein deposition [13, 60, 82, 90–93]. Study of early AD-stages reveals that in the majority of cases formation of intraneuronal alterations precedes that of insoluble (aggregated) amyloid-β protein [15, 16, 34, 81]. The development of amyloid-β plaques, in turn, is necessary for that of neuritic plaques, which are the last of the hallmark AD-related lesions to develop. Topographically, plaque pathology evolves independently of the intraneuronal lesions in sporadic AD; thus, the distribution of plaques differs considerably from that of the neurofibrillary lesions [13].
In this sample under the age of 30, 41/42 cases did not have amyloid-β plaques or neuritic plaques (Table 1). The absence of amyloid-β deposition in these individuals is not compatible with the amyloid cascade hypothesis, which assumes that amyloid-β drives AD pathogenesis and secondarily induces intraneuronal tau changes “downstream” [35, 46–49, 54, 75, 87]. Further biochemical studies are required to test for the presence of soluble oligomeric amyloid-β and its consequences in a comparable cohort of young subjects. Here, however, it appears that it is the development of abnormally phosphorylated tau material in select nerve cell types within brainstem nuclei and, more specifically, in the coeruleus/subcoeruleus complex, which marks the beginning of the AD-associated pathological process; occasionally even before puberty.
A considerable number of cases here displayed AT8-immunoreactivity confined to portions of axons (“a” in Table 1). Thus, in the subcortical nuclei under consideration, axonal changes may precede those in the somatodendritic compartment. The abnormal AT8-ir material in axons appeared in full intensity without visible precursors, whereas the initial axon segments were devoid of such material. Because a transfer of AT8-ir material from the soma via the initial segment into the axon has not been reported, it is usually assumed that abnormal tau within axons originates from the tau protein bound to axonal microtubules. Investigations by previous groups have reached the conclusion that in its hyperphosphorylated state the tau protein becomes detached from axonal microtubules with a resultant destabilization of the microtubules and purported failure of their essential transport functions [22, 85]. In addition, according to the tau-microtubule hypothesis, it has been argued that soluble hyperphosphorylated tau is in itself highly toxic and incompatible with physiological cell functioning, whereas a better compatibility for the insoluble neurofibrillary lesions purportedly exists [2, 22, 53, 59, 61].
In AD, affected nerve cells are not peracutely endangered at first, i.e., they do not face immediate cell death: under the light microscope their aspect remains bland and without strong signs of reactive responses in their cell nuclei or somatodendritic compartments. On the other hand, the neuronal cytoskeleton abnormalities are not spontaneously reversible but undergo a series of morphological permutations during the decades that follow [12]. Although evidence from previous studies [11, 68] is compatible with the notion that AD-related cytoskeletal alterations are remarkably well tolerated for an inordinately long period of time by involved nerve cells, it still has to be shown that the cytoskeletal lesions are also subjected at the end of their development to age-dependent influences. Unfortunately, however, it is also presently unknown what developmental phase the AD-associated intraneuronal lesions have to reach before physiologically measurable cell dysfunctions occur, which, in turn, once sufficiently large percentages of principal nerve cells in a given brain region have become impaired, are capable of triggering clinically recognizable symptoms [95].
Morphologically, the young cases here had not undergone premature loss of neuromelanin-containing neurons in the coeruleus/subcoeruleus complex [10, 36, 79]. Extraneuronal deposits of neuromelanin granules are useful cell loss markers because they are not easily eliminated by macrophages and, as a result, remain in the tissue surrounding the dead parent neurons. However, to the extent that extraneuronal neuromelanin (so-called “pigment incontinence”) was seldom encountered in Campbell–Switzer stained sections and because such granules accompanied by AT8-ir remnants of abnormal tau material did not occur in the brains of the individuals under the age of 30, it is indeed questionable whether soluble hyperphosphorylated tau is, in fact, highly toxic or that the postulated toxic effect can be ameliorated by polymerization into insoluble fibrillary material. Similarly, whether the cytoskeletal alterations are well tolerated in the long run is a further open-ended issue. They seem to cause premature death in at least some nerve cell types, e.g., in projection neurons of the upper raphe nuclei or those of the entorhinal layer II [25]. Thus, the intraneuronal material seen here in young persons may be neither acutely toxic nor chronically benign: initially compatible with nerve cell survival, it may eventually result in cellular or axonal dysfunction [39] and, ultimately, even premature loss of involved neurons at certain sites.
