
Abstract
Alzheimer’s disease (AD) is, at the neuropathological level, characterized by the accumulation and aggregation of misfolded proteins. The presence of misfolded proteins in the endoplasmic reticulum (ER) triggers a cellular stress response called the unfolded protein response (UPR) that may protect the cell against the toxic buildup of misfolded proteins. In this study we investigated the activation of the UPR in AD. Protein levels of BiP/GRP78, a molecular chaperone which is up-regulated during the UPR, was found to be increased in AD temporal cortex and hippocampus as determined by Western blot analysis. At the immunohistochemical level intensified staining of BiP/GRP78 was observed in AD, which did not co-localize with AT8-positive neurofibrillary tangles. In addition, we performed immunohistochemistry for phosphorylated (activated) pancreatic ER kinase (p-PERK), an ER kinase which is activated during the UPR. p-PERK was observed in neurons in AD patients, but not in non-demented control cases and did not co-localize with AT8-positive tangles. Overall, these data show that the UPR is activated in AD, and the increased occurrence of BiP/GRP78 and p-PERK in cytologically normal-appearing neurons suggest a role for the UPR early in AD neurodegeneration. Although the initial participation of the UPR in AD pathogenesis might be neuroprotective, sustained activation of the UPR in AD might initiate or mediate neurodegeneration.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is, at the neuropathological level, characterized by the accumulation and aggregation of misfolded proteins: intracellular aggregates of tau in the neurofibrillary tangles and extracellular aggregates of β-amyloid (Aβ) in the senile plaques [20]. The presence of misfolded proteins in the endoplasmic reticulum (ER) triggers a cellular stress response called the unfolded protein response (UPR) that may protect the cell against the toxic buildup of misfolded proteins [6, 15]. The UPR is initiated by the binding of the ER chaperone BiP/GRP78 to the misfolded proteins. Under normal conditions, BiP/GRP78 forms a complex with three key proteins at the ER membrane; pancreatic ER kinase (PERK) [8], transcription factor ATF-6, and endoribonuclease IRE-1. The binding of BiP/GRP78 to unfolded proteins releases BiP/GRP78 from PERK, ATF-6 and IRE-1, which consequently are activated. The activation of the UPR results in an overall decrease in translation, increased protein degradation and increased levels of ER chaperones, including BiP/GRP78 [11], which consequently increases the protein folding capacity of the ER. Eventually, the cell might return to normal ER homeostasis or, under prolonged ER stress, continue towards apoptosis. As neurons are highly susceptible to the toxic effects of misfolded proteins, and AD has features of misfolded proteins, ER-stress-mediated cell death might have an important role in the pathogenesis of this disease [1].
A first indication of a role for ER stress in AD came from a report showing increased expression of BiP/GRP78 in AD brain in neurons that remained cytologically normal [7]. This is in contrast with later studies that reported a decrease of BiP/GRP78 in AD patients or showed no difference [10, 16]. Thus, to date, there is no clear picture of how activation of the UPR fits into AD pathogenesis. In the present study we investigated the activation of the UPR in different stages of AD pathology according to the Braak staging [2]. The localization and expression levels of BiP/GRP78 and phosphorylated (activated) PERK (p-PERK), as markers for UPR activation, were investigated in AD and non-demented control patients using Western blotting and immunohistochemistry.
Materials and methods
Postmortem brain tissue
Human brain specimens of probable AD and age-matched non-demented control cases were obtained at autopsy with a short postmortem interval (The Netherlands Brain Bank, Amsterdam, The Netherlands; coordinator Dr. R. Ravid). Clinical diagnosis was defined according to DSM-III-R criteria and the severity of dementia was evaluated according to the Global Deterioration Scale of Reisberg (GDS) [14]. Neuropathological evaluation was performed on formalin-fixed, paraffin-embedded tissue from different sites, including the frontal cortex (F2), temporal pole cortex, parietal cortex (superior and inferior lobule), occipital pole cortex and the hippocampus (essentially CA1 and entorhinal area of the parahippocampal gyrus). The distribution and the density of neurofibrillary tangles was determined in Bodian-stained sections, while senile plaques were stained with the methenamine silver method [21]. Staging of AD was evaluated according to Braak and Braak [2, 3]. Age, sex, clinical diagnosis and Braak scores of the AD cases and non-demented controls used in this study are listed in Table 1.
