
Abstract
It has been reported that immunoglobulins (IgG) in sera of patients with amyotrophic lateral sclerosis (ALS) kill cultured motoneurones (MN), but whether they also cause MN degeneration in vivo is unclear. To test this, protein-A affinity purified and dialysed IgGs were prepared from sera of 44 ALS patients without paraproteinemias, 20 healthy controls and 15 disease controls. Control and ALS-IgGs were injected intraperitoneally into groups of mice for 5 consecutive days and examined at day 8. IgG was localised immunocytochemically and spinal MN were characterised histologically and ultrastructurally and by comparative counts of Ca2+ containing organelles revealed with oxylate-pyroantimonate histochemistry. ELISA revealed no differences in IgG concentration between ALS patients and control subjects. Immunocytochemistry showed IgG was present in MN of mice injected with control or ALS-IgG, but densitometry showed immunostaining in MN was stronger in mice injected with ALS-IgG. Compared to MN of non-injected mice, control-IgG-treated mice showed near normal MN morphology and numbers of Ca2+-containing organelles. Disease control IgGs evoked negligible or minor morphological changes according to disease, but normal numbers of Ca2+ containing organelles. Ultrastructurally, about 70% of ALS-derived IgGs induced a population of MN with electron lucent cytoplasm, distended Golgi, disrupted Nissl and mitochondria (i.e., necrosis). However 30% of ALS-IgGs additionally induced electron-dense degeneration in 40% of the MN. These MN exhibited shrinkage, condensed nuclear chromatin and ill-defined nuclear membranes and resembled preliminary stages of apoptosis. We conclude that passive transfer of ALS-derived, but not control IgGs, does result in MN degeneration in the recipient mice. This appears to be associated with abnormal calcium homeostasis, but the exact target of ALS-IgG remains conjectural, and the possibilities are discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Amyotrophic lateral sclerosis (ALS) is characterised by the progressive death of motoneurones (MN) in the motor cortex, certain cranial nuclei, and in the spinal cord. While 5–10% of patients have a family history of the condition, with a proposed genetic basis entailing mutations of the SOD1 gene in approximately 2% of all patients, 90–95% of patients are sporadic with an unproven aetiology, which is probably multifactorial. Possible mechanisms include oxidative damage, excitotoxicity, protein aggregation affecting cytoskeletal function, neurotrophic deficit and autoimmunity. The argument for an immune-mediated component in the pathogenesis of ALS remains controversial. Circumstantial supporting evidence includes: abnormal immunostaining for IgG in cortical and spinal motoneurones of patients with ALS [15], the presence of macrophages and infiltrates of activated T-cells in spinal cords of some patients with ALS [12, 20, 37] and the identification of serum antibodies targeting GM1 antigen [30], neurofilaments [9], proapoptotic FAS [41] and voltage gated Ca2+ channels (VGCC) [4, 38].
The case for an immune-mediated assault on voltage-gated calcium channels rests on both in vitro and in vivo evidence. In vitro, IgG of ALS patients binds to L-, P- and N-type Ca2+ channels [10, 17, 21], enhances Ca2+ influx into cultured motoneurones [8] and synaptic terminals of Purkinje cells [23] and increases transmitter release at the neuromuscular junction [3, 38, 39]. In vivo, ultrastructural abnormalities with Ca2+ enhancement have been described in neuromuscular junctions of patients with ALS [35] and in mice after passive transfer of ALS-derived IgG [13].
The contra-argument for autoimmunity in ALS rests chiefly on the failure of immunosuppression or plasmaphoresis to alter disease progression [11]. Some authors expressed doubts [40] when others reported difficulty identifying anti-Ca2+ channel antibodies in significant numbers of ALS cases [5]. Anti-Ca2+ channel antibodies were identified in only 8% of patients [5] as opposed to 75% reported by Appel’s group [17]. In addition, it was found that IgG from ALS patients only blocks binding of specific antibodies to the alpha-1 subunit of voltage-gated Ca2+ channels in synaptosomes in the presence of serine protease inhibitors [27], suggesting protease action may explain the physiological effects attributed to IgG.
The original in vitro physiological findings have been verified in studies showing that isolated IgG from at least 50% of ALS patients causes increases in spontaneous and evoked potential frequency in muscle preparations [28] and that 70% of sera possess antibodies responsive to the dihydropyridine-sensitive L-type Ca2+ channel [22]. In a previous study [32], we demonstrated that passive transfer of purified IgG in sera of ALS patients to mice promoted a distinctive ultrastructural cytopathology in the murine motoneurones and increased the numbers of Ca2+-containing organelles, confirming the original morphological observations by Engelhardt et al. [13, 14]. The cytological abnormalities did not occur after passive transfer of IgG from healthy individuals. Prompted by our findings we undertook a more detailed analysis of the neurones of mice receiving IgG from ALS patients and healthy controls with the aims of firstly, characterising the range of neuronal response, and secondly, determining whether passive transfer of ALS-IgG to mice resulted in degeneration of the murine motoneurones, as is reported to occur in vitro [2, 8]. This morphometric study was complemented by immunochemical investigations to determine possible activation of pro-apoptotic proteases and retrograde labelling experiments sought evidence of alterations in MN number. Results from these studies will be reported separately.
Materials and methods
Sera
Sera were collected with prior local Ethical Committee and UCLH Trust approval and the informed written consent of patients and control subjects. In some ALS patients, limited blood samples were taken in the interest of the patient. Sera were obtained from three groups: (1) The sporadic ALS without paraproteinemia group (n=40) had an age range of 28–80 years (mean ± SD: 57.9±11.4) and was made up of 17 females and 27 males. Disease duration ranged between 1.25–14 years from presentation. All patients had a clinical diagnosis of probable or definite ALS according to World Federation of Neurology criteria (40). Six patients had bulbar onset; in all others onset was in the limbs. One patient had had poliomyelitis infection 50 years prior to onset of ALS. No patients had a notable inflammatory disease. (2) The normal healthy controls (n=20) had an age range of 26–70 years (mean ± SD: 50.6±13.1) and was made up of 14 females and 10 males. (3) For the disease controls (n=15), Sera were collected from a range of patients with diseases unrelated to ALS, including multiple sclerosis (MS), Guillain-Barré syndrome (GBS), Lambert-Eaton Myasthenic Syndrome (LEMS), myeloma and dystonia.
