
Abstract
We report the synthesis of semi-interpenetrating polymer network (semi-IPN) hydrogel composed of carboxymethyl cellulose (CMC), linear polyvinylpyrrolidone (PVP), acrylic acid (AA) and α-cyclodextrin (CD) by free-radical solution polymerization in presence of hydrogen peroxide as the initiator and its characterization by FTIR, SEM and Rheology. The formulated hydrogel is thermoreversible, and its rheological parameters like gelation temperature, gelation time, elastic modulus and stress were studied as a function of CD concentrations. Swelling ability of the hydrogel decreases while as the drug encapsulation and loading capacities enhance with increase in CD wt%. The partitioning of ibuprofen between regions of different polarity within the hydrogel was studied by spectrofluorimetry. The ability of hydrogel with 0.04 wt% of CD to release ibuprofen spanned over the time period of 2 h and was modelled in light of various kinetic models.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Hydrogels are three-dimensional polymer network structures obtained from a class of synthetic and/or natural polymers that immobilises a large amount of water besides maintaining integrity [1]. Multi-component hydrogels are produced by semi-interpenetrating polymer network (Semi-IPN) technology involving cross-linking a variety of polymers/monomers via free radical polymerisation, and there are practically little chances of residual monomer/polymer which can act as a potential toxic agent [2]. Multi-component polymer-based hydrogels exhibit surprising properties superior to constituent polymers [2, 3]. The commercially available multi-component hydrogels are comprised of petroleum-based synthetic polymers having high production cost in addition to being less eco-friendly limiting their application domain [4]. Multi-component hydrogels derived from natural polymers and biocompatible synthetic polymers containing biodegradable additives are gaining interest in a number of new research areas due to the unique physicochemical, biological, industrial, and environmental advantages [4]. Synthetic polymers have many attractive qualities like they can be processed into a variety of shapes and have much better mechanical and thermal properties than most of naturally occurring polymers. These demanding properties increase the flexibility of their usage in biomedical field. Therefore, the current thrust of research is to develop multi-component polymeric hydrogels based on the blends of natural and synthetic polymers which are biocompatible and at the same time possess good thermal and mechanical properties [3]. Moreover, many of the synthesized multi-component hydrogels are stimuli responsive where a minor change in its chemical structure in response to external stimuli is symbiotically augmented to bring about impressive transformations in macroscopic properties of the material [3, 4]. The resemblance of physical properties of stimuli responsive multi-component hydrogels to human tissues, minimal toxicity and excellent tissue compatibility has rendered them potential candidates as biomaterials like in soft contact lenses [5], chemical sensors [6], controlled drug delivery materials [7], tissue engineering [8], wound dressing [9], dental applications, injectable polymers, implants, ophthalmic applications and diagnostics [10–12]. There are two important desirable but antagonistic properties of multi-component hydrogels viz self-healing ability and mechanical strength which are needed to be taken into consideration during their synthesis. The autonomous damage-repair and resulting healing involves energy dissipation mechanism created by reversible bonds which prevent the fracture of the molecular backbone, while the mechanical strength is necessary for their use in stress bearing applications. The efficiency of self-healing increases by decreasing the lifetime of dynamic cross-links due to the favourable chain diffusion across fractured surfaces of hydrogels, but the hydrogels with short-living cross-links become weak at experimental time scales [13]. So, the design of multi-component hydrogels with damage-repair ability and a good mechanical strength is crucially important in many existing and potential applications fields [14, 15].
