
Abstract
Argyrophilic grain disease (AGD) is characterized by the occurrence of argyrophilic grains and coiled bodies in brain tissue, mainly in limbic areas located in the temporal lobe. Recent biochemical data have shown that inclusions in AGD consist of aggregates of pathological microtubule-associated tau protein isoforms of 64/69 kDa. We report here a study on two AGD patients, belonging to a series of demented patients affected by several tauopathies, prospectively followed until death. In both patients, clinical, neuropathological and biochemical investigations clearly demonstrated AGD. Diffuse tau pathology was shown by Gallyas’ silver stain, tau immunohistochemistry and tau protein variant biochemical analysis, not only in temporal lobes but also in all cortical and subcortical areas that were assessed. Primary motor, primary sensory, and associative brain cortices were involved, as well as brain stem, but not cerebellum. We suggest that “diffuse” AGD might be a subgroup of AGD, the specific profile of which is different from that of “limbic” AGD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In 1987, Braak and co-workers reported a series of eight patients with a non-Alzheimer, late onset dementia [3], characterized by the occurrence of argyrophilic grains (ArGs) on light microscopy of the brain tissue, and therefore referred to as argyrophilic grain dementia (AGD). ArGs are neuronal inclusions stained by silver dyes, the most sensitive stain being Gallyas [4]. The diagnosis of dementia with ArGs is based on widespread occurrence of minute, spindle or comma-shaped, argyrophilic, tau-immunoreactive structures distinct from neuropil threads and predominantly located in the hippocampus and related limbic areas [18]. On silver stains, ArGs are associated with coiled bodies (CBs), which are oligodendroglial inclusions mostly located within deep cortical layers and white matter [4, 15]. Under light microscopy, ArGs are often associated with neurofibrillary tangles (NFTs) and neuritic plaques and are therefore difficult to distinguish from other neurofibrillary lesions in people with Alzheimer changes over stage IV, according to the Braak staging system [5, 21].
Ultrastructurally, ArGs occur in the dendritic compartment of the affected neurons, they consist of 9- to 18-nm straight filaments [17], or 25-nm rods, but without paired helical filaments [15]. Phosphorylated tau proteins are the major component of ArGs [34]. Aggregated tau proteins are also the main component of the neurofibrillary lesions found in numerous neurodegenerative disorders, termed tauopathies. A biochemical classification of these neurological disorders, based on the electrophoretic profile of tau protein isoforms extracted from autopsy-derived brain tissue samples, distinguishes four subclasses. Class I includes Alzheimer’s disease and several rare tauopathies, and class II includes progressive supranuclear palsy (PSP) and corticobasal degeneration (CBD); Pick’s disease and myotonic dystrophy have the typical profiles for class III and IV subtypes, respectively [9, 10].
Very recently, the biochemical profile of tau proteins in AGD has been clearly linked to PSP and CBD tau proteins [32]. Tau aggregates in AGD are four-repeat tau isoforms of 64 and 69 kDa [37]. Together, neuropathological and biochemical studies showed that tau pathology in AGD is clearly restricted to the limbic areas. Furthermore, all studies reveal the gap between well-documented neuropathological findings and the description of related clinical signs.
A series of ninety consecutive brains obtained at autopsy from demented patients, prospectively followed for several years in the Memory Unit in the University Hospital of Lille, was assessed neuropathologically and biochemically for tau and related proteins after death [12]. Two cases that fitted the neuropathological criteria for AGD were characterized by the occurrence of widespread cortical and subcortical ArGs and CBs. We suggest that diffuse AGD might be a new subclass of four-repeat tauopathies, distinct from PSP, CBD and limbic AGD.
Patients and methods
Patients
Case 1
A 70-year-old woman was referred to the memory unit of University hospital of Lille. The mini mental state examination (MMSE) was 19/30, and the dementia rating scale (DRS) was 72 [23]. The patient had aphasia, apraxia, agnosia, and visuospatial disability. This woman had neither hypertensive nor stroke disease in her medical personal history. Her parents were not consanguineous and there was no positive familial history of dementia or Parkinsonism. Her family noticed behavioral disturbances 8 years before the cognitive impairment. The very first signs were obsessions and changes of eating habits, followed by apathy. She never developed self-neglect or flat affect. At first examination, her behavioral frontotemporal scale (BFS) was 2/4 [19]. Although she displayed carbohydrate craving, there was no hyperphagia, alcoholism, loss of sexual inhibition or hypermetamorphosis. She did not have visual hallucinations. There was no parkinsonism, gaze palsy or motor impairment at neurological examination. The biological data were normal. On CT scan, both anterior temporal lobes were atrophic, and MRI showed only little foci of leukoaraiosis. The HmPAO-Tc single photon emission computed tomography showed decreased perfusion of the anterior frontal areas, most severe in the left hemisphere. She progressively worsened and died when 76.