In the fetal period as well as in newborns, phosphorylation of the tau protein is transiently up-regulated during axonal development [40, 58, 64]. Axonal growth cones, including their microtubules, require maximum flexibility for guidance to their target regions and, once there, to be capable of forming synapses. They achieve this flexibility primarily by means of their highly phosphorylated tau protein [64]. Axons from nerve cells of non-thalamic nuclei with diffuse cortical projections, including those of the coeruleus/subcoeruleus complex, are exceptionally long and poorly myelinated. It is conceivable that they could more easily revert to an earlier undifferentiated state than strongly myelinated axons, and their tau protein thereby achieve a higher degree of phosphorylation; for instance, in buds of new collaterals sprouting from proximal axons. Alone such axonal sprouting [89] could serve as an initial event in a youthful brain that might trigger an irreversibly abnormal hyperphosphorylation of tau in axons.
An important issue with respect to the pathogenesis of AD is not only how the destructive process begins but also where and when [88]. The results here complement the data from an earlier comprehensive report regarding the development of cortical AD-associated lesions in a cohort of individuals with an age span of 26–95 years [15]. In that study, hyperphosphorylated tau protein was seen primarily in specialized types of pyramidal cells, and the available evidence indicated that cortical intraneuronal lesions begin, as a rule, in the transentorhinal region (NFT stage I) and progress from there in a systematic manner into primary neocortical areas (NFT stage VI) [13]. Up to now, however, the focus of staging protocols has been confined to the hallmark tau lesions in neurons of the cerebral cortex [1, 13, 20].
In the current study, we report that in 22/42 young persons AD-associated AT8-ir pretangle material was present in select subcortical nuclei without involvement of the cerebral cortex. Nuclei prone to develop these lesions are non-thalamic nuclei with diffuse projections to the cerebral cortex [36], including the magnocellular nuclei of the basal forebrain [16, 66, 67, 77, 78, 80], upper raphe nuclei [42, 72, 83], and the coeruleus/subcoeruleus complex [5, 6, 16, 21, 37, 38, 44, 45, 69, 72, 79, 94–96]. This study not only extends the findings of the earlier effort by providing an overview of the age spectrum under 30 that was missing there [15], it also suggests that it is precisely during the first decades of life that the probability of encountering brainstem AD-associated intraneuronal lesions without involvement of cortical predilection sites is high. It is striking that in 19/22 cases the AT8-ir pretangle material occurred at a single subcortical site and, at the same time, in cases with some of the most subtle tau pathology. Here, the focal point of the lesions was the coeruleus/subcoeruleus complex, which may represent the starting point for the pathological process underlying AD. These results must be tested on a much larger number of young individuals, including subjects with head trauma, whereby the presence of the latter in 5/42 cases does not appear to have been sufficient to induce the development of the pretangle material seen here [56, 65, 71, 84] (Table 1).
In Parkinson’s disease, early Lewy pathology usually occurs in anterior olfactory structures and/or the dorsal motor nucleus of the vagal nerve [8, 19, 26, 31]. Early damage to the olfactory system has also been described in AD [23, 27, 33, 46, 50, 70]. Inasmuch as none of our cases revealed the presence of abnormal tau or amyloid-β protein in the olfactory bulb or olfactory tract, we could rule out these structures as sites where the AD-associated process is likely to commence.