Acrylic acid (AA)-based hydrogels mediate H-bonding across rupture, and a strong self-healing is reported in such hydrogels [13]. The chemically cross-linked AA-based hydrogels show mechanical hysteresis for a range of deformation which is less pronounced in salt solutions, as the existence of ionic associations due to electrostatic interactions seems to be responsible for the energy dissipation under strain [13]. In the current study, 65 % of acrylic acid, as mentioned in the experimental section, was neutralized so that hydrogel with balanced damage-repair property and mechanical strength is obtained. AA is also one of the popular and extensively used chemical cross-linking agent for carboxymethylcellulose (CMC) [16–18]. The esterification reaction of CMC with AA in a solution of NaOH results in the formation of diether cross-links between the hydroxyl groups of CMC and AA [16]. Besides the formation of chemical bonds, there is possibility of H bonding, electrostatic and other physical interactions which may balance the cohesive solid-like properties and diffusive transport liquid-like properties of the synthesized hydrogel. Hydrogels based on CMC have been, in particular, the subject of intense investigation due to their ability to display enhanced swelling capacity and undergo external stimuli-induced phase transitions making the synthesized hydrogels extremely important carriers for controlled drug release [19]. In order to change the gelling properties (solubility and swellability), rheology and release properties of a typical CMC hydrogel, we tried to induce porosity into its structure by its simultaneous cross-linking with non-ionic polyvinyl pyrollidine (PVP) to formulate a macroporous hybrid hydrogel. Owing to their unique swelling properties, macroporous hybrid hydrogels have been found to be attractive entities for more specific pharmaceutical and biomedical applications [18]. Hydrogels in general and macroporous hydrogels in particular suffer from weak mechanical strength in their wet or hydrated state, so attempts have been made to enhance their mechanical properties, and hydrocolloid-containing hydrogels in particular have been found to be most effective [20–22]. Alternatively, various concentrations of hydrocolloid can be used to manipulate the mechanical properties of hydrogel. Therefore, the hydrogel prepared from the blend of CMC, PVP and AA with cyclodextrin (CD) as a hydrocolloid could be interesting to study for its mechanical properties, encapsulation and release ability. The unique swelling capacity of CMC and PVP, increased adhesive ability of AA [23] and increased solubilization capability towards drugs due to CD could be simultaneously envisaged in such multi-component polymeric network.
Therefore, in this work, we report the synthesis of macroporous CMC-based hydrogel by the free-radical copolymerization and semi-IPN technology. The structure and morphologies of the semi-IPN hydrogels were characterized by Fourier transformation infrared (FTIR) spectroscopy, scanning electron microscopy (SEM) and rheology. The mechanical properties of the synthesized hydrogel were studied as a function of CD concentration. Further, the synthesized hydrogel was explored for its encapsulation capacity, partitioning and release behaviour of the poorly water-soluble drug ibuprofen (IBU). IBU is an important water insoluble non-steroidal anti-inflammatory drug [24] and has been studied extensively with respect to its encapsulation and delivery [25, 26]. The experimental results of this study will provide insights into the structure-property relationship of the hydrogel at a range of CD concentrations in order to assess its potential for use in biomedicine.
Experimental section
Materials
Ibuprofen (IBU) was obtained from Himedia laboratories (India >98 %). CMC, AA and PVP were Aldrich products. H2O2 used was of analytical grade. All the chemicals were used as received, and the solutions were prepared in triple distilled water.
Preparation of hydrogel
For the preparation of hydrogel, the procedure of Wang et al. was followed [16]. CMC (5 wt%) and PVP (5 wt%) dissolved in three-necked round-bottom flask (100-mL capacity) containing 30 mL of water fitted with a reflux condenser, a nitrogen line and a thermometer were continuously stirred on a magnetic stirrer to form a sticky solution. The polymeric solution was heated to 60 °C on an oil bath and purged with N2 for 30 min to remove the dissolved oxygen. For generation of radicals, 5 mL of H2O2 (50 mM) was added drop-wise under continuous stirring, and the solution was kept at 60 °C for 10 min. A 7.2 g of acrylic acid (AA) was neutralized using 7.6 mL of 8.8 mol L−1 NaOH solution to reach a total neutralization degree of 65 %, and then 0.01 wt% of CD was dissolved in the partially neutralized acrylic acid (NaAA) under magnetic stirring. The polymeric solution in the flask was cooled to 50 °C, and then mixture of NaA and CD was added. The temperature was then slowly raised to 70 °C and kept for 3 h to complete polymerization. The reaction mixture was continuously purged with nitrogen throughout the reaction period. The same procedure was repeated to prepare a series of solution with a range of CD concentrations (0.01 to 0.1 wt%).