Case 2
This woman was first referred to the memory unit when she was 74 years old. She did not have any noticeable medical history and there was no family history of dementia or parkinsonism. She had apraxia, and visuospatial disability. There was only very mild cognitive impairment: MMSE was 25/30 and DRS was 113/144. Her husband first noticed cognitive slowing and slight memory impairment when she was 72 years old. Cooking recipes seemed to be simplified, and her husband had to plan her daily life. She had got aggressive and irritable. She had partially lost inhibition, and had carbohydrate craving for 2 years. Voluntary movements had got slow. She did not have any noticeable personal medical history. Amongst the whole family there was only one demented aunt.
The initial examination disclosed apathy, anosodiaphoria, loss of inhibition, and logorrhoea. There was no aphasia and prosopagnosia. There was very slight parkinsonism, including micrography and loss of expression, but she was receiving neuroleptics. There were small hyperintense foci in the hemispheric white substance and striatum on MRI, suggesting an ischemic disorder. The whole brain was mildly atrophic, except the hippocampus that had shrunk. All serum levels were normal. When neuroleptic treatment was stopped, the behavioral impairment decreased.
Later on, the cognitive impairment progressively worsened. She developed rest tremor and an akineto-rigid parkinsonian syndrome. There were neither fluctuation nor hallucinations at any time. Although she received Tacrine and Doneprezil, the cognition never improved, MMS was 12/30 3 years after the first examination, at age 77.
MRI was performed shortly before death, showing diffuse atrophy of the brain cortex, calcium deposits in the diencephalon, and a lacuna in the right thalamus. She died at age 79.
Neuropathological methods
Post-mortem delay at autopsy was 7 and 8 h, respectively. For both patients, one cerebral hemisphere was immediately frozen and kept at –80°C until biochemical investigations could be performed. Most of the other hemisphere was fixed in buffered formalin for 6 weeks before sampling and embedding in paraffin. Sections from formalin-fixed, paraffin-embedded brain tissue were stained with hematoxylin-eosin, Klüver-Barrera method for myelo-architecture and Bodian and Gallyas stains for neurofibrillary changes. Immunohistochemistry was performed using the AD2 anti-tau antibody (kindly given by Dr Mourton Gilles, Montpellier, France; monoclonal antibody raised against the phosphorylated Ser 396/Ser 404 residues on tau), and commercially available antibodies directed against ubiquitin, α-synuclein, neurofilaments, glial fibrillary acidic protein (GFAP), synaptophysin, chromogranin A, vimentin, and β amyloid. Labeling was achieved with a two-step procedure amplified by a peroxidase-coupled, streptavidin-biotin complex, either in a Ventana ES200 automate, using 3,3’-diaminobenzidine as chromogen, or manually with amino ethyl carbazole. All primary antibodies were used following standard retrieval methods (autoclaving in citrate buffer for all antibodies and formic acid for amyloid labeling). Transmissible spongiform encephalopathy was excluded by a negative immunolabeling of prion protein following guanidine thiocyanate and proteinase K incubation, performed on samples from the frontal cortex, caudate nucleus and cerebellum.
Quantification of tau and amyloid precursor protein pathologies
Brain tissue samples from the right hemisphere of both patients were processed for tau and insoluble Aβ40 and -42 deposits, as previously described [12, 37]. Available frozen samples taken from the left hemisphere of patient 1 before fixation were also processed to assess the symmetry of the tau protein biochemical profile.
Immunoblot was performed from samples obtained in the hippocampus, amygdala and temporal cortex [Brodmann’s area (BA) 38 and BA20), associative and primary motor frontal cortices (BA8, BA10 and BA4), cingulate (BA24), parietal angular gyrus (BA39), secondary visual occipital cortex (BA18), substantia nigra, red nucleus, and caudate nucleus.