Purification of IgG
IgG was isolated and purified by affinity chromatography on Protein A-4% agarose columns (Pierce Inc.). IgG was bound with 10 mM TRIS buffer (pH 7.5), and 1 ml fractions were eluted with 0.2 M glycine (pH 2.7). The approximate protein content of each fraction was determined using a Camlab 3000 spectrophotometer at λ280 nm. Fractions with highest optical densities were pooled and dialysed against 0.1 M phosphate buffered saline (PBS), pH 7.2. The presence of IgG and absence of contaminants in the dialysed samples, in particular serine protease, was verified by Western blotting as described above.
Determination of serum IgG levels
IgG (H+L) levels were measured in whole sera from 40 ALS patients, 20 healthy controls and 15 disease controls by ELISA using WHO-approved IgG calibration standards (Calibrator 4, DeSoirin, UK) and verified by Western blotting. Ninety-six-well ELISA plates (Nunc-Maxisorp surface, Nunc, Denmark ) were coated with 1:200 monoclonal anti-human IgG (H+L) (Vectorlabs, UK) and blocked with 3% bovine serum albumin-20 mM Tris buffered saline (3% BSA-TBS), washed, then incubated with diluted patient sera (1:100–1:10, 000, in duplicate series). Bound IgG was detected with 1:1,000 anti-human IgG conjugated to alkaline phosphatase (Vectorlabs, UK) and visualised with 1 mg/ml p-nitrophenylphosphate (pNPP). Optical densities were read at λ405 nm using a Camlab 300 spectrophotometer, and values were interpolated against an IgG standard curve. For Western blots, both sera diluted 1:100 and purified IgGs were separated by SDS-PAGE using 5% polyacrylamide gels and transferred to nitrocellulose membranes. Membranes were blocked with 3% BSA-TBS, washed, and incubated in rabbit anti-human IgG (H+L); (antibody from Vectolabs, UK). Bound antibody was detected with 1:300 biotinylated anti-rabbit IgG (Amersham, UK) followed by 1:200 streptavidin-HRP (Amersham, UK). HRP was visualised with 1.3 mM 3,3 diaminobenzidine (DAB), 0.02% H2O2.
In vivo experiments
In vivo experiments were undertaken with British Home Office licensed approval and local veterinary supervision.
Passive transfer of IgG
Purified IgG from ALS patients, healthy and disease-control subjects were injected intraperitoneally (i/p) into groups of adult male BalbC mice (25 g body weight). Mice received twice daily i/p injections of IgG for 5 days (3–4 mg in total) and were perfusion fixed on day 8. Before injection, the IgG content of the purified dialysed samples was re-verified by spectrophotometry, and the pH adjusted to 7.2 if required. Samples typically, contained 3–4 mg in 2 ml of buffer, which provided 10 aliquots of 0.2 ml for injection over the 5 days. Our original experiments (32) found this dose range evoked well-defined neuronal lesions, which showed an identical range of cytological abnormalities. Prior to terminal perfusion-fixation mice were deeply anaesthetised by intraperitoneal (i/p) injection of a mixture of Domitor (Pzfizer, UK; 1 mg/ml medetomidine HCl, 100 μg/kg) and Ketaset (Fortdodge, UK; 100 mg/ml ketamine HCl, 0.6 mg/kg).
Electronmicroscopy and localisation of Ca2+
Anaesthetised mice were perfusion-fixed with 50 mM PBS buffered 3% glutaraldehyde containing 90 mM K oxalate. Spinal cords were removed and cut into 2-mm transverse slices. These were post-fixed in 1% osmium tetroxide containing 2% Na+-pyroantimonate to localise intracellular Ca2+ as described by Engelhardt, Siklos et al. [13] and processed for electronmicroscopy (EM).
Immunocytochemical localisation of IgG in motoneurones
To localise IgG in MN, three groups of three mice were injected i/p with ALS-IgG, control IgG and disease control IgG as described above and perfusion-fixed under anaesthesia with 4% paraformaldehyde-0.1% glutaraldehyde-PBS. At least five 40-μm vibroslice sections of spinal cord were cut from each animal, blocked in 5% normal goat serum-1% bovine serum albumin and incubated with 1:300 goat-anti-human IgG (H+L) (Vectorlabs, UK). This antibody has <0.1% cross-reactivity with mouse IgG. Tissue-bound antibody was detected with a routine streptavidin-biotin-HRP method. To test for any differences between levels of immunostaining in MN of mice treated with control and ALS-IgG, sections were analysed by densitometry using a procedure devised in previous studies of MN in ALS [7]. Images of immunostained sections were captured via a microscope-mounted Sony CCTV camera, under constant conditions of section thickness, microscope illumination, condenser aperture and magnification. The digitalized images were re-acquired in SigmaScan (Jandel, Germany) and converted to greyscale. Greyscale values were measured in several regions of the grey matter and used to determine mean grey matter background. Greyscale values measured in at least four regions of the cytosol in individual immunostained motoneurones were used to calculate the mean greyscale value for the motoneurone. The absorbance value for each motoneurone was then calculated according to the formula:
The values were used to calculate (1) the mean absorbance value for each animal and (2) the overall mean absorbance values for control-IgG- and ALS-IgG-treated groups.
Analysis and morphometry
Neuronal and organelle structure and pyroantimonate deposition in motoneurons of mice injected with ALS and control IgGs were compared using morphometric techniques. The methods and their underlying principles are given with the appropriate results.
Statistical analyses
ALS-IgG-control IgG and ALS-IgG-disease control IgG differences in mean values of measured parameters were tested with a two-tailed unpaired Student’s t-test, and inter-sample and inter-group differences were tested with a two-way ANOVA. Distribution profiles of IgG staining levels in MN were tested with a Kolmogorov-Smirnow procedure and F-ratio analysis assuming samples from populations with unequal variances.