Immunohistochemistry
For immunohistochemical staining, formalin-fixed (4%, 24 h) paraffin-embedded tissue from the mid-temporal cortex and hippocampus was used. Sections (5 μm thick) were mounted on superfrost plus tissue slides (Menzel-Gläser, Germany) and deparaffinized. Subsequently, sections were immersed in 0.3% H2O2 in methanol for 30 min to quench endogenous peroxidase activity, and treated in 10 mM pH 6.0 citrate buffer heated by microwave for 10 min for antigen retrieval. Normal serum and antibodies were dissolved in phosphate-buffered saline (PBS) containing 1% (w/v) bovine serum albumin (BSA, Boehringer Mannheim, Germany).
Sections were pre-incubated for 10 min with either normal rabbit or swine serum (DAKO, Glostrup, Denmark). Rabbit anti-GRP78/BiP was obtained from Santa Cruz Biotechnology (Santa Cruz, CA, 1:100 dilution) and rabbit anti-p-PERK (Thr980, 1:100 dilution) was obtained from Cell Signaling Technology (Beverly, MA). Mouse monoclonal AT8 antibody (dilution 1:200), recognizing the phosphorylated Ser202 residue of the tau protein, was obtained from Innogenetics (Ghent, Belgium), SMI311 (dilution 1:100), a cocktail of monoclonal antibodies to non-phosphorylated neurofilaments, was obtained from Sternberger Monoclonals Inc. (Luhterville, MD), and rabbit anti-α-synuclein was obtained from SanverTech (Boechout, Belgium). Antibodies were incubated for 1 h at room temperature. After washing with PBS, slides were incubated with a biotin-conjugated secondary antibody [rabbit anti-mouse F(ab’)2, 1:500 dilution; swine anti-rabbit F(ab’)2, 1:300 dilution, DakoCytomation, Glostrup, Denmark] for 30 min. Slides were incubated with streptavidin-biotin horseradish peroxidase complex (streptABComplex/HRP, 1:200 dilution, DakoCytomation) for 60 min. Color was developed using 3,3’-diaminobenzidine (0.1 mg/ml, 0.02% H2O2, 3 min) as chromogen. Sections were mounted with Entellan (Merck, Darmstadt, Germany).
Double immunohistohemistry
To determine the co-localization of BiP/GRP78, p-PERK, with AT8 immunoreactivity, double-immunostaining experiments were performed. Rabbit polyclonal antibodies were detected using biotin-conjugated swine anti-rabbit F(ab’)2 and with either alkaline phosphatase-conjugated streptavidin (1:100 dilution, Boehringer) or streptABComplex/HRP. The mouse monoclonal antibodies were detected using goat anti-IgG1 mouse conjugated with either HRP [1:100, Southern Biotechnology Associates (SBA), Birmingham, AL] or alkaline phosphatase (1:20, SBA). Color was developed using 3-amino-9-ethyl-carbazole (AEC, Zymed, San Francisco, CA) and Fast Blue BB base (Sigma, St. Louis, MO) as chromogen. Slides were mounted in Aquamount (BDH, Poole, England).