DNA mutational analysis
Genomic DNA was extracted from brain tissue with the Nucleospin Tissue Kit (Macherey-Nagel, Germany). Exons 9, 10, 11, 12 with flanking intronic sequences were amplified from genomic DNA of the two patients. To amplify the 3’ intronic region of exon 10, the reverse primer was chosen 97 nucleotides downstream the splice site. The primers used are as followed: exon 9: forward primer 5’-CTTTTCTGACCCCACCCACT-3’ and reverse primer 5’-CCTACCCTTCCAGGCACAG-3’ ; exon 10: forward primer 5’-ATGTCACTCATCGAAAGTGGAG-3’ and reverse primer 5’-ATCCTGAGAGCCCAAGAAGG-3’; exon 11: forward primer 5’-GTCTTCCTCTCTCTCTGCCTTTC-3’ and reverse primer 5’-CCTTCTGAAGTCTGGAGCAGTT-3’; exon 12: forward primer 5’-TGTTAAGTCCACAGAACCACAGA-3’ and reverse primer 5’-TGATTAATGCCCATATACCCAAG-3’. PCR products were subsequently sequenced on an automated CEQ8000 DNA sequencer (Beckman, USA) using the CEQ Dye terminator Cycle Sequencing “Quick Start” kit (Beckman) in both forward and reverse directions.
Tau H1 haplotype analysis
Tau H1 haplotype analysis was determined on DNA samples by amplifying a 490 bp sequence in intron 9 using: forward primer 5’-ACCCAAAGCACACTGTTTCC-3’ and: reverse primer 5’-CACACAGCCAGGTTTGAGAA-3’. Amplified products were then analyzed on 5% polyacrylamide gels: alleles with haplotype H1 give a 490-bp fragment and alleles with haplotype H2, characterized by a 238-bp deletion give a 252-bp fragment [1].
Results
Case 1
Neuropathological findings
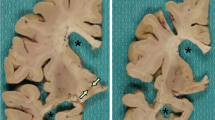
There were non-occlusive, fibro-lipidic plaques on the medial and posterior cerebral arteries. All gyri, including the pre and post central ones, were diffusely and mildly shrunken, but the temporal lobe was deeply atrophied (Fig. 1A). When cut coronally, the lateral ventricle appeared widened, mostly the temporal horn, which had a cystic appearance (Fig. 1B). The ambient gyrus and the Ammon’s horn were flattened.

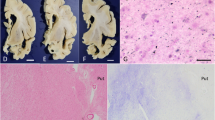
A Posterior view of coronal sections of the left hemisphere from patient 1 shows thinning of the hippocampus, entorhinal cortex and adjacent isocortical areas of the temporal lobe (arrows), with relative sparing of the frontal and parietal lobes. B The atrophy of the amygdala and prehippocampal gyrus is extreme. The ventricle shows pseudo-cystic dilatation. Under light microscopy, normal ependymal cells line the lumen without any true cyst formation. Only scarce and small lacunae are seen on macroscopic examination (B, asterisks). C Internal view of the right hemisphere from patient 2, showing the selective atrophy of the anterior part of the rhinal cortex and Ammon’s horn (arrows). D As in patient 1, the hippocampal region is severely atrophied. A few small lacunae are seen, for example in the white matter from the frontal lobe (asterisk)
Under light microscopy, there was a diffuse neuron loss extending to all cortical areas, almost complete in the medial portion of the temporal lobe. There were rare ballooned neurons in the nuclei of the amygdala, as well as in the ambient gyrus and the hippocampus. Atrophy was associated with mild spongiosis in the temporal lobe and astrocytic gliosis, as shown by GFAP staining (data not shown). Gallyas stain displayed prominent ArGs and CBs in the amygdala, the prehippocampal and hippocampal areas, and the temporal isocortex (Fig. 2A). Gallyas and tau immunohistochemistry also showed such inclusions, but to a lesser extent, in the insula and the cingulate gyrus, the frontal and parietal associative cortices, the motor areas of the precentral gyrus, the primary and secondary visual cortices of the occipital lobe, and the pallidum, striatum and capsules (Fig. 3, left part). In addition, the basal ganglia and subthalamic nucleus contained scarce ArGs and CBs, but the substantia nigra, cerebellum, pontine base and medulla were devoid of staining (data not shown). Rare neuropil threads and NFTs occurred mainly in the pyramidal cell layer of the hippocampus and the associative temporal areas. There were no Pick bodies. The neurons were not rarefied in the substantia nigra; neither Gallyas nor anti-tau displayed PSP-type NFTs in the mesencephalon, tufted astrocytes or glial plaques.