References
Alafuzoff I, Arzberger T, Al-Sarraj S et al (2008) Staging of neurofibrillary pathology in Alzheimer’s disease: a study for the Brain Net Europe Consortium. Brain Pathol 18:484–496
Alonso AC, Li B, Grundke-Iqbal I, Iqbal K (2008) Mechanism of tau-induced neurodegeneration in Alzheimer disease and related tauopathies. Curr Alzheimer Res 5:375–384
Amieva H, Le Goff M, Millet X et al (2008) Prodromal Alzheimer’s disease: successive emergence of clinical symptoms. Ann Neurol 64:492–498
Arnold SE, Hyman BT, Flory J, Damasio AR, van Hoesen GW (1991) The topographical and neuroanatomical distribution of neurofibrillary tangles and neuritic plaques in the cerebral cortex of patients with Alzheimer’s disease. Cereb Cortex 1:103–116
Aston-Jones G, Cohen JD (2005) An integrative theory of locus coeruleus–norepinephrine function: adaptive gain and optimal performance. Ann Rev Neurosci 28:403–450
Baker KG, Törk I, Hornung JP, Halasz P (1989) The human locus coeruleus complex: an immunohistochemical and three dimensional reconstruction study. Exp Brain Res 77:257–270
Bancher C, Brunner C, Lassmann H et al (1989) Accumulation of abnormally phosphorylated tau precedes the formation of neurofibrillary tangles in Alzheimer’s disease. Brain Res 477:90–99
Beach TG, White CL 3rd, Hladik CL et al (2009) Olfactory bulb alpha-synucleinopathy has high specificity and sensitivity for Lewy body disorders. Acta Neuropathol 117:169–174
Beach TG, Adler CH, Sue SI et al (2010) Multi-organ distribution of phosphorylated alpha-synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathol 119:689–702
Benarroch EE (2009) The locus ceruleus norepinephrine system. Neurology 73:1699–1704
Bobinski M, Wegiel J, Tarnawski M et al (1998) Duration of neurofibrillary changes in the hippocampal pyramidal neurons. Brain Res 799:156–158
Braak E, Braak H, Mandelkow EM (1994) A sequence of cytoskeleton changes related to the formation of neurofibrillary tangles and neuropil threads. Acta Neuropathol 87:554–567
Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82:239–259
Braak H, Braak E (1991) Demonstration of amyloid deposits and neurofibrillary changes in whole brain sections. Brain Pathol 1:213–216
Braak H, Braak E (1997) Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging 18:351–357
Braak H, Del Tredici K (2004) Alzheimer’s disease: intraneuronal alterations precede insoluble amyloid-β formation. Neurobiol Aging 25:713–718
Braak H, Braak E, Yilmazer D, Schultz C, Bohl J (1995) Age-related changes of the human cerebral cortex. In: Cruz-Sanchez FF, Ravid R, Cuzner ML (eds) Neuropathologic diagnostic criteria for brain banking. IOS Press, Amsterdam, pp 14–19
Braak H, Del Tredici K, Braak E (2003) Spectrum of pathology. In: Petersen R (ed) Mild cognitive impairment. Oxford University Press, New York, pp 149–189
Braak H, Del Tredici K, Rüb U, de Vos RAI, Jansen Steur ENH, Braak E (2003) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24:197–210
Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K (2006) Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 112:389–404
Busch C, Bohl J, Ohm TG (1997) Spatial, temporal and numeric analysis of Alzheimer changes in the nucleus coeruleus. Neurobiol Aging 18:401–406
Cowan CM, Bossing T, Page A, Shepherd D, Mudher A (2010) Soluble hyper-phosphorylated tau causes microtubule breakdown and functionally compromises normal tau in vivo. Acta Neuropathol 120:593–604
Davies DC, Brooks JW, Lewis DA (1993) Axonal loss from the olfactory tracts in Alzheimer’s disease. Neurobiol Aging 14:353–357
Delacourte A, Défossez A (1986) Alzheimer’s disease: tau proteins, the promoting factors of microtubule assembly, are major antigenic components of paired helical filaments. J Neurol Sci 76:173–186
Del Tredici K, Braak H (2008) Neurofibrillary changes of the Alzheimer type in very elderly individuals: neither inevitable nor benign. Commentary on “No disease in the brain of a 115-year-old woman”. Neurobiol Aging 29:1133–1136