Results
IgG concentration in sera
The mean (±SD) IgG concentration obtained for 40 ALS sera was 13.1±8.2 mg/ml (range 3–37.5 mg/ml), and for 20 healthy control sera it was 13.0±5.7 mg/ml (range 7.5–50.0 mg/ml), and for 15 disease control sera it was 11.4±4.6 mg/ml (range 6.6–20.0 mg/ml). Student’s t-tests showed that the ALS-control IgG difference (t=0.03, P>0.1) and the ALS-disease control IgG difference (t=0.5, P>0.1) was not significant. To examine the effect of disease duration, the mean IgG concentration for a sample of five ALS patients with a duration of less than 2 years was compared with that for a group of four patients with a duration of 8–14 years. The difference was not significant (<2 years mean 20.0±10.2 mg/ml, >8 years mean 15.2±2.3 mg/ml; t=0.996, P>0.1).
IgG after affinity purification
Western blots identified purified human IgG in protein-A affinity purified samples of control and ALS sera (IgG-H bands at ~100 kDa and IgG-L at ~50 kDa in Fig. 1a). Parallel blots also identified the presence of plasminogen in affinity-purified IgG samples (Fig. 1b), confirming the observation that serine proteases can co-purify with IgG [27]. These proteases were eliminated prior to injection by dialysis of samples overnight in 100 mM PBS (lane 5 in Fig. 1b).

Analyses of control and ALS sera. The western blot in (a) demonstrates the presence of IgG (H) and (L) in sera of control (CON) and ALS subjects, and in (b) reveals serine protease plasminogen in control and ALS sera (lanes 2–4) and in non-dialysed affinity purifed IgG (lane 6), but not in purified dilaysed ALS-IgG (lane 5)
Localisation of human IgG in mice
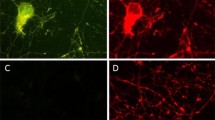
Mice were injected with dialysed affinity-purified IgGs as described above. Vibroslice sections of spinal cord from three mice injected with control IgG, three injected with disease control IgG and three ALS-IgG-treated mice all exhibited cytosolic immunostaining in MN with the antibody to human IgG (H+L). MN in spinal cords from non-injected mice did not immunostain. Since the primary antibody has <0.1% cross-reactivity with mouse IgG, immunostaining in these MN represents human IgG. Using densitometry, levels of immunostaining in MN of the mice injected with ALS-IgG were compared with those in MN of the mice injected with IgG from healthy individuals. The data were pooled to provide two samples, ALS-IgG (n=154 MN), and Control-IgG (n=180 MN). The distributions of absorbance values for spinal MN in ALS-IgG-treated mice were greater than those in mice injected with control-IgG (Fig. 2). Paired Student’s t-test and F-ratio tests proved the distribution means to be significantly different (P<0.0002 and P<0.0001, respectively).

Immunocytochemical localisation of human IgG in MN in spinal cord of mice following passive transfer of IgG. (a) No immunostaining occurs in MN of normal mice when immunostained for mouse IgG. (b) Immunostaining in MN of mice injected with ALS-IgG detected using antibodies specific for human IgG. (c) Distribution profiles of levels of immunostaining measured by densitometry and expressed as absorbance values for MN of mice injected with control or ALS-IgG reveal higher levels of staining in MN with ALS-IgG
Morphology and ultrastructural response to IgG
Spinal MN in non-injected mice (n=3)
MN were examined in toluidine blue-stained 0.5-μm semi-thin plastic sections and in the EM. In the semi-thin sections, MN exhibited smooth somal membranes, pale or unstained nuclei and cytoplasm, intensely stained nucleoli and variously sized prominent basophilic cytosolic structures representing Nissl bodies. EM revealed all MN had similar ultrastructure (Fig. 3a) and were round or oval in shape, with a round central nucleus with dispersed chromatin. Mitochondria were intact, and Golgi complexes consisted of parallel or curvilinear ER lamellae with narrow lumens (40–60 μm; Fig. 3b). Nissl bodies comprised rough endoplasmic reticulum (rER) lamellae and polyribosomes organised in three broad types of structure, which have been defined in previous studies as type 1, type 2 and type 3 [32, 33]. The predominant structure was type 1, defined by a stack of alternating parallel rER lamellae and arrays of polyribosomes (Fig. 3c). Type 2 Nissl bodies were characterised by shorter rER lamellae organised in random orientation within a pool of polyribosomes (Fig. 3d). Type 3 Nissl bodies chiefly comprised groups of closely packed polyribosomes containing few, if any, short rER lamellae (Fig. 3e). No electron-dense or vacuolated MN occurred.

Electron micrographs of representative MN in a non-injected mouse (a–e), and in mice injected with control IgG (f, g) and disease control IgG (h, i). Normal MN have dispersed chromatin, non-vacuolated lucent cytoplasm (a), multilayered Golgi complexes and intact mitochondria (b). The multilayered rER type 1 Nissl body structure in (c) contrasts with the less ordered rER and numerous polyribosomes in type 2 (d) and the predominantly ribosomal composition of type 3 (e). Near normal MN and organelle morphology following treatment with control-IgG (f and g) and IgG from a case of Lambert-Eaton-Myasthenic syndrome (LEMS; h, i). Scale bar =5 μm in a, f and h; 1 μm in b–e, g and i
Spinal MN in mice injected with IgG from healthy controls (n=10 controls)
MN viewed by light microscopy and EM (Fig. 3f) possessed a normal somal shape, pale staining nucleus with dispersed chromatin and pale non-vacuolated cytoplasm. A small number of MN exhibited stronger staining with toludine blue because of the diffuse nature of the Nissl substance, but no abnormal intensely stained MN were seen. Ultrastructurally, Golgi complexes had a multi-lamellar structure (Fig. 3g), Nissl bodies exhibited all three types of composition and structure, and mitochondria were intact. With 3/10 control IgGs ~10% of MN were mildly vacuolated due to slight ER distension, but otherwise displayed normal morphology. No abnormal electron-dense MN occurred.