A, B Patient 1, C, D patient 2. Gallyas stain displays argyrophilic grains (A) and coiled bodies (C) in the hippocampus of both patients. On hematoxylin and eosin staining, mild cribriform changes are seen in the claustrum and capsules from patient 1 (B), and in the striatum in patient 2 (D). Bar in C represents: A, C 25 μm; B 100 μm, D 1,000 μm

Tau immunolabeling shows argyrophilic grains and coiled bodies in the hippocampus and amygdala from both patients, and in isocortical areas, i.e. primary (frontal motor, BA4), secondary (visual, BA18), and polymodal associative cortices (BA39 and BA40). Insula, capsules, pallidum, striatum and basal nuclei are also involved (BA Brodmann’s area). Bars 10 μm
Small scarce lacunae were seen in the deep white matter, as well as in the claustrum and capsules (Figs. 1B, 2B). Anti-amyloid disclosed focal deposits in the BA18, BA10, BA20, BA39, and hippocampus, with rare vascular deposits in the pia matter. Very rare Lewy bodies (LBs) were seen in the substantia nigra and the dorsal nucleus of the vagus nerve (data not shown). Anti-synuclein and anti-ubiquitin did not show cortical LBs or Lewy-related neurites. The cerebellum was normal.
Biochemical analysis of tau and Aβ
Western blot analysis displayed the characteristic tau protein doublet of 64/69 kDa in BA4, 9, 10, 17, 39, 44, 47 from the right cerebral hemisphere (Fig. 4). Tau aggregates were also quantified in BA10, 17, and 39 from the left hemisphere, showing no asymmetry. In the hippocampus, BA20 and 38, a pathological tau component at 60 kDa was detected and the 64 kDa component appeared doubled, giving rise to an Alzheimer-type triplet, suggesting stage 5/10 grade Alzheimer-type tauopathy superimposed on AGD pathology (according to the recently proposed CEBAD grading of tau aggregates in Alzheimer’s disease, corresponding to a mild involvement [11]). Very mild β amyloid deposits were observed on homogenates from the same brain samples (using the 21F12 antibody from Athena Neurosciences, USA).

Topographical distribution of pathological tau in brain. The same amount of brain tissue protein (50 µg) was loaded onto 8–16% SDS-PAGE followed by Western blotting with the monoclonal antibody AD2. The brain regions analyzed are indicated according to Brodmann’s nomenclature. The substantia nigra (LN), red nucleus (RN) and caudate nucleus (CN) were analyzed for AGD case 2. The relative amount of pathological tau isoforms are indicated under each lane and expressed as the percentage of the signal in an Alzheimer’s brain homogenate (Alz). Case 1: A Brain tissue homogenate of the right hemisphere; B the amounts of pathological tau are shown for the brain tissue homogenates obtained from the same brain region in the right (R) and the left (L) hemisphere. Arrowheads indicate pathological tau components (AGD argyrophilic grain disease)
Case 2
Neuropathological findings
After formalin fixation, the right cerebral and cerebellar hemispheres weighted 450 and 75 g, respectively. There were atherotic plaques in the mid cerebral arteries. The temporal lobe was very atrophic (Fig. 1C). When cut, the lateral ventricle was seen to be widened; the white matter was also atrophic. The lobar white matter, as well as the thalamic nuclei and striatum displayed some lacunas (Fig. 1D).
The main histological changes were neuron loss, predominantly in the temporal lobe, mild spongiform change and gliosis in the same areas on the standard stains. Gallyas staining showed ArGs and CBs in the hippocampus, amygdala, associative temporal areas, and to a lesser extent in the frontal and parietal associative cortices, and primary motor frontal cortex (Fig. 2C). Myelin was pale on Luxol staining, many striatal vessels were calcified, perivascular spaces were dilated, and some hemosiderinic deposits were seen (Fig. 2D).