Western blotting
Brain tissue was lysed in buffer (100 mM TRIS pH 7.5, 100 mM KCl, 1 mM EDTA, 1% NP-40 and protease inhibitor cocktail (Roche, Indianapolis, MA)) at 4°C for 30 min, and centrifuged at 12,000 g at 4°C for 10 min to obtain total soluble cell lysate. Protein concentration was determined accordingly with a Bio-Rad (Hercules, CA) protein assay using BSA as standard. Equal amounts of proteins were separated on 10% or 12% polyacrylamide gel and blotted onto Immobilon membranes (Millipore Corporation, Billerica, MA). After blocking nonspecific binding, membranes were incubated sequentially with primary antibodies (dilution 1:1,000), and secondary antibodies conjugated with HRP (DakoCytomation, dilution 1:2,000). Primary antibodies used were directed against BiP/GRP78 (rabbit polyclonal, Santa Cruz) and nonphosphorylated eukaryotic elongation factor 2 (eEF2, Cell Signaling Technology). The blots were visualized using Lumi-LightPLUS Western blotting substrate (Roche) and a LAS-3000 luminescent image analyzer [Fuji Photo Film (Europe), Düsseldorf, Germany]. Western blot data were evaluated and quantified using Advanced Image Data Analyzer (AIDA, version 3.45.039, Raytest, Straubenhardt, Germany).
Results
Expression levels of BiP/GRP78 are increased in AD temporal cortex and hippocampus
The expression of BiP/GRP78 was investigated in protein lysates of temporal cortex and hippocampus derived from AD and non-demented control patients. BiP/GRP78 expression levels were normalized for eEF2 expression levels. In both temporal cortex and hippocampus, the levels of BiP/GRP78 were significantly increased in AD cases compared to non-demented control cases (Fig. 1A, B). We also compared the BiP/GRP78 expression levels obtained from the temporal cortex to the AD pathology, as determined by the Braak score for neurofibrillary changes and amyloid deposits (Fig. 2). BiP/GRP78 expression levels increase progressively with increasing scores for both neurofibrillary changes and amyloid pathology. This indicates increased activation of the UPR with AD pathology.

BiP/GRP78 protein expression levels in non-demented and AD brain lysates. A The protein expression levels of BiP/GRP78 were evaluated in protein lysates derived from the temporal cortex of non-demented control and clinical AD patients using Western blot analysis. The levels of BiP/GRP78 and eEF2 are shown from representative cases. A protein sample derived form SK-N-SH cells treated with tunicamycin (Tun) was used as a positive control. B Expression levels of BiP/GRP78 were normalized to the expression levels of eEF2. Shown are the mean levels (± SD) of relative BiP/GRP78 expression in the temporal cortex and hippocampus of non-demented control and AD cases. Asterisks indicate significant difference (P<0.05, Student’s t-test) compared to non-demented control patients (eEF2 eukaryotic elongation factor 2, AD Alzheimer’s disease)

BiP/GRP78 expression levels in the temporal cortex in relation to the Braak score for neurofibrillary changes and amyloid deposits. Shown are mean levels (± SD) of relative BiP/GRP78 expression in the different pathological Braak stages for neurofibrillary changes and amyloid deposits. Asterisks indicate significant difference (P<0.05, Student’s t-test) compared to stage 0/O
Immunohistochemical localization of BiP/GRP78 and p-PERK in neurons in the temporal cortex and hippocampus
To determine which cells show increased UPR activation, the cellular localization of BiP/GRP78 was assessed in both temporal cortex and hippocampus of AD and non-demented control cases by immunohistochemistry. BiP/GRP78 immunoreactivity was prominent in neurons in both control and AD subjects, whereas very low signals were seen in glial cells in all investigated areas.
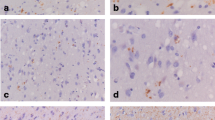
Neurons in AD brain, especially neurons in the CA1 (compare Fig. 3A and B) and CA2 (compare Fig. 3C and D) region of the hippocampus, exhibited stronger immunoreactivity for BiP/GRP78 compared to neurons in non-demented control brain. A difference in immunoreactive intensity for BiP/GRP78 between control and AD cases was difficult to observe in the temporal cortex (Fig. 4E). However, in most AD cases, BiP/GRP78-immunoreactive inclusion bodies were occasionally observed throughout the temporal cortex (inset, Fig. 4E). These inclusions were absent in the controls.