Spinal MN in mice injected with IgG from disease controls (n=10 controls)
Light microscopy revealed no discernible abnormalities in MN with 7/10 disease control IgGs, and EM confirmed morphology was similar to that seen with control IgG. Of special note, MN of mice exposed to LEMS-IgG had normal morphology (Fig. 3h). However, 3/10 disease control IgGs prompted irregular somal contour in 30% of MN, or irregularly contoured nuclei. Chromatin in these MN was universally dispersed with no abnormal increase in density, Golgi complexes and Nissl bodies had normal composition and ultrastructure (Fig. 3i), and both cytoplasm and nuclei were electron-lucent. No electron-dense MN were encountered in this group of mice.
Spinal MN in mice injected with IgG from ALS patients (n=20 patients)
In toluidine blue-stained sections, ALS-IgGs evoked a spectrum of abnormalities ranging between MN with pale stained cytoplasm and nucleus, exhibiting variable degrees of vacuolation, to MN with dark or intensely stained cytoplasm, abnormally dense nuclei containing chromatin aggregates and extensive vacuolation.
The complete spectrum of MN abnormalities was not evoked by every IgG. With 70% of the ALS-IgGs tested, all MN exhibited a pale toludine blue-stained cytoplasm and nucleus and normal shaped soma. Electronmicroscopy showed these MN to have an electron-lucent cytoplasm and electron-lucent nucleus (Fig. 4a). Invariably the cytoplasm was vacuolated (Fig. 4b, c) due to highly distended Golgi ER and damaged mitochondria (Fig. 4b). The mitochondria had reduced numbers of cristae. There was an increased prominence of type 2 and type 3 Nissl body structure, but a reduced prominence of type 1 Nissl bodies, indicating dispersal of polyribosomes. These MN showed signs of necrosis.

Morphology of motoneurones (MN) after treatment with ALS-IgG (a–g). Some MN possess lucent nuclei (a), and lucent but vacuolated cytoplasm, in (b) with distended Golgi ER, damaged mitochondria and ribosomal dispersion. Others are electron-dense (d and e) with highly distended ER (Golgi in e) and indistinct nuclear membrane (Nucl in e), or are electron-dense with pre-apoptotic signs of perinuclear chromatin clumps and multiple condensed chromatin bodies (f, arrowed in g). Scale bar =5 μm in a, c, d and f; 1 μm in b, e and g
However, 30% of the ALS-IgGs evoked two populations of MN in mice. About 60% of the MN conformed to the pale stained/electron-lucent population described above. Approximately 40% of MN were characterised by a darkly stained basophilic vacuolated cytoplasm containing an intensely stained nucleus and enlarged dense nucleolus, and some MN were shrunken. Ultrastructurally, both nucleus and cytoplasm of these MN were abnormally electron-dense (Fig. 4d, e), and in some instances the nucleus was denser than the cytoplasm. While chromatin in some MN was diffuse, in others it formed accumulations in the nucleoplasm and immediately subjacent to the nuclear membrane (arrowed in Fig. 4g). The cytoplasm of all electron-dense MN contained highly distended swollen Golgi ER, and lysed mitochondria, and some were surrounded by an abnormally wide extracellular space indicative of shrinkage (Fig. 4d, f). These MN showed clear signs of neurodegeneration.
To provide a more detailed analysis, MN were examined in 0.5-μm ‘plastic’ sections and classified into groups distinguished by their relative levels of cytoplasmic and nuclear staining (pale, dark, very dark), the size and distribution of basophilic Nissl bodies (large-discrete, small-diffuse) and the absence or presence of vacuolation. Preliminary examinations identified five sub-types of neuronal appearance (A to E in Table 1, Fig. 5). Two hundred MN were studied in mice injected with control IgG and 400 MN in mice injected with ALS-IgG. Over 85% of MN in mice injected with control IgG had normal pale staining cytoplasm and nuclei, with large Nissl bodies and no vacuolation (group A), while 11% (group B) had normal levels of cytoplasmic and nuclear staining, but smaller, more diffuse Nissl bodies. Staining was stronger in about 4% of MN (groups C, D), but no intensely stained and highly vacuolated MN occurred (group E). By contrast 60% of MN of mice exposed to ALS IgG were classified within groups A and B on the basis of pale cytoplasm and nucleus with variable degrees of vacuolation. Approximately 40% were abnormally dark-stained with intensely stained nuclei, exhibited diffuse Nissl and extensive vacuolation (groups D and E).

Light microscopical examinations of MN in semi-thin sections exhibited a spectrum of characteristics that enabled them to be grouped and their relative proportions determined. The characteristics of each group (A to E) are tabulated, and the relative proportions of each are illustrated below. Data from individual experiments have been pooled to provide overall profiles
Analysis of organelle responses
To further characterise MN responding to ALS and control IgGs, in particular those undergoing necrosis and electron-dense type degeneration, alterations in the Golgi complex, mitochondrial and Nissl body structures were analysed in a random, but smaller sample of MN. We previously found these features to be sensitive indicators of injury in a number of situations, including ALS [32, 33].
In MN of non-injected mice, Golgi complexes comprised a stack of narrow, closely spaced ER cisternae. The average width of the ER lumens for each complex was determined on electronmicrographs by drawing a line perpendicular to the plane of the ER through the entire ER stack. The lumen width was measured for each ER lamella intersecting the line. Average lumen widths were calculated for each MN, for all MNs examined for each animal, and finally from these data the overall mean value was calculated for individual treatment groups. To investigate complex fragmentation, alterations in overall Golgi complex size, measured as cross-sectional area, and number (number/100 μm2 cytoplasm) were determined for each MN, for each animal and for each treatment group.
Mitochondrial size, expressed as cross-sectional area (μm2), and mitochondrial number, expressed as number/100 μm2 of cytoplasm, were determined on micrographs. The relative proportions of type 1, type 2 and type 3 Nissl bodies were calculated from counts made on micrographs.
Results are summarised in Table 2. Values given are the overall means (±SD) of the mean values calculated for each animal in a group. In non-injected mice (n=3 mice, 40 MN) Golgi lumens in individual MN ranged from 27.0 nm to 92.6 nm, and Golgi complex areas ranged between 0.13 and 2.31 μm2. There were approximately 3.70 complexes per 100 μm2 cytoplasm. Mitochondria had a narrow range of area, 0.15 μm2 to 0.25 μm2, and numbered about 53 per 100 μm2. Type 1 and type 2 structures dominated Nissl bodies, with only 10% possessing type 3 ultrastructure (Table 2, Fig. 6).