Tau immunolabeling displayed huge number of ArGs and CBs in the amygdala, hippocampus, and temporal isocortex. Moreover, they were seen on samples from the associative parietal and occipital, and from the primary motor and sensory areas as well (Fig. 3, right part). Insular cortex and deep gray nuclei (subthalamic nucleus, pallidum, striatum, basal nucleus) also contained ArGs and CBs. There were no NFTs, neuropil threads, Pick bodies, LBs or Lewy-related neurites in tau and synuclein labeling. Anti-amyloid displayed only few tiny, focal deposits in the occipital lobe. Amyloid labeling was negative in other isocortical areas, in the hippocampus, and in the subarachnoidal and subpial vessels. The cerebellum, medulla and pontine base were spared on each technique.
Biochemical analysis
Pathological tau isoforms with the characteristic doublet of 64 and 69 kDa were found in the homogenates from BA4, 10, 17, 20, 24, 44 from the hippocampus, caudate nucleus, substantia nigra, and red nucleus. The 60-kDa pathological tau component was not seen in any cortical area, including the entorhinal and hippocampal areas. The amounts of the detected doublet ranged from 5% to 40% of the signal observed in an Alzheimer’s brain extract. The most affected region was the hippocampus followed by cingulate gyrus, the substantia nigra, the temporal and the frontal cortex. Moreover, there was no trace of β amyloid aggregates in neocortical areas.
Tau gene studies
Tau gene mutations are the cause of the inherited disease frontotemporal dementia with Parkinsonism linked to chromosome 17 (FTDP-17T). To date, 29 mutations have been described in the tau gene, and 25 of them are located in exons 9, exon 10 and its flanking intron and exons 11 and 12. We screened all these exons for mutations, and did not find any new or known mutation for the two patients.
The tau gene is also characterized by numerous polymorphisms. Some polymorphisms show strong linkage disequilibrium with each other and define two haplotypes (H1 and H2) that extend over the entire tau gene [1]. The observed tau genotype was H1/H1 for both patients.
Discussion
Here we report two cases of diffuse AGD, which were remarkable for the diffuse extent of ArGs and CBs, as well as the characteristic tau doublet in all cortical and subcortical brain areas.
Clinically, these AGD patients fit with the spectrum that has been previously reported: behavioral disturbances had preceded memory and cognitive impairment [38]. Both patients exhibited moderate memory disturbances, and dietary changes (carbohydrate craving), and patient 2 showed loss of inhibition, reminiscent of the Klüver-Bucy syndrome [16]. The early behavior disturbances were not typical of Alzheimer’s disease [24]. Moreover, the features did not fit the clinical criteria for frontotemporal dementia: there was neither physical neglect nor affective changes (BFS <3) [8, 25, 26].
On macroscopic examination, the brain was not obviously atrophied, except the temporal lobe; of interest was the huge enlargement of the temporal horn of the lateral ventricle in patient 1, which is unusual [36]. Ventricle enlargement was not related to ischemic, parasitic or an arachnoidal cyst in this patient, and could be considered as a final atrophy of the amygdala.
All neuropathological techniques, especially silver stains and tau immunochemistry were consistent with the final diagnosis of AGD, and neither clinical nor pathological data fulfilled consensus criteria of CBD or PSP [13, 14]. In our patients, the behavioral and cognitive impairment, which were very mild, could not be attributed to ischemia or Alzheimer changes. Mild ischemic changes have been noticed previously in AGD [20]. NFTs restricted to limbic and rhinal areas as well as amyloid deposits (seen in up to 73% of AGD cases) are very frequent findings [5, 35]. Autopsy examinations shows that AGD occurs in patients over 65; pure disease is seen in a limited number of cases (4 out of 15 in a recently reported series) and can be associated not only with Alzheimer and ischemic changes, but also with LBs, hippocampal sclerosis or PSP lesions [31]. In addition to ArGs, anti-tau antibodies may demonstrate diffusely immunoreactive astrocytes in the amygdala and the entorhinal cortex of AGD patients without tufted astrocytes or glial plaques [2]. Our patients lacked such glial lesions. Moreover, the tau isoform aggregates in brain tissue homogenates displayed a 64/69-kDa doublet, common to PSP and CBD, as recently described in brain tissue samples from AGD patients [32, 37]. Although Alzheimer-type tau aggregates of 60/64/69 kDa were found in the limbic areas from patient 1, as well as discrete amyloid deposition, such extension is not correlated with significant cognitive decline in large populations [11].