Characterization of hydrogels by IR spectroscopy
The structural properties of the hydrogels were examined using a Perkin-Elmer Spectrum Two FT-IR spectrophotometer fitted with a Perkin-Elmer Universal ATR sampling accessory. Spectra were recorded in 4000–400 cm−1 range using KBr pellets, acquiring 64 scans with a resolution of 1 cm−1.
SEM
The morphology of synthesized gels was characterized by Hitachi S-3000H Scanning Electron Microscope equipped with a digital camera. Samples of the xerogels were prepared by placing a small aliquot of hydrogel on carbon tape placed over sample stub, coated with gold in a Hitachi Fine Coat Ion Sputter.
Rheology
Steady and dynamic rheological experiments were performed on Anton Paar MCR-102 Rheometer equipped with a peltier temperature control system with an accuracy of ±0.01 °C. Cone-plate geometry (diameter of 25 mm with cone angle of 1.998°) was used for the measurements. The experiments were carried out in duplicate to ensure reproducibility. To prevent dehydration during rheological measurements, a thin layer of silicone oil was placed on the periphery surface of the hydrogel held between the plates. The test methods employed were oscillatory temperature, time, stress, and frequency sweeps at a range of CD concentrations (0.01–0.1 %). The thermal behaviour of hydrogel was studied by temperature sweep. The hydrogels were subjected to a temperature ramp in the range of 0–80 °C with a heating rate of 2 °C/min, and in order to check its thermo-reversibility, it was cooled from 80 to 0 °C at the same rate. To ensure that the experimental conditions did not interfere with the gelation process, the temperature ramp was performed at a low oscillation frequency (1 Hz) and a small deformation (0.01). In linear viscoelastic region (LVR), the evolution and dynamics of elastic storage modulus ‘G′’ and the viscous loss modulus ‘G″’ with time were monitored, and time sweep was performed at constant shear frequency (1 Hz) to observe in situ gelation behaviour and gelation time of the hydrogels. Stress sweep was performed on the hydrogels at a constant frequency of 1 Hz to determine the linear viscoelastic region by shearing them until its structural breakdown occurs. The hydrogels were also subjected to a frequency sweep in the linear viscoelastic region (stress of 20 Pa) to study the viscoelastic performance over a wide range of frequencies (0.01 to 100 Hz).
Swelling studies
The weight of the completely dry hydrogel sample (W d ) was measured, and the sample was immersed in phosphate buffer medium of physiological pH (7.4) at room temperature (25 ± 0.5 °C). The surface of the hydrogel was dried with a filter paper and the weight of the swollen hydrogel was measured (Ws). The swelling ratio (SR) of polymer networks was then determined gravimetrically using Anamed weighing balance (Mumbai, India) having an accuracy of ±0.0001 g by applying the following relation:
The values of SR reported are average of three independent measurements.