Comparative analysis of organelles in motoneurones (MN) of non-injected mice and mice injected with IgG from healthy controls, disease controls and ALS subjects. Following ALS-IgG treatment, MN were grouped according to whether they were electron-lucent or electron-dense. Golgi ER lumen width (a) and Golgi complex area (b) are significantly increased after ALS-IgG treatment, but the complex number is unchanged (c),particularly in electron-dense MN. Mitochondrial swelling (d) with loss of mitochondria (e) occurs in electron-dense MN, but not electron-lucent MN. Marked differences occur in Nissl body composition in MN exposed to ALS-IgG. Nissl body composition is compared in (f). Values given are the overall means ± SD of the means calculated for individual animals in each group. Significance points above bars denote control-ALS difference (Student’s t-test; **P=<0.01>0.005, ***P<0.0001). Significance points within bars denote disease control-ALS difference (Student’s t-test; **P=<0.01>0.005, ***P<0.0001)
Golgi complexes in 42 MN from mice injected with control-IgGs had a similar range of lumen widths to those in untreated mice (21.04–118.4 nm), and the non-injected-control IgG difference was not significant (unpaired Student’s t-test, P>0.1) (Table 2). Similarly, no significant differences were found for Golgi complex areas (t=0.649, P>0.1) or complex number (t=0.005, P>0.1). No significant differences occurred in mitochondrial areas (t=1.943, P>0.1) or number (t=0.522, P>0.1), and Nissl body composition was normal with 10.2% type 3 Nissl bodies. Application of a two-way ANOVA confirmed variation both within the sample and between samples was not significant. Despite slight Golgi ER distension seen in MN of mice treated with some disease control IgGs, which resulted in a small increase in individual complex area, the overall mean Golgi ER lumen width for the group (88.8±30.7 nm) was not significantly different from that in normal or control IgG injected animals (t=0.735, P>0.1; Fig. 4).
In contrast to the MN in control IgG-treated mice, vacuolated electron-lucent MN in ALS-IgG-treated mice exhibited significant Golgi ER distension (62 MN, 590 complexes, range 65.6–262 nm), which was significant when compared to lumens in MN of mice treated with control-IgG (t=2.51, P<0.01). A two-way ANOVA revealed that within sample variation was not significant (P=0.13) and further verified a significant control-ALS difference (F=22.9, P=0.003). While the overall range of Golgi complex area in MN of the ALS-IgG-treated mice was similar to that in control-IgG-treated animals (0.51–2.8 μm2), the mean area was larger due to greater numbers of larger complexes. The control-IgG-ALS-IgG difference in complex area was significant (Student’s t-test, P<0.01, two-way ANOVA, P<0.0001). There was no associated alteration in complex number (Student t-test, t=1.913, P>0.1; two-way ANOVA, P=0.16), implying ER distension was not accompanied by complex fragmentation. Mitochondria were swollen in the ALS-IgG group, possessed a greater upper limit of area (0.54 μm2) and were significantly bigger than those in control IgG-treated MN ( Student t-test, P<0.002, two-way ANOVA, P=0.005). However, numbers of mitochondria were not significantly different from those in control-IgG-treated MN (mean 49.8±13.5/100 μm2, difference t=0.596, P=0.57; Fig. 6). Ribosomal dispersion observed in micrographs was reflected in an approximately 20% reduction in type 1 Nissl bodies, with a concomitant increase in type 3 to 20%, confirming the loss of Nissl ER with polyribosomal proliferation (Table 2).
Further differences occurred between electron-lucent MN of mice treated with ALS-IgG undergoing necrosis and those exhibiting electron-dense degeneration. Golgi ER were significantly more distended when compared to the electron-lucent MN, with lumens ranging up to 580 nm, (t=4.03, P<0.0007). The Golgi complexes were significantly larger (up to 2.86 μm2, t=4.09, P=0.0003), but no more numerous (t=1.438, P>0.1). Mitochondrial swelling was greater in the electron-dense MN than in electron-lucent MN (P=0.076, see Table 2), but mitochondria were also significantly fewer (t=4.789, P<0.0001). Electron-dense degenerating MN had near normal numbers of type 1 Nissl bodies, but fewer type 2 Nissl bodies (Table 2).
Ca2+ histochemistry
Tissue fixation in an aldehyde-oxylate mixture followed by post-fixation with an osmium-pyroantimonate mixture results in electron-dense deposits in organelles, such as vesicles of presynaptic terminals and Golgi ER [35]. Application of energy dispersive X-ray spectroscopy (EDAX) showed these deposits have secondary emission energy (Kα1) values for pyroantimonate and Ca2+. We previously [32] employed the same technique to demonstrate increased frequency of Ca2+ containing organelles in MN of ALS-IgG-treated mice, and further verified a Kα1 value for Ca2+ (3.6 keV) to correlate with sites of electron-dense reaction product in Golgi ER, mitochondria and presynaptic terminals of IgG-treated MN. We used the technique in this study to compare Ca2+ in MN exhibiting different types of morphological response to control and ALS-IgGs. Results are given in Fig. 7. Values given are the overall means (±SD) of the mean values calculated for each animal in a group. In confirmation of our earlier study [32], significantly greater numbers of Ca2+ containing Golgi complexes and presynaptic terminals occurred in MN of animals injected with ALS-IgG relative to numbers in mice treated with control or disease control IgGs (Fig. 5), but differences in number of Ca2+ containing mitochondria and Nissl rER were not significantly different (P>0.1). These MN all exhibited electron-lucent nuclei and cytoplasm, vacuolation and distended Golgi ER, and a variable degree of mitochondrial damage. None exhibited electron-dense type degeneration. In contrast, MN from ALS-IgG-treated mice undergoing electron-dense neurodegeneration showed a significant further increase in the number of Ca2+-containing Golgi complexes and terminals, but also a significant abnormal increase in Ca2+-containing mitochondria and rER (Fig. 7).