Immunohistochemical detection of BiP/GRP78 and p-PERK in non-demented control and AD cases. A Representative immunohistochemical staining for BiP/GRP78 in the CA1 region of the hippocampus of a non-demented control case (Braak 1/O). B Immunohistochemical staining for BiP/GRP78 in the CA1 region of the hippocampus of an AD case (Braak 5/C, arrow indicates neuritic plaque). C Staining for BiP/GRP78 in the CA2 region of the hippocampus of a non-demented control case (Braak 1/O). D BiP/GRP78 in the CA2 region of the hippocampus of an AD case (Braak 6/C). E BiP/GRP78-immunoreactive neurons in the temporal cortex of an AD patient (Braak 6/C). The inset shows a BiP/GRP78-immunoreactive inclusion body, which is occasionally observed in the temporal cortex of AD cases. F Representative immunohistochemical staining for p-PERK in the temporal cortex of an AD case (Braak 5/C). G Double immunohistochemistry for p-PERK (blue) and SMI311 (red) in the CA1 region of the hippocampus in an AD case (Braak 5/C). H Higher magnification of p-PERK (blue) and SMI311 (red) immunostaining in the CA1 region of the hippocampus in an AD case (Braak 5/C), showing the intracellular localization of p-PERK. In A–E, DAB (brown) was used as chromogen; in F, AEC (red) was used as chromogen. A–F hematoxylin staining of the nucleus [ER, endoplasmic reticulum, p-PERK phosphorylated (activated) pancreatic ER kinase]. Bar C–F, H 30 μm; A 60 μm; B, G 120 μm

Double-immunohistochemical detection of BiP/GRP78 and p-PERK with AT8 in AD cases. A Representative double-immunohistochemical staining for BiP/GRP78 (blue) and AT8 (red) in the CA1 region of the hippocampus of an AD case (Braak 6/C). Arrow indicates double-labeled neuron, arrowhead indicates single-labeled AT8-positive tangle. B Double immunolabeling for BiP/GRP78 (blue) and AT8 (red) in the CA1 region of the hippocampus of an AD case (Braak 6/C) (higher magnification). Arrow indicates double-labeled neuron, arrowhead indicates single-labeled AT8-positive tangle. C Double immunohistochemistry for BiP/GRP78 (blue) and AT8 (red) in the temporal cortex of an AD case (Braak 5/C). Arrow indicates single-labeled BiP/GRP78-positive neuron, arrowhead indicates single-labeled AT8-positive tangle. D Double immunolabeling for BiP/GRP78 (blue) and AT8 (red) in the CA1 region of the hippocampus of an AD case (Braak 6/C). Arrow indicates a single-labeled BiP/GRP78-positive neuron. E–H Pictures of CA2 region of the hippocampus taken from adjacent sections of the same AD case (Braak 6/C). E BiP/GRP78 immunostaining using DAB (brown) as chromogen and hematoxylin staining of the nucleus. F p-PERK immunostaining using AEC (red) as chromogen and hematoxylin staining of the nucleus. Arrows indicate p-PERK-containing neurons. G Double immunolabeling for BiP/GRP78 (blue) and AT8 (red). Arrows indicate single-labeled AT8-positive tangles. H Double immunolabeling for p-PERK (blue) and AT8 (red). Arrow indicates single-labeled AT8-positive tangle. E, F Hematoxylin staining of the nucleus. Bar B, C 30 μm; E–H 60 μm; A, D 120 μm
To confirm the activation of the UPR in AD cases, the localization of p-PERK was investigated. No immunoreactivity for p-PERK was observed in either hippocampus or temporal cortex of non-demented control cases (results not shown). p-PERK immunoreactivity was occasionally observed in neurons in AD temporal cortex (Fig. 3F), whereas a substantial number of neurons in AD hippocampus clearly stained for p-PERK (Fig. 3G). Especially neurons in the CA1 (Fig. 3G) and CA2 (Fig. 4F) region of the hippocampus were immunoreactive for p-PERK. p-PERK immunoreactivity was observed as a granular staining pattern, but clearly present inside the neurons as shown by p-PERK and SMI-311 double immunostaining (Fig. 3G, H). The increased presence of BiP/GRP78 in AD neurons and the presence of p-PERK only in AD cases confirm activation of the UPR in AD.