Drug encapsulation and in vitro release
The ibuprofen (IBU) encapsulation capacity of hydrogel and its release kinetics from the hydrogel were determined spectrophotometrically with a Schimazdu Spectrophotometer (model UV-1650PC). Briefly, 400 mg of IBU (L 1 ) was dissolved in 20-mL vial containing 5 mL of ethanol, and then, 200 mg (L) of hydrogel was added. It is pertinent to mention that the hydrogel was insoluble in ethanol. The vials were sealed with screw caps and then agitated for a period of 5 h on a magnetic stirrer at a temperature of (37 ± 0.5 °C) using magnetic Teflon pieces previously placed in the vials. The solution was filtered, and the precipitate (drug-loaded gel) was washed with 2 mL of ethanol to remove the drug adsorbed on the surface of hydrogel. The concentration of IBU in the filtrate ethanol solution (L 2 ) was determined by measuring absorbance at 235 nm [25] using a predetermined calibration curve. The IBU encapsulation efficiency (EC) and drug loading capacity (LC) [25] of the hydrogel was determined as:
The drug release test was performed by adding the drug-loaded hydrogel to a beaker containing 100 mL of pH 7.4 phosphate buffer sealed with parafilm as release medium. The solution was continuously stirred at 37 ± 0.5 °C. Aliquots of release medium were withdrawn at specific time intervals to measure the drug concentration, and equal volumes of fresh buffer were immediately added to the medium to maintain a constant volume. The drug release profile was obtained by plotting a curve of M t /M against t, where M t is the amount of drug released at time t, and M is the amount of drug released once the equilibrium state is reached.
Fluorescence measurements
The fluorescence emission spectra of IBU in hydrogel and different solvents were obtained on Schimazdu Spectrofluorimeter (Model RF-5301) operating in the steady-state mode at 37 ± 0.1 °C. Measurements were made in quartz cuvette using a 3-mm excitation/3-mm emission slit width. The excitation wavelength used to excite IBU was 228 nm. Fluorescence emission spectra were recorded over the range of 250–400 nm.
Results and discussion
Hydrogel synthesis
The hydrogel was synthesized in aqueous solution by the method described in Experimental section which involves generation of macro-radicals followed by chain propagation and finally cross-linking resulting in the formation of three-dimensional gel network (Scheme 1). Briefly, the initiator H2O2 decomposes upon heating to produce hydroxyl radicals which possibly abstract hydrogen radical from CMC to form macro-radical chains. The radical chain reacts synchronously and most probably with the amide groups of PVP (being the reactive site) resulting in further chain propagation, and PVP might also get interpenetrated with the growing network through hydrogen-bonding interaction in accordance with the findings of Wang et al. [16]. They proposed that the macro-radical chain of sodium alginate combines with amide group of PVP to propagate the growing chain in addition to hydrogen bonding. Further, the active radical sites of the different growing polymer radical chains (CMC/PVP) may combine with vinyl group of the NaAA and hydroxyl groups of CD to proceed with chain propagation and get cross-linked to form three-dimensional polymeric hydrogel networks. The proposed mechanism is depicted in Scheme 1.

One possible proposed reaction mechanism for the formation of hydrogel
Structural characterization
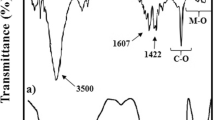
The IR spectra of NaCMC, PVP, AA, CD, hydrogel and drug-loaded hydrogel are shown in Fig. 1a. NaCMC shows bands at 3452 cm−1 due to O–H stretching vibrations, 2920 cm−1 due to C–H stretching vibrations, 1607/1424 cm−1 due to the asymmetric/symmetric stretching of the carboxylate group and 1157/1034 cm−1 due to C–O–C stretching vibrations in accordance with earlier reports [27]. PVP shows the band at 2955 cm−1 due to –CH stretching, 1659 cm−1 due to –C = O stretching, 1438 cm−1 due to –CH3 scissoring and 1285 cm−1 due to the tertiary amine –C–N stretching in accordance with the earlier reports [1]. In the case of CD, O–H stretching band is observed at 3412 cm−1, while C–H stretching band appears at 2928 cm−1. The appearance of band at 1639 cm−1 is due to O–H bending and at 1034 cm−1 due to C–O–C stretching [28]. Acrylic acid shows the band at 3443 cm−1 due to O–H stretching, 1730 cm−1 due to C = O stretching, 1639 cm−1 due to C = C stretching, while at 1413 cm−1 due to CH2 bending [29]. After formation of hydrogel by all these components, we observed weakening of some bands like the bands corresponding to C = C of AA and the C = O of the amide group in PVP, as these functional groups participate in hydrogel formation as shown in Scheme 