Comparative proportions of Golgi complexes, mitochondria, Nissl rER and presynaptic boutons containing histochemical Ca2+ pyroantimonate reaction product in motoneurones (MN) of non-injected mice, and mice injected with control, disease-control and ALS-IgGs. MN following ALS-IgG treatment are grouped according to whether they are electron-lucent or electron-dense. Significantly more Golgi and boutons contained Ca2+-product after ALS-IgG injection, and electron-dense degenerating MN of these mice showed a greater degree of response than MN undergoing necrosis. Values given are the overall means ± SD of the means calculated for individual animals in each group. Significance points above bars denote control-ALS difference (Student’s t-test; **P=<0.01>0.005, ***P<0.0001). Significance points within bars denote disease control-ALS difference (Student’s t-test; **P=<0.01>0.005 ***P<0.0001)
In summary, mice injected with control and disease control IgGs exhibited a single population of MN with no or negligible cytopathology, but mice injected with ALS-IgG displayed two populations of MN, one with the morphological profile of necrosis and the other showing clear signs of degeneration.
Discussion
Using histological and ultrastructural studies, we have demonstrated that the passive transfer of ALS-IgG evokes a spectrum of neuronal abnormalities. including electron-lucent acute necrosis and electron-dense type degeneration. The morphological abnormalities are associated with an increase in the number of calcium-containing organelles. MN exhibiting electron-dense degeneration have ultrastructural features strongly resembling those defining the attritional phase prior to apoptosis [31], although Golgi enlargement and mitochondrial damage more closely correspond to paraptosis. Passive transfer of IgG from healthy individuals and disease control IgGs does not evoke significant cytopathology in motoneurones and does not promote an enhancement of intraneuronal calcium.
The major difference between this and previous investigations of ALS-IgG is the unequivocal in vivo morphological evidence of MN degeneration in mice injected with ALS-IgG, but not in those treated with control IgG. Previously, neuronal degeneration has only been reported in vitro [2, 18]. Published in vivo studies focus on the physiological effects of IgG on channel operation, transmitter release at the neuromuscular junction or Ca2+-related ultrastructural abnormalities in the muscle synapses of patients with ALS [34] or of ALS-IgG-treated mice [13, 14]. In the single study examining neurones after passive transfer of ALS IgG, no degenerative changes were found [13]. In vitro, neuronal death followed application of IgGs from six ALS patients to a VSC4.1 hybrid MN cell line, but not with IgGs from five neurological controls. [2]. Apoptotic-like features were identified morphologically by shrinkage and membrane blebbing, and biochemically by in situ end labelling of DNA and DNA laddering. In a second study [18], ALS-IgG induced death in 40–70% of cells in a hybrid MN cell line by a mechanism requiring extracellular Ca2+ and mediated by Ca2+-influx at neuronal-type N- or P-type, but not L-type channels. However, while IgG from patients with Lambert-Eaton-Myasthenic-syndrome (LEMS) was also found to be cytotoxic and bind to L-type VGCC, it reduced, and did not enhance, Ca2+ influx, indicating a different mode of toxicity from that of ALS-IgG [18]. IgG from patients with Guillain-Barré syndrome also bound L-type channels, but was not cytotoxic, implying that IgG binding to L-type channels does not necessarily result in neuronal toxicity. Other investigations found antibodies in ALS-sera, which reacted with non-identified epitopes on A172 glioblastoma cells [34]. Using dye exclusion techniques, significant increases in dying cultured glioblastoma cells exposed to both ALS IgG and IgG from Alzheimer’s patients have been reported [29], suggesting a wider link between autoimmunity and neuronal degeneration, rather than exclusivity with ALS. The targets of the IgGs in this study were also unidentified.
Direct comparison between the neuronal lesion in mice receiving purified IgG from ALS patients and the abnormalities assigned to motor neurones in ALS patients at this juncture is difficult for two reasons. Firstly, the cytopathology of ALS is based on studies of tissues taken post-mortem, which represent the end-stage of a progessive disease rather than the earliest stages of abnormality seen in these animal-based experiments. Secondly, the role of autoimmunity in ALS remains uncertain. Similarity between the IgG-evoked lesion and that seen in ALS is more likely if autoimmunity is a primary cause of ALS. A body of opinion suggests it may not be [5, 11, 40], in which case similarity is less likely. Moreover, further experiments of longer duration are required to elucidate the continuing evolution and time-course of the immune mediated lesion.
Regarding possible modes of toxicity, it has been postulated that ALS-IgG selectively targets the alpha-1 calcium channel subunit at the neuromuscular junction and motoneurone membrane, causing an abnormal influx of Ca2+ into the neurone, increase in transmitter release and enhancement of Ca2+ in organelles (reviewed in [4]). The alternative suggestion [27] of a non-immune effect attributed to serine proteases co-purifying with ALS-IgG can be excluded in our experiments since affinity purified IgG preparations were dialysed before injection into mice to remove proteases-like plasminogen. The finding of Ca2+ enhancement in Golgi ER and synaptic terminals in mice treated with ALS-IgG is consistent with calcium enhancement seen in mice carrying a mutated SOD1 gene [36], in kainate-induced excitotoxicity [1], as well as an IgG-mediated assault on Ca2+ channels [8, 13, 23]. While SOD1 toxicity and kainate excitotoxicity can be discounted in the current experiments, an exclusive assault on calcium channels needs further investigation, since the experiments were not designed to identify IgG targets. The possibility of multiple IgGs in patient sera that target a variety of epitopes, one of which is the VGCC, cannot be excluded.