To investigate the relation between ER stress and the occurrence of neurofibrillary changes, double immunohistochemistry was performed using an antibody that detects hyperphosphorylated tau protein (AT8). In the hippocampus, the majority of the BiP/GRP78-positive neurons showed no AT8 immunoreactivity (Fig. 4A). However, occasionally some AT8 immunoreactivity in single BiP/GRP78-positive neurons (arrow Fig. 4A, B) was observed. AT8-positive neurofibrillary tangles did not show BiP/GRP78 immunoreactivity (arrowhead Fig. 4A, B). Also, in the temporal cortex of AD cases, BiP/GRP78 was observed in neurons without AT8 immunoreactivity, while AT8-positive tangles did not show BiP/GRP78 immunoreactivity (Fig. 4C).
The immunohistochemical staining of BiP/GRP78 and AT8 in AD hippocampus and temporal cortex, suggests that increased BiP/GRP78 expression precedes tangle formation. In addition, the observation that most neurons that show increased immunoreactivity for BiP/GRP78 do not show AT8 immunoreactivity suggests that BiP/GRP78 might have a neuroprotective role. This idea is nicely illustrated by the double immunostaining for BiP/GRP78 and AT8 of an advanced AD case (Braak 6C, hippocampus CA1), which shows a lot of neurofibrillary degeneration. Although all tangle-bearing neurons are negative for BiP/GRP78, a small number of BiP/GRP78-positive normal-looking neurons are still present (arrows Fig. 4D).
Interestingly, neurons in the CA2–3 region of the hippocampus showed the strongest immunoreactvity for BiP/GRP78, while, compared to other regions, the CA2–3 showed less neurofibrillary changes. Figure 4E–H shows pictures of the same AD case taken from adjacent sections. BiP/GRP78 is present in almost every neuron (Fig. 4E), while p-PERK is only observed in a few neurons (arrows Fig. 4F). Thus, although high levels of BiP/GRP78 are present in CA2–3 neurons, only a few show activation of the UPR. Double immunohistochemistry shows that BiP/GRP78- and p-PERK-positive neurons are negative for AT8 (Fig. 4G, H, respectively), while AT8-positive tangles do not show BiP/GRP78 and p-PERK immunoreactivity (arrows Fig. 4G, H, respectively). Together, these data suggest that high levels of BiP/GRP78 and activation of the UPR precede or even postpone neurofibrillary degeneration.
In the cases and areas investigated in this study, no α-synuclein-positive inclusions were observed (data not shown). This suggests that in these cases the activation of the UPR is not caused or mediated by accumulation of α-synuclein. However, this does not exclude that α-synuclein accumulation can induce ER stress in other AD cases or neurodegenerative disorders. The correlation between UPR activation and the accumulation of α-synuclein as well as intracellular Aβ in AD pathology needs to be addressed in future studies.
Discussion
In the present study, we investigated the activation of the UPR in different stages of AD pathology. The localization and expression levels of BiP/GRP78 and p-PERK, as markers for UPR activation, were increased in neurons in AD compared to non-demented control patients. A first indication of a role for ER stress in AD came from a report showing increased expression of BiP/GRP78 in healthy neurons in AD brain material [7]. This is in contrast with other studies that either reported a decrease of BiP/GRP78 in AD patients [10], or no changes at all between controls, sporadic or presenilin 1-mediated familial AD patients [16]. These differences may be explained by the use of different antibodies (a generic anti-KDEL antibody [10] and BiP/GRP78-specific antibodies in the other studies) or analysis of different brain areas (parietal lobe [16] versus temporal cortex and hippocampus in the other studies). Our results on BiP/GRP78 immunohistochemistry are consistent with the study by Hamos et al. [7]. In addition, we quantified the total BiP/GRP78 levels, which we compared to the clinical status and stage of pathology (Figs. 1, 2) and immunostainings for p-PERK, showing that the UPR is activated in AD hippocampus and temporal cortex.