1. Further, there occurs shifting of already existing bands like one of the very important band in AA corresponding to carbonyl group shifts to lower wave number (from 1730 to 1701 cm−1) as the resonance which occurs in AA is absent in the proposed product hydrogel. In addition, some strong absorption bands corresponding to the functionalities like peroxo linkage (918 cm−1 due to O–O symmetric stretching, 993 cm−1 due to O–O asynchronous vibration) [30] and ether groups (1296, 1235 and 1127 cm−1) [31] which were not present in constituent polymers but are being formed during the course of reaction are present in the proposed product hydrogel (Fig. 1b). These experimental observations support the formation of hydrogel as per proposed mechanism given in Scheme 1. Comparison of IR spectra of hydrogel and IBU-loaded hydrogel clearly shows that IBU is associated with hydrogel through non-covalent interactions as the IR spectrum of hydrogel and drug-loaded hydrogel is the same and only the reshuffling of some characteristic peaks occurs like bands corresponding to C = O shifts from 1701 to 1693 cm−1 and those of hydroxyl groups shift from 3445 to 3433 cm−1 due to hydrogen bonding between the corresponding hydroxyl groups of the hydrogel and carboxyl group of IBU molecules.

FTIR spectra of a CMC, PVP, AA, hydrogel and drug-loaded hydrogel, and b hydrogel at lower wavenumbers
Morphological analyses
SEM micrographs of synthesized hydrogel reveal that the surface of gel is dense and smooth except for some light cavities (Fig. 2). This is in contrast with the hydrogels of CMC, Chitosan, PVP, etc. reported in other studies [28, 32, 33] having rougher surfaces. The smoothness and experimentally observed lesser swelling capacity of the synthesized hydrogel could be attributed to extensive cross-linking which renders the hydrogel hydrophobic, as the roughness and swelling ability is directly related with the hydrophilicity of the hydrogel surface [31]. In a densely cross-linked hydrogel network, hydrophobic interaction prevail, chances of H-bonding with the solvent are reduced and is relatively smoother with less swelling ability.

SEM micrographs of hydrogel
Rheological measurements
Oscillatory temperature sweep
A representative plot of the temperature sweep of the hydrogel with 0 wt% CD is given in Fig. 3. At lower temperatures, loss modulus (G″) is more than that of elastic storage modulus (G′) indicating the polymeric network is in liquid state. Both of the moduli (G′ and G″) decrease slightly with increase in temperature up to ca. 60 °C probably due to breaking of H bonds and then increase abruptly due to growing hydrophobic interactions at higher temperatures. However, G′ increases more rapidly than G″ resulting in crossover at about 62 °C which also represents the gelation temperature where the sol-gel transition occurs. Beyond this temperature, the polymeric network is in gel-phase at all higher temperatures (G′ > G″). The hydrogel has tendency to revert back to the sol state on cooling at around 42 °C (cooling curve, Fig. 3) and reveals a certain degree of thermo-reversibility being important from fundamental and industrial point of view. The hydrogels prepared are inverse thermoresponsive, and their behaviour is a result of delicate balance between H bonding and hydrophobic interactions which depends on temperature. The gelation temperature of the hydrogels decreases with increase in CD concentration recorded both during heating as well as cooling as shown in inset of Fig. 3. The gelation temperature is, therefore, adjustable at a desired value by manipulating the CD wt% in polymeric network to make the gel workable in broad range of temperature. The gelation mechanism may involve a range of molecular interactions like electrostatic repulsion between the like charged CMC and AA chains, hydrogen bonding and polar interactions between the carboxyl, carbonyl and hydroxyl groups of CMC, PVP, AA and CD. However, the prime factors responsible for gelation are hydrophobic and steric interactions between the residual carboxyl groups and reduced solubility at higher temperatures. Further, the increased degree of cross-linking of the polymeric chains at higher concentrations of CD seems to play a pivotal role in bringing about the gelation within the practically important temperature range. Cross-linking results in the formation of high molecular weight three-dimensional polymer networks with less provision for polar interactions thus decreasing its solubility even at lower temperatures and leading to low temperature gelation. Since the gelation temperature is sensitive to CD wt%, all subsequent rheological tests were performed over a range of CD concentrations at a constant temperature of 37 °C.