Del Tredici K, Rüb U, de Vos RAI, Bohl JRE, Braak H (2002) Where does Parkinson pathology begin in the brain? J Neuropathol Exp Neurol 61:413–425
Devanand DP, Tabert MH, Cuasay K et al (2010) Olfactory identification deficits and MCI in a multi-ethnic elderly community sample. Neurobiol Aging 31:1593–1600
Dickson DW (1997) Neurodegenerative diseases with cytoskeletal pathology: a biochemical classification. Ann Neurol 42:541–544
Dickson DW (1998) Aging in the central nervous system. In: Markesbery WR (ed) Neuropathology of dementing disorders. Arnold, London, pp 56–88
Dickson DW, Crystal HA, Mattiace LA et al (1991) Identification of normal and pathological aging in prospectively studied nondemented elderly humans. Neurobiol Aging 13:179–189
Dickson DW, Uchikado H, Fujishiro H, Tsuboi Y (2010) Evidence in favor of Braak staging of Parkinson’s disease. Mov Disord 25(Suppl. 1):S78–S82
Dickson DW, Ahmed Z, Algom AA, Tsuboi Y, Josephs KA (2010) Neuropathology of variants of progressive supranuclear palsy. Curr Opin Neurol 23:394–400
Doty RL (2003) Odor perception in neurodegenerative disease. In: Doty RL (ed) Handbook of olfaction and gustation, 2nd edn. Marcel Dekker, New York, pp 479–501
Duyckaerts C, Hauw JJ (1997) Prevalence, incidence and duration of Braak’s stages in the general population: can we know? Neurobiol Aging 18:362–369
Frautschy SA, Cole GM (2010) Why pleiotropic interventions are needed for Alzheimer’s disease. Mol Neurobiol 41:392–409
German DC, White CL, Sparkman DR (1987) Alzheimer’s disease: neurofibrillary tangles in nuclei that project to the cerebral cortex. Neuroscience 21:305–312
German DC, Walker BS, Manaye K, Smith WK, Woodward DJ, North AJ (1988) The human locus coeruleus: computer reconstruction of cellular distribution. J Neurosci 8:193–203
German DC, Manaye KF, White CL 3rd (1992) Disease-specific patterns of locus coeruleus cell loss. Ann Neurol 32:667–676
Goedert M, Spillantini MG (2006) A century of Alzheimer’s disease. Science 314:777–781
Goedert M, Jakes R, Crowther RA et al (1993) The abnormal phosphorylation of tau protein at Ser-202 in Alzheimer disease recapitulates phosphorylation during development. Proc Natl Acad Sci USA 90:5066–5070
Goedert M, Trojanowski JQ, Lee VMY (1997) The neurofibrillary pathology of Alzheimer’s disease. In: Rosenberg RN (ed) The molecular and genetic basis of neurological disease, 2nd edn. Butterworth-Heinemann, Boston, pp 613–627
Grinberg LT, Rüb U, Ferretti REL, Brazilian Brain Bank Study Group et al (2009) The dorsal raphe nucleus shows phospho-tau neurofibrillary changes before the transentorhinal region in Alzheimer’s disease. A precocious onset? Neuropathol Appl Neurobiol 35:406–416
Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI (1986) Abnormal phosphorylation of the microtubule-associated protein tau in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci USA 83:4913–4917
Grudzien A, Shaw P, Weintraub S, Bigio E, Mash DC, Mesulam MM (2007) Locus coeruleus neurofibrillary degeneration in aging, mild cognitive impairment and early Alzheimer’s disease. Neurobiol Aging 28:327–335
Haglund M, Sjöbeck M, Englund E (2006) Locus ceruleus degeneration is ubiquitous in Alzheimer’s disease: possible implications for diagnosis and treatment. Neuropathology 26:528–532
Hardy JA (1992) An anatomical cascade hypothesis for Alzheimer’s disease. Trends Neurosci 15:200–201
Hardy JA (2006) The amyloid hypothesis: history and alternatives. In: Jucker M, Beyreuther K, Haass C, Nitsch R, Christen Y (eds) Alzheimer: 100 years and beyond. Springer, Berlin, pp 151–156
Hardy JA, Higgins GA (1992) Alzheimer’s disease: the amyloid cascade hypothesis. Science 256:184–185
Hardy JA, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297:353–356
Hawkes CH, Doty RL (2010) The neurology of olfaction. Cambridge University Press, Cambridge, pp 153–214
Hyman BT, Goméz-Isla T (1994) Alzheimer’s disease is a laminar, regional, and neural system specific disease, not a global brain disease. Neurobiol Aging 15:353–354