The results presented in this study are consistent with the reports that phosphorylation of eukaryotic initiation factor-2α (eIF2α) and RNA-dependent protein kinase (PKR) are increased in AD [4, 12]. PERK as well as PKR are involved in the phosphorylation of eIF2α. Phosphorylated eIF2α inhibits the assembly of the 80S ribosome, and thereby facilitates the translational block of the UPR. In another study we found that the small GTPase Rab6A is up-regulated in AD neurons, and is strongly correlated with BiP/GRP78 levels (Scheper et al., submitted for publication). Rab6a may be involved in protein quality control by providing a retrograde trafficking route from the Golgi to the ER. In summary, these studies show that pathways involved in ER stress and protein quality control are increased in AD pathogenesis.
There is also indirect evidence that Aβ is involved in UPR activation during AD pathogenesis. In vitro studies have demonstrated that Aβ1–42 is generated in the ER of neuronal cells [5, 9] and, although it is unclear how extracellular Aβ signals to the ER, it has been shown that extracellular Aβ can induce the UPR [18, 22] (data reproduced in our lab, Chafekar and Scheper, unpublished). Aβ induces Ca2+ release from the ER [19], and inhibition of Ca2+ release, which concomitantly attenuates the up-regulation of BiP/GRP78, partly relieves Aβ toxicity [18]. In contrast, up-regulation of BiP/GRP78 was suggested to be protective in Aβ toxicity [22]. These apparently contradicting observations may relate to the balance between protective and destructive responses of the UPR [17]. A direct correlation between intracellular Aβ and activation of the UPR during AD pathology needs to be addressed in future studies.
The levels of BiP/GRP78 in the temporal cortex are increased with the overall AD pathology. Double immunohistochemistry for BiP/GRP78 or p-PERK and AT8 suggests that an increase in UPR precedes the formation of neurofibrillary tangles. The increased levels of BiP/GRP78 and p-PERK in cytologically normal neurons is consistent with the previous findings on BiP/GRP78 expression in AD by Hamos et al. [7]. This could indicate a protective role for the UPR, possibly in response to amyloid precursor protein (APP) mismetabolism and the concomitant increase of Aβ in the ER. Alternatively, ER stress may also aggravate the severity and/or accelerate the onset of the disease by favoring amyloidogenic processing of APP [13].
In summary, we found that overall protein levels of BiP/GRP78 are increased in AD temporal cortex and hippocampus compared to non-demented control cases. At the immunohistochemical level BiP/GRP78 and p-PERK are increased in healthy neurons in AD and do not co-localize with neurofibrillary tangles. The initial participation of the UPR in AD pathogenesis might be neuroprotective. Whether a sustained activation of the UPR in AD initiates or accelerates neurodegeneration needs to be addressed in future studies.
References
Benn SC, Woolf CJ (2004) Adult neuron survival strategies—slamming on the brakes. Nat Rev Neurosci 5:686–700
Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82:239–259
Braak H, Braak E (1995) Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol Aging 16:271–8; discussion 278–284
Chang RC, Wong AK, Ng HK, Hugon J (2002) Phosphorylation of eukaryotic initiation factor-2alpha (eIF2alpha) is associated with neuronal degeneration in Alzheimer’s disease. Neuroreport 13:2429–2432
Cook DG, Forman MS, Sung JC, Leight S, Kolson DL, Iwatsubo T, Lee VM, Doms RW (1997) Alzheimer’s A beta(1–42) is generated in the endoplasmic reticulum/intermediate compartment of NT2N cells. Nat Med 3:1021–1023
Forman MS, Lee VM, Trojanowski JQ (2003) ‘Unfolding’ pathways in neurodegenerative disease. Trends Neurosci 26:407–410