The most striking feature in both patients was the diffusion of the tau pathology all over the cerebral cortex, very distant from the limbic temporal region, in brain areas that have been considered to be spared in AGD. These diffuse inclusions were the only causative lesions of dementia in these two cases.
Previous studies have established that the density of ArGs is particularly high in the pre-α (II) and pre-β (III) layers of the entorhinal cortex, perirhinal cortex, CA1 subfield of the hippocampus, basolateral nuclei of the amygdala, anterior insula, lateral tuberal nuclei of the hypothalamus and layer III of the mediobasal temporal isocortex and gyrus rectus [4]. The ambient gyrus is constantly and severely involved in AGD [28], but not specifically [6, 7]. The periaqueductal gray matter, anterior raphe, locus ceruleus and dorsal nucleus of the vagus nerve were shown to be involved in several reports. In the absence of any other neurodegenerative disorder, the posterior CA1 is spared in non-demented ArG-bearing patients and involved in demented ArG-bearing patients [33]. However, previous reports have mentioned occasional and isolated tau pathology in the precentral gyrus [17] and insula [16], but the BA7, 40, 17 and 19 were thought to be devoid of ArGs in AGD patients [17, 21, 33].
So far, the 64/69-kDa tau isoforms have not been found outside the limbic areas [32, 37]. In particular, we have never found tau-doublet aggregates in the frontal, parietal and occipital cortices of patients suffering from classical AGD [37].
Conversely, the large extent of tau pathology in AGD might be a further feature common to PSP and CBD, as well as a clue to a pathological continuum between PSP, CBD, limbic AGD and diffuse AGD. Many features are shared by all four-repeat tauopathies. For instance, as in PSP and CBD, the subthalamic nuclei are selectively involved in AGD patients, displaying tau aggregates [22]. The H1/H1 genotype is more frequent in PSP/CBD patients than in controls or other tauopathies. H1/H1 may be more frequent in AGD patients than in controls, although recent studies have failed to establish a statistically significant difference [32]. ArGs are preferentially observed in PSP and CBD cases, although they are also seen in Pick disease [21], and in multiple system atrophy, which is a synucleinopathy [39]. Unlike that seen in CBD patients, in whom tau pathology is more or less asymmetrical on immunohistochemical and biochemical examination, the semiquantified 64/69-kDa tau aggregates from patient 1 were similar in right and left hemispheres.
Cases of multisystem tauopathy with dementia have been reported. Sporadic late-onset cases were similar to CBD, whereas early-onset cases occurred in individuals suffering from FTDP-17T [27]. Of particular interest is a familial multisystem tauopathy (with a substitution at position 3 in the intron flanking the exon 10, referred to as E10+3 mutation) found in a large pedigree spanning seven generations [29, 30]. Although the pattern of degeneration and tau aggregation in our cases was reminiscent of this family, all known intronic mutations were excluded in our patients, as were most of the exonic mutations leading to FTDP-17T, which lie between exons 9 and 12 of the tau gene.
Conclusion
In our prospective and multidisciplinary study comprising more than 90 cases, two cases of non-familial “diffuse” AGD were observed. One of them was a “pure” form of AGD, as demonstrated at the clinical (absence of PSP or CBD features), neuropathological (occurrence of ArGs and CBs, in the absence of tangles and amyloid deposits) and biochemical (characteristic tau doublet in all brain areas, without Aβ42 or -40 aggregates) levels. The other case displayed associated changes of Alzheimer type, confined to the hippocampal area. Tau gene mutations in exons 9–12 and intronic mutations in the 3’ sequence flanking exon 10 were excluded.
Clinical features were reminiscent of “limbic” AGD and showed frontotemporal impairment. From our study, these two cases demonstrate that diffuse AGD is a significant subgroup of AGD and probably not rare.
Together, we characterize here a dementing disease that has a very specific profile and that enlarges the already important group of tau-related dementias.