Hyman BT, Trojanowski JQ (1997) Editorial on consensus recommendations for the postmortem diagnosis of Alzheimer disease from the National Institute on Aging and the Reagan Institute Working Group on diagnostic criteria for the neuropathological assessment of Alzheimer disease. J Neuropathol Exp Neurol 56:1095–1097
Iqbal K, Liu F, Gong CX, Alonso AC, Grundke-Iqbal I (2009) Mechanisms of tau-induced neurodegeneration. Acta Neuropathol 118:53–69
Jack CR, Knopman DS, Jagust WJ et al (2010) Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol 9:119–128
Klosen P, Maessen X, de Aguilar P (1993) PEG embedding for immunocytochemistry: application to the analysis of immunoreactivity loss during histological processing. J Histochem Cytochem 41:455–463
Kok E, Haikonen S, Luoto T et al (2009) Apolipoprotein E-dependent accumulation of Alzheimer disease-related lesions begins in middle age. Ann Neurol 65:650–657
Kovacech B, Skrabana R, Novak M (2010) Transition of tau protein from disordered to misordered in Alzheimer’s disease. Neurodegener Dis 7:24–27
Lee VM-Y, Goedert M, Trojanowski JQ (2001) Neurodegenerative tauopathies. Ann Rev Neurosci 24:1121–1159
Li B, Chohan MO, Grundke-Iqbal I, Iqbal K (2007) Disruption of microtubule network by Alzheimer abnormally hyperphosphorylated tau. Acta Neuropathol 113:501–511
Li S, Shankar GM, Selkoe DJ (2010) How do soluble oligomers of amyloid beta-protein impair hippocampal synaptic plasticity? Front Cell Neurosci 4:5
Mandelkow EM, Thiers E, Biernat J, Mandelkow E (2006) Influence of tau on neuronal traffic mechanisms. In: Jucker M, Beyreuther K, Haass C, Nitsch R, Christen Y (eds) Alzheimer: 100 years and beyond. Springer, Berlin, pp 345–354
Mandelkow E, von Bergen M, Biernat J, Mandelkow EM (2007) Structural principles of tau and the paired helical filaments of Alzheimer’s disease. Brain Pathol 17:83–90
Mattson MP (2006) Molecular and cellular pathways towards and away from Alzheimer’s disease. In: Jucker M, Beyreuther K, Haass C, Nitsch R, Christen Y (eds) Alzheimer: 100 years and beyond. Springer, Berlin, pp 371–378
Mattsson N, Sävman K, Osterlundh G, Blennow K, Zetterberg H (2010) Converging molecular pathways in human and neural development and degeneration. Neurosci Res 66:330–332
McKee AC, Cantu RC, Nowinsky CJ, Hedley-White ET et al (2009) Chronic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol 68:709–735
Mesulam MM (1996) The systems-level organization of cholinergic innervation in the human cerebral cortex and its alterations in Alzheimer’s disease. Prog Brain Res 109:285–297
Mesulam MM, Shaw P, Mash D, Weintraub S (2004) Cholinergic nucleus basalis tauopathy emerges early in the aging-MCI-AD continuum. Ann Neurol 55:815–828
Morsch R, Simon W, Coleman PD (1999) Neurons may live for decades with neurofibrillary tangles. J Neuropathol Exp Neurol 58:188–197
Mouton PR, Pakkenberg B, Gundersen HJ, Price DL (1994) Absolute number and size of pigmented locus coeruleus neurons in young and aged individuals. J Chem Neuroanat 7:185–190
Ohm TG, Braak H (1987) Olfactory bulb changes in Alzheimer’s disease. Acta Neuropathol 73:365–369
Omalu BI, Bailes J, Hammers JL, Fitzsimmons RP (2010) Chronic traumatic encephalopathy, suicides, and parasuicides in professional American athletes: the role of the forensic pathologist. Am J Forensic Med Pathol 31:130–132
Parvizi J, Van Hoesen GW, Damasio A (2001) The selective vulnerability of brainstem nuclei to Alzheimer’s disease. Ann Neurol 49:53–66
Pearson J, Goldstein M, Markey K, Brandeis L (1983) Human brainstem catecholamine neuronal anatomy as indicated by immunocytochemistry with antibodies to tyrosine hydroxylase. Neuroscience 8:3–32
Pikkarainen M, Martikainen P, Alafuzoff I (2010) The effect of prolonged fixation time on immunohistochemical staining of common neurodegenerative disease markers. J Neuropathol Appl Neurol 69:40–52
Pimplikar SW (2009) Reassessing the amyloid cascade hypothesis of Alzheimer’s disease. Int J Biochem Cell Biol 41:1261–1268