Other antibodies reported in ALS sera include those directed towards neurofilaments [6, 9] and FAS [41]. Anti-neurofilament antibodies might be expected to compromise cytoskeletal structure and function rather than raise intraneuronal calcium, and any elevation would therefore be a secondary effect. The relationship between anti-neurofilament antibodies and intraneuronal calcium, if any, is unclear. Anti-FAS antibody binding to FAS leads to apoptosis via FAS activation of caspases and disturbances of intracellular Ca2+ [41]. Unfortunately, the ultrastructural effects of FAS activation have not been reported, precluding a comparison with the morphological changes in MN exposed to ALS-IgG. In vitro, ALS-IgG increases Ca2+ currents through P-channels of cerebellar Purkinje cells [23], leading to cell death [25]. In vivo, it is envisaged that IgG accesses MN where the blood nerve barrier is absent, principally at muscle (see review, [4]), although certain muscles appear resistant to IgG [26]. Binding of IgG to the VGCCs is considered to enhance Ca2+ influx into the neuromuscular junction. However, Fab fragments of IgG act on the external (extracellular) α1 subunit of the VGCC, but not on its internal (cytosolic) β subunit [24], precluding direct IgG action on Ca2+ channels from within the cytoplasm of the nerve terminal or MN. This implies that to access Ca2+ channels, IgG translocates across either the neuronal membrane or capillary epithelia to the extracellular space. However, the case for selective translocation of ALS-IgG, but not control IgG, across epithelia forming the blood-nerve barrier, or blood-brain barrier, remains unproven. Ca2+ influx subsequent to binding of ALS-IgG to the calcium channel followed by interaction between Ca2+ and synaptic vesicle associated proteins may explain the IgG-induced enhancement of transmitter release from the NMJ [28, 38]. How this leads to neuronal pathology remains to be explained. While endocytotic uptake of IgG into the NMJ followed by its retrograde axonal transport to the MN soma may account for the observed presence of human IgG in the mouse MN and the unusual presence of IgG in cortical and spinal MN of ALS patients [16], no intraneuronal target for IgG has been identified so far to explain a direct IgG-mediated assault from within the MN soma. Further studies are therefore needed to clarify these issues.
References
Adalbert R, Engelhardt JI, Siklos L (2002) DL-homocysteic acid application disrupts calcium homeostasis and induced degeneration of spinal motor neurons in vivo. Acta Neuropathol 103:428–436
Alexianu ME, Mohamed AH, Glenn Smith R, Colom LV, Appel SH (1994) Apoptotic cell death of hybrid motoneuron cell line induced by immunoglobulins from patients with amyotrophic lateral sclerosis. J Neurochem 63:2365–2368
Appel SH, Engelhardt JI, Garcia J, Stefani E (1991) Immunoglobulins from animal models of motor neuron disease and from human amyotrophic lateral sclerosis patients passively transfer physiological abnormalities to the neuromuscular junction. Proc Nat Acad Sci USA 88:647–651
Appel SH, Glenn-Smith R, Alexianu M, Siklos L, Engelhardt J, Colom LV, Stefani E (1996) Increased intracellular calcium triggered by immune mechanisms in amyotrophic lateral sclerosis. Clin Neurosci 3:368–374
Arsac C, Raymond C, Martin-Moutot N, Dargent B, Courand F, Pouget J, Seagar M (1996) Immunoassays fail to detect antibodies against neuronal calcium channels in amyotrophic lateral sclerosis serum. Ann Neurol 60:695–700
Bahmanyar S, Moreau-Dubois M-C, Brown P, Cathala A, Gajdusek DC (1983) Serum antibodies to neurofilament antigens in patients with neurological and other diseases and in healthy controls. J Neuroimmunol 5:191–196
Baxter RG, Martin JE, Pullen AH (2002) Reduced neuronal nitric oxide synthase (NOS1) antigen in sacral motor neurones in motor neurone disease. Acta Neuropathol 104:391–397
Colom LV, Alexianu ME, Mosier DR, Glenn-Smith R, Appel SH (1997) Amyotrophic lateral sclerosis immunoglobulins increase intracellular calcium in a motoneuron cell line. Exp Neurol 146:354–360
Couratier P, Yi FH, Preud’homme JL, Clavelou,P, White A, Sindou P, Vallat JM, Jauberteau MO (1998) Serum autoantibodies to neurofilament proteins in sporadic amyotrophic lateral sclerosis. J Neurol Sci 154:137–145
Delbono O, Garcia J, Appel SH, Stefani E (1991) Calcium current and charge movement of mammalian muscle: action of amyotrophic lateral sclerosis immunoglobulins. J Physiol (Lond) 444:723–744
Drachman DB, Fishman PS, Rothstein JD, Motomura M, Lang B, Vincent A, Mellits ED (1995) Amyotrophic lateral sclerosis—an autoimmune disease? In: Serratrice G, Munsat T (eds) Pathogenesis and therapy of amyotrophic lateral sclerosis. Adv Neurol 68:59–65
Engelhardt JI, Tajti J, Appel SH (1993) Lymphocytic infiltrates in the spinal cord in amyotrophic lateral sclerosis. Arch Neurol 50:30–36
Engelhardt JI, Siklos L, Komuves L, Glenn Smith R, Appel SH (1995) Antibodies to calcium channels from ALS patients passively transferred to mice selectively increase intracellular calcium and induce ultrastructural changes in motoneurons. Synapse 20:185–199
Engelhardt JI, Siklos L, Appel SH (1997) Altered calcium homeostasis and ultrastructure in motoneurones of mice caused by passively transferred anti-motoneuron IgG. J Neuropathol Exp Neurol 56:21–39
Fabian RH, Petrov G (1987) Intraneuronal IgG in the CNS: uptake by retrograde axonal transport. Neurology 37:1780–1784
Fratantoni SA, Dubrovsky AL, Uchitel OD (1996) Uptake of immunoglobulin G from amyotrophic lateral sclerosis patients by motor nerve terminals in mice. J Neurol Sci 137:97–102