References
Baker M, Litvan I, Houlden H, Adamson J, Dickson D, Perez-Tur J, Hardy J, Lynch T, Bigio E, Hutton M (1999) Association of an extended haplotype in the tau gene with progressive supranuclear palsy. Hum Mol Genet 8:711–715
Botez G, Probst A, Ipsen S, Tolnay M (1999) Astrocytes expressing hyperphosphorylated tau protein without glial fibrillary tangles in argyrophilic grain disease. Acta Neuropathol 98:251–256
Braak H, Braak E (1987) Argyrophilic grains: characteristic pathology of cerebral cortex in cases of adult onset dementia without Alzheimer changes. Neurosci Lett 76:124–127
Braak H, Braak E (1989) Cortical and subcortical argyrophilic grains characterise a disease associated with adult onset dementia. Neuropathol Appl Neurobiol 15:13–26
Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82:239–259
Braak H, Braak E (1992) The human entorhinal cortex: normal morphology and lamina-specific pathology in various diseases. Neurosci Res 15:6–31
Braak H, Del Trecidi K, Bohl J, Bratzke H, Braak E (2000) Pathological changes in the parahippocampal region in select non-Alzheimer’s dementias. Ann N Y Acad Sci 911:221–239
Brun A, Englund B, Gustafson L (1994) Clinical and neuropathological criteria for frontotemporal dementia. J Neurol Neurosurg Psychiatry 57:416–418
Buée L, Bussière T, Buée-Scherrer V, Delacourte A, Hof PR (2000) Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Rev 33:95–130
Delacourte A, Buée L (2000) Tau pathology: a marker of neurodegenerative disorders. Curr Opin Neurol 13:371–376
Delacourte A, David JP, Sergeant N, Buée L, Wattez A, Vermersch P, Ghozali F, Fallet-Bianco C, Pasquier F, Lebert F, Petit H, Di Menza C (1999) The biochemical pathway of neurofibrillary degeneration in aging and Alzheimer’s disease. Neurology 52:1158–1165
Delacourte A, Sergeant N, Champain D, Wattez A, Maurage CA, Lebert F, Pasquier F, David JP (2002) Nonoverlapping but synergistic tau and APP pathologies in sporadic Alzheimer’s disease. Neurology 59:398–407
Dickson DW, Bergeron C, Chin SS, Duyckaerts C, Horoupian D, Ikeda K, Jellinger K, Lantos PL, Lippa CF, Mirra SS, Tabaton M, Vonsattel JP, Wakabayashi K, Litvan I (2002) Office of rare diseases neuropathologic criteria for corticobasal degeneration. J Neuropathol Exp Neurol 61:935–946
Hauw JJ, Daniel SE, Dickson D, Horoupian DS, Jellinger K, Lantos PL, McKee A, Tabaton M, Litvan I (1994) Preliminary NINDS neuropathologic criteria for Steele-Richardson-Olszewski syndrome (progressive supranuclear palsy). Neurology 44:2015–2019
Ikeda K, Akiyama H, Kondo H, Haga C (1995) A study of dementia with argyrophilic grains. Possible cytoskeletal abnormality in dendrospinal portion of neurons and oligodendroglia. Acta Neuropathol 89:409–414
Ikeda K, Arai T, Matsushita M, Tsuchiya K, Miyazaki H (2000) Clinical aspects of argyrophilic grain disease. Clin Neuropathol 19:278–284
Itagaki S, McGeer PL, Akiyama H, Beattie BL, Walker DG, Moore GR, McGeer EG (1989) A case of adult onset dementia with argyrophilic grains. Ann Neurol 26:685–689
Jellinger KA (1998) Dementia with grains (argyrophilic grain disease). Brain Pathol 8:377–386
Lebert F, Pasquier F, Souliez L, Petit H (1998) Frontotemporal behavioural scale. Alzheimer Dis Assoc Disord 12:335–339
Markesbery WR (1998) Dementia with argyrophilic grains. In: Markesbery WR (ed) Neuropathology of dementing disorders. Arnold, London, pp 14–15
Martinez-Lage P, Munoz DG (1997) Prevalence and disease associations of argyrophilic grains of Braak. J Neuropathol Exp Neurol 56:157–164
Mattila P, Togo T, Dickson DW (2002) The subthalamic nucleus has neurofibrillary tangles in argyrophilic grain disease and advanced Alzheimer’s disease. Neurosci Lett 320:81–85