Sandmann-Keil D, Braak H, Okochi M, Haass C, Braak E (1999) Alpha-synuclein immunoreactive Lewy bodies and Lewy neuritis in Parkinson’s disease are detectable by an advanced silver-staining technique. Acta Neuropathol 98:461–464
Saper CB (1987) Diffuse cortical projection systems: anatomical organization and role in cortical function. In: Plum F (ed) Handbook of physiology, vol. 5, The nervous system. Am Physiol Soc, Bethesda, pp 169–210
Saper CB (1990). Cholinergic system. In: Paxinos G (ed) The human nervous system. Academic Press, New York, pp 1095–1013
Sara SJ (2009) The locus coeruleus and noradrenergic modulation of cognition. Nat Rev Neurosci 10:211–223
Sassin I, Schultz C, Thal DR et al (2000) Evolution of Alzheimer’s disease-related cytoskeletal changes in the basal nucleus of Meynert. Acta Neuropathol 100:259–269
Schönheit B, Zarski R, Ohm TG (2004) Spatial and temporal relationships between plaques and tangles in Alzheimer-pathology. Neurobiol Aging 25:697–711
Selkoe DJ (2003) Aging, amyloid, and Alzheimer’s disease: a perspective in honor of Carl Cotman. Neurochem Res 28:1705–1713
Simic G, Stanic G, Mladinov M, Jovanov-Milosevic N, Kostovic I, Hof PR (2009) Does Alzheimer’s disease begin in the brainstem? Neuropathol Appl Neurobiol 35:532–554
Smith C, Graham DI, Murray LS, Nicoll JA (2003) Tau immunohistochemistry in acute brain injury. Neuropathol Appl Neurobiol 29:496–502
Stamer K, Vogel R, Thies E, Mandelkow E, Mandelkow EM (2002) Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J Cell Biol 156:1051–1063
Takeda S, Hashimoto M, Mallory M et al (1998) Abnormal distribution of the non-Aβ component of Alzheimer’s disease and amyloid precursor/α-synuclein in Lewy body disease as revealed by proteinase K and formic acid pretreatment. Lab Investig 78:1169–1177
Tanzi RE, Bertram I (2005) Twenty years of the Alzheimer’s disease amyloid hypothesis: a genetic perspective. Cell 120:545–555
Terry RD (2000) Where in the brain does Alzheimer’s disease begin? Ann Neurol 47:421
Teter B, Ashford JW (2002) Neuroplasticity in Alzheimer’s disease. J Neurosci Res 70:402–437
Thal DR, Rüb U, Schultz C et al (2000) Sequence of Aβ-protein deposition in the human medial temporal lobe. J Neuropathol Exp Neurol 59:733–748
Thal DR, Rüb U, Orantes M, Braak H (2002) Phases of Aβ-deposition in the human brain and its relevance for the development of AD. Neurology 58:1791–1800
Tolnay M, Probst A (1999) Review: tau protein pathology in Alzheimer’s disease and related disorders. Neuropathol Appl Neurobiol 25:171–187
Trojanowski JQ, Lee VMY (2000) “Fatal attractions” of proteins. A comprehensive hypothetical mechanism underlying Alzheimer’s disease and other neurodegenerative disorders. Ann NY Acad Sci 924:62–67
Weinshenker D (2008) Functional consequences of locus coeruleus degeneration in Alzheimer’s disease. Curr Alzheimer Res 5:342–345
Zarow C, Lyness SA, Mortimer JA, Chui HC (2003) Neuronal loss is greater in the locus coeruleus than nucleus basalis and substantia nigra in Alzheimer and Parkinson diseases. Arch Neurol 60:337–341
Zweig RM, Ross CA, Hedreen JC et al (1988) The neuropathology of aminergic nuclei in Alzheimer’s disease. Ann Neurol 24:233–242
Acknowledgments
This study was supported by the German Research Council (Deutsche Forschungsgemeinschaft, DFG grant number TR 1000/1-1). Autopsy material was supplied by the Braak Collection (Goethe University Frankfurt). The skillful technical assistance of Ms. Siegrid Baumann, Ms. Verena Hofmann, Ms. Gabriele Ehmke, Ms. Irina Lundgrin (immunohistochemistry), and Mr. Stephan Mayer (graphics) from the University of Ulm is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Additional information
For Professor Kurt Jellinger, in honor of his 80th birthday.
Rights and permissions
About this article
Cite this article
Braak, H., Del Tredici, K. The pathological process underlying Alzheimer’s disease in individuals under thirty. Acta Neuropathol 121, 171–181 (2011). https://doi.org/10.1007/s00401-010-0789-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-010-0789-4