Glenn-Smith R, Hamilton S, Hofmann F, Schneider T, Nastainczyk W, Birnbaumer L, Stefani E, Appel SH (1992) Serum antibodies to L-type calcium channels in patients with amyotrophic lateral sclerosis. N Eng J Med 327:1721–1728
Glenn-Smith R, Alexianu ME, Crawford G., Nyormoi O, Stefani E, Appel SH (1994) Cytotoxicity of immunoglobulins from amyotrophic lateral sclerosis patients on a hybrid motoneuron cell line. Proc Nat Acad Sci USA 91:3393–3397
Johnson IP, Pullen AH, Sears TA (1985) Target dependence of Nissl body ultrastructure in cat thoracic motoneurones. Neurosci Lett 61:201–205
Kawamata T, Akiyama H, Yamada T, McGeer PL (1992) Immunologic reactions in amyotrophic lateral sclerosis. Brain and spinal cord. Am J Pathol 140:691–707
Kimura F, Glenn-Smith R, Delbono O, Nyormoi O, Schneider T, Nastainczyk W, Hofmann F, Stefani E, Appel SH (1994) Amyotrophic lateral sclerosis patient antibodies label Ca2+ channel alpha1 subunit. Ann Neurol 35:164–171
Lindsey NJ, Jamadar K, O’Brien MR, Parkin SM, Mitchell JD (2000) Sera from patients with ALS contain antibodies reactive with the dihyropyridine binding site on L-type calcium channels and inhibit the growth of A172 cells (abstract). Proc Int Symp ALS/MND P80:105
Llinas R, Sugimori M, Chersky BD, Glenn-Smith R, Delbono, O, Stefani E, Appel SH (1993) IgG from amyotrophic lateral sclerosis patients increases current through P-type calcium channels in mammalian cerebellar Purkinje cells and in isolated channel protein in lipid bilayer. Proc Nat Acad Sci USA 90:11743–11747
Magnelli V, Sawada T, Delbono O, Glenn-Smith R, Appel SH, Stefani E (1993) The action of amyotrophic lateral sclerosis imunoglobulins on mammalian single skeletal muscle Ca2+ channels. J Physiol (Lond) 461:103–118
Mosier DR, Baldelli P, Delbono O, Smith RG, Alexianu ME, Appel SH, Stefani E (1995) Amyotrophic lateral sclerosis immunoglobulins increase Ca2+ currents in a motoneurone cell line. Ann Neurol 37:102–109
Mosier DR, Siklos L, Appel SH (2000) Resistance of extraocular motoneuron terminals to effects of amyotrophic lateral sclerosis sera. Neurology 54:252–255
Nyormoi O (1996) Proteolytic activity in amyotrophic lateral sclerosis IgG preparations. Ann Neurol 40:701–706
O’shaughnessy TJ, Yan H, Kim J, Middlekauf EH, Lee KW, Phillips LH, Kim J, Kim YI (1998) Amyotrophic lateral sclerosis: serum factors enhance spontaneous and evoked transmitter release at the neuromuscular junction. Muscle Nerve 21:81–90
Parkes AB, Rickards CR, Rees P, Scanlon MF (1995) Cytotoxic changes in A172 glioblastoma cells exposed to serum IgG from patients with motor neurone disease. J Neurol Sci 129 [Suppl]:136–137
Pestronk A, Adams RN, Cornblath D, Kuncl RW, Drachman DB, Clawson L (1989) Patterns of serum antibodies to GM1 and GD1a gangliosides in ALS. Ann Neurol 25:98–102
Portera-Cailliau C, Price DL, Martin LJ (1997) Non-NMDA and NMDA receptor mediated excitotoxic neuronal deaths in adult brain are morphologically distinct: further evidence for an apoptotic-necrosis contiuum. J Comp Neurol 378:88–104
Pullen AH, Humphreys P (2000) Ultrastructural analysis of spinal motoneurones from mice treated with IgG from ALS patients, healthy individuals, or disease controls. J Neurol Sci 180:35–45
Pullen AH, Martin JE (1995) Ultrastructural abnormalities with inclusions in Onuf’s nucleus in motor neuron disease (amyotrophic lateral sclerosis). Neuropathol Appl Neurobiol 21:327–340
Rickards CR, Parkes AB, Williams H, Ham J, Scanlon MF (1992) Antibodies against a human glioblastoma cell line in patients with chronic neurological disease. Euro J Neurosci 37:
Siklos L, Engelhardt J, Harati Y, Glenn-Smith R, Joo F, Appel SH (1996) Ultrastructural evidence for altered calcium in motor nerve terminals in amyotrophic lateral sclerosis. Ann Neurol 39:203–216
Siklos L, Engelhardt JI, Alexianu ME, Gurney ME, Siddique T, Appel SH (1998) Intracellular calcium parallels motoneuron degeneration in SOD1 mutant mice. J Neuropath Exp Neurol 57:571–587
Troost,D, Van Den Ord JJ, Vianney De Jong JMB (1990) Immunohistochemical characterisation of the inflammatory infiltrate in amyotrophic lateral sclerosis. Neuropath Appl Neurobiol 16:401–410
Uchitel OD, Appel SH, Crawford F, Sczcupak L (1988) Immunoglobulins from amyotrophic lateral sclerosis patients enhance spontaneous transmitter release from motor nerve terminals. Proc Nat Acad Sci USA 85:7371–7374
Uchitel OD, Scornik F, Protti DA, Fumberg CG, Alvarez V, Appel SH (1992) Long-term neuromuscular dysfunction produced by passive transfer of amyotrophic lateral sclerosis immunoglobulins. Neurology 42:2175–2180
Vincent A, Drachman DB (1996) Amyotrophic lateral sclerosis and antibodies to voltage-gated calcium channels—new doubts. Ann Neurol 40:691–692
Yi F H, Lautrette C, Vermot-Desroches C, Bordessoule D, Couratier P, Wijdenes J, Preud’homme JL, Jauberteau MO (2000) In vitro induction of neuronal apoptosis by anti-Fas antibody-containing sera from amyotrophic lateral sclerosis patients. J Neuroimmunol 109:211–220
World Federation of Neurology Research Group of Neuromuscular Diseases (1994) El Escorial World Federation of Neurology criteria for diagnosis of amyotrophic lateral sclerosis. J Neurol Sci 124 [Suppl]:S96–107
Acknowledgements
We thank the patients and other individuals who kindly donated samples for this study. We are indebted to Action Research for their generous support of this project. We also thank Heather Brooks for her help in reading the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Pullen, A.H., Demestre, M., Howard, R.S. et al. Passive transfer of purified IgG from patients with amyotrophic lateral sclerosis to mice results in degeneration of motor neurons accompanied by Ca2+ enhancement. Acta Neuropathol 107, 35–46 (2004). https://doi.org/10.1007/s00401-003-0777-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-003-0777-z