Mattis S (1976) Mental status examination for organic mental syndrome in the elderly patients. In: Bellak L and Karasu TB (eds) Geriatric psychiatry: a handbook for psychiatrists and primary care physicians. Grune & Stratton, New York, pp 77–101
McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM (1984) Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s disease. Neurology 34:939–944
McKhann GM, Albert MS, Grossman M, Miller B, Dickson D, Trojanowski JQ (2001) Clinical and pathological diagnosis of frontotemporal dementia. Report on the Work Group on Frontotemporal Dementia and Pick’s Disease. Neurology 58:1803–1809
Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, Freedman M, Kertesz A, Robert PH, Albert M, Boone K, Miller BL, Cummings J, Benson DF (1998) Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 51:1546–1554
Reed LA, Wszolek ZK, Hutton M (2001) Phenotypic correlations in FTDP-17. Neurobiol Aging 22:89–107
Saito Y, Nakahara K, Yamanouchi H, Murayama S (2002) Severe involvement of ambient gyrus in dementia with grains. J Neuropathol Exp Neurol 61:789–796
Spillantini MG, Goedert M, Crowther RA, Murrell JR, Farlow MR, Ghetti B (1997) Familial multiple system tauopathy with presenile dementia: a disease with abundant neuronal and glial tau filaments. Proc Natl Acad Sci USA 94:4113–4118
Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B (1998) Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci USA 95:7737–7741
Togo T, Cookson N, Dickson DW (2002) Argyrophilic grain disease: neuropathology, frequency in a dementia brain bank and lack of relationship with apolipoprotein E. Brain Pathol 12:45–52
Togo T, Sahara N, Yen S-H, Cookson N, Ishizawa T, Hutton M, Silva R de, Lees A, Dickson DW (2002) Argyrophilic grain disease is a sporadic 4-repeat tauopathy. J Neuropathol Exp Neurol 61:547–556
Tolnay M, Schwietert M, Monsch AU, Staehelin HB, Langui D, Probst A (1997) Argyrophilic grain disease: distribution of grains in patients with and without dementia. Acta Neuropathol 12:45–52
Tolnay M, Spillantini MG, Goedert M, Ulrich J, Langui D, Probst A (1997) Argyrophilic grain disease: widespread hyperphosphorylation of tau protein in limbic neurons. Acta Neuropathol 93:477–484
Tolnay M, Calhoun M, Pham HC, Egensperger R, Probst A (1999) Low amyloid (Aβ) plaque load and relative predominance of diffuse plaques distinguish argyrophilic grain disease from Alzheimer’s disease. Neuropathol Appl Neurobiol 25:295–305
Tolnay M, Monsch AU, Probst A (2001) Argyrophilic grain disease. A frequent dementing disorder in aged patients. Adv Exp Med Biol 487:39–58
Tolnay M, Sergeant N, Ghestem A, Chalbot S, Vos RAI de, Jansen Steur ENH, Probst A, Delacourte A (2002) Argyrophilic grain disease and Alzheimer’s disease are distinguished by their different distribution of tau protein isoforms. Acta Neuropathol 104:425–434
Tolnay M, Ghebremedhin E, Probst A, Braak H (2003) Tauopathies. Argyrophilic grain disease. In: Dickson DW (ed) Neurodegeneration. The molecular pathology of dementias and movement disorders. ISN Neuropath Press, Basel, pp 132–136
Wakabayashi K, Kawachi I, Toyoshima Y, Takahashi H (1999) Occurence of argyrophilic grains in multiple system atrophy: histopathological examination of 26 autopsy cases. No To Shinkei 51:433–437
Acknowledgements
This study was supported by the Ministère de l’Enseignement National, de la Recherche et de la Technologie (MENRT/EA 2691) and INSERM. The authors wish to thank Monique Henneron, Véronique Dumetz, Annick Wattez and Véronique Vervaeck for technical assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Maurage, CA., Sergeant, N., Schraen-Maschke, S. et al. Diffuse form of argyrophilic grain disease: a new variant of four-repeat tauopathy different from limbic argyrophilic grain disease. Acta Neuropathol 106, 575–583 (2003). https://doi.org/10.1007/s00401-003-0762-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-003-0762-6