Hamos JE, Oblas B, Pulaski-Salo D, Welch WJ, Bole DG, Drachman DA (1991) Expression of heat shock proteins in Alzheimer’s disease. Neurology 41:345–350
Harding HP, Zhang Y, Ron D (1999) Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397:271–274
Hartmann T, Bieger SC, Bruhl B, Tienari PJ, Ida N, Allsop D, Roberts GW, Masters CL, Dotti CG, Unsicker K, Beyreuther K (1997) Distinct sites of intracellular production for Alzheimer’s disease A beta40/42 amyloid peptides. Nat Med 3:1016–1020
Katayama T, Imaizumi K, Sato N, Miyoshi K, Kudo T, Hitomi J, Morihara T, Yoneda T, Gomi F, Mori Y, Nakano Y, Takeda J, Tsuda T, Itoyama Y, Murayama O, Takashima A, St George-Hyslop P, Takeda M, Tohyama M (1999) Presenilin-1 mutations downregulate the signalling pathway of the unfolded-protein response. Nat Cell Biol 1:479–485
Kozutsumi Y, Segal M, Normington K, Gething MJ, Sambrook J (1988) The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature 332:462–464
Onuki R, Bando Y, Suyama E, Katayama T, Kawasaki H, Baba T, Tohyama M, Taira K (2004) An RNA-dependent protein kinase is involved in tunicamycin-induced apoptosis and Alzheimer’s disease. EMBO J 23:959–968
Piccini A, Fassio A, Pasqualetto E, Vitali A, Borghi R, Palmieri D, Nacmias B, Sorbi S, Sitia R, Tabaton M (2004) Fibroblasts from FAD-linked presenilin 1 mutations display a normal unfolded protein response but overproduce Abeta42 in response to tunicamycin. Neurobiol Dis 15:380–386
Reisberg B, Ferris SH, Leon MJ de, Crook T (1982) The Global Deterioration Scale for assessment of primary degenerative dementia. Am J Psychiatry 139:1136–1139
Rutkowski DT, Kaufman RJ (2004) A trip to the ER: coping with stress. Trends Cell Biol 14:20–28
Sato N, Urano F, Yoon Leem J, Kim SH, Li M, Donoviel D, Bernstein A, Lee AS, Ron D, Veselits ML, Sisodia SS, Thinakaran G (2000) Upregulation of BiP and CHOP by the unfolded-protein response is independent of presenilin expression. Nat Cell Biol 2:863–870
Scheper W, Hol E (2005) Protein quality control in Alzheimer’s disease: a fatal saviour. Curr Drug Targets CNS Neurol Disord 4:283–292
Suen KC, Lin KF, Elyaman W, So KF, Chang RC, Hugon J (2003) Reduction of calcium release from the endoplasmic reticulum could only provide partial neuroprotection against beta-amyloid peptide toxicity. J Neurochem 87:1413–1426
Suen KC, Yu MS, So KF, Chang RC, Hugon J (2003) Upstream signaling pathways leading to the activation of double-stranded RNA-dependent serine/threonine protein kinase in beta-amyloid peptide neurotoxicity. J Biol Chem 278:49819–49827
Taylor JP, Hardy J, Fischbeck KH (2002) Toxic proteins in neurodegenerative disease. Science 296:1991–1995
Yamaguchi H, Haga C, Hirai S, Nakazato Y, Kosaka K (1990) Distinctive, rapid, and easy labeling of diffuse plaques in the Alzheimer brains by a new methenamine silver stain. Acta Neuropathol 79:569–572
Yu Z, Luo H, Fu W, Mattson MP (1999) The endoplasmic reticulum stress-responsive protein GRP78 protects neurons against excitotoxicity and apoptosis: suppression of oxidative stress and stabilization of calcium homeostasis. Exp Neurol 155:302–314
Acknowledgements
The authors thank the Netherlands Brain Bank for supplying the human brain tissue (coordinator Dr. R. Ravid), Dr. W. Kamphorst for the neuropathological diagnosis of control and AD tissue, and Marlies Jacobs for her technical assistance. This study was supported by the Internationale Stichting Alzheimer Onderzoek (ISAO grant 02501 to J.H., grant 02504 to W.S.). W.S. is a fellow of the Anton Meelmeijer Center for Translational Research.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hoozemans, J.J.M., Veerhuis, R., Van Haastert, E.S. et al. The unfolded protein response is activated in Alzheimer’s disease. Acta Neuropathol 110, 165–172 (2005). https://doi.org/10.1007/s00401-005-1038-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-005-1038-0