Sol-gel transition of the hydrogel without CD during heating and cooling cycle between 0 and 100 °C (inset shows the variation of transition temperature with CD wt%)
Oscillatory time sweep
The time sweep profiles of G′ and G″ at 37 °C over a range of CD concentrations for the semi-IPN hydrogel are given in Fig. 4. For the hydrogels without CD and at lower concentrations of CD up to 0.01 wt%, viscous properties dominate as G″ has larger values than G′ throughout the time sweep, which is also expected since the applied temperature is lower than the gelation temperature of these samples. When the CD concentration is increased to 0.02 wt%, both the moduli increase with time indicating the solution prepares to turn into a gel-like state due to the increase in cross-linking density at the experimental temperature (Fig. 4) which is much lower than gelation temperature of these samples. These results imply that the gelation has dependence on time as well so that samples having higher gelation temperature can even undergo gelification over time at lower temperatures which is in agreement with earlier studies reported for different gels [34]. As the concentration of CD is further increased up to 0.03 wt% having gelation temperature of about 42 °C, the rate of increase of G′ is higher than G″, so the elastic properties start to dominate resulting in cross-over of G′ and G″ at about 1050 s referred to as gelation time. Therefore, a delayed hydrogel is obtained at 37 °C for the sample with 0.03 wt% CD. The gelation time is reduced to around 307 s when the concentration of CD is raised up to 0.035 wt%. Further reduction in gelation time to 30 s is observed for the sample with 0.04 wt% of CD (Fig. 4) representing a very significant and expected reduction as the working temperature lies above the gelation temperature of the sample. These results show that in the CD concentration range of 0.03 to 0.04 wt%, gelation time varies inversely with the CD concentration as the cross-linking density increases with increase in CD wt% at a constant temperature. Hydrogels with longer gelation time pose serious concerns like in drug delivery; it leads to drug diffusion [35], while in tissue engineering, heterogeneous cell distribution within matrix is observed [36]. Therefore, designing gels with optimal gelation temperature and gelation time prove highly beneficial for their practical applicability. In this context, the gelation time and the gelation temperature of the synthesized hydrogel with 0.04 wt% CD seem to be ideal for in vivo applications. The synthesised semi-IPN hydrogel remains in gel state at or above 0.05 wt% of CD throughout the time sweep (G′ > G″) as shown in Fig. 4. The results obtained are expected as the experimental temperature is much higher than the gelation temperature of these samples, no sol-gel transition is observed. Since for practical purposes, the solution is injected into the body and allowed to gellify in situ; hence, the hydrogel with 0.04 wt% of CD could prove to be an appropriate system for drug delivery and other pharmaceutical applications.

Dynamics of G′ and G″ with time at a range of CD concentrations
Oscillatory stress sweep
The stress sweep for all cross-linked hydrogels and the polymeric network without CD was carried out at 37 °C and are presented in Fig. 5. At low shear stress, there appears the linear viscoelastic region (LVR) in which G′ is almost independent of the applied shear stress, but at higher shear stress, structural breakdown occurs. The applied stress and the resulting strain remains in phase in the LVR region, and beyond it, the elastic modulus abruptly decreases due to the imposition of large deformations resulting in structural breakdown. As evident from the curve (Fig. 5), G′ and critical shear stress (stress at which polymeric system begins to show nonlinear viscoelastic behaviour) increase with increase in the percentage concentration of CD. Since G ′ = n e RT 37 [37], where n e is the number of cross-links in the network. Therefore, with increase in the concentration of CD, the proportion of number of effective intermolecular cross-links formed in the hydrogel network increases causing G′ to increase. Also, an increase in cross-linking density increases the tensile strength of the hydrogel with consequent extension in the LVR region.

Variation of G′ with applied stress for a range of CD concentrations
Oscillatory frequency sweep
Frequency sweep experiments are very important for attaining the information regarding the stability of three-dimensional hydrogel networks [38]. The dynamics of G′ and G″ for the studied semi-IPN hydrogels with frequency at 37 °C are presented in Fig. 6 which shows that the gelation depends upon applied frequency in accordance with earlier studies on different hydrogels [34]. Some very important results were obtained from the frequency sweep tests. For the semi-IPN without CD, the polymer network is in sol-phase throughout the sweep (G′ < G″), and although both the moduli increased with increase in frequency, no gelification occurred in response to change in frequency (Fig. 6). Further, no crossover was observed in the experimentally investigated frequency range. When 0.01 wt% of CD is added to the polymeric network, the rate of increase of G′ is more than that of G″ resulting in crossover at around frequency of 27.5 Hz (Fig. 6). Since crossover frequency (ω c) is related to the relaxation time (τ R) as

Evolution of elastic modulus as a function of the applied frequency for hydrogel with varying CD concentrations
The relaxation time for the resulting hydrogel with 0.01 wt% CD comes out to be 3.6 × 10−2 s which is too small indicating poor viscoelasticity [38] and more liquid-like nature of the polymeric hydrogel (Fig. 6) [39, 40]. Increasing the concentration of CD up to 0.02 wt% increases the relaxation time up to 1.8 × 10−1 s confirming elasticity of the polymer network increases with increase in CD wt% (Fig. 6). With further increase in CD wt% from 0.03 to 0.04 %, the relaxation time continuously increases from 3.98 × 10−1 to 8.77 × 10−1 s indicating that the solution changes from semi-dilute polymeric solutions to entangle melts with more desirable viscoelasticity [39, 40]. When the concentration of CD is increased beyond 0.04 %, we observe that the polymeric network is throughout in gel form (G′ > G″), and both the moduli appears to be essentially independent of a wide range of frequency (Fig. 6). This indicates that the viscoelastic relaxation of the polymeric network occurs in low frequency region inferring that reorganisation time required by network to reach equilibrium and form well defined network is quite large. Since, it is difficult to introduce hydrogels with high elasticity directly into the body without surgical intervention which is undesirable for routine clinical use, it is need of the hour to develop in situ gellable hydrogel formulations. Therefore, the polymeric network with 0.04 % could prove to be perfect hydrogel with ideal relaxation time much longer than polymeric network systems with less significant viscoelasticity and much smaller than those with rigid solid-like behaviour.
All the hydrogels with CD percentage concentration greater than 0.01 % presented an increase in G′ at higher frequencies. For the samples with lesser CD wt%, we observed a rapid increase in G′ at lower frequencies when compared to the samples with more amounts of CD concentrations. The length of the flexible polymer chain and the nature of imposed mechanical motions determine the viscoelastic response of a polymer network [41–43]. The polymeric systems with low concentration of CD have less cross-linked polymeric networks. Hence, the polymer chain segments between cross-links are longer and unable to rearrange in the time scale of the imposed motion, and therefore, these stiffen up and assume a more solid-like behaviour characterized by a sharp increase in G′. The polymeric system with higher CD concentration requires higher applied frequencies to obtain a similar response, leading to a gradual rise in G′ at higher frequencies.
Swelling degree measurements
The swelling ability of the synthesized hydrogels is much lower than other reported hydrogels of CMC and AA [16–18], so it is expected that these hydrogels show less water absorption as well as less absorption capability particularly toward hydrophilic drugs. The high water solubility of pure hydrogels of CMC and AA limits their use as a drug delivery system, as these may dissolve in the buccal mucous membrane before the desired duration for the drug to permeate across the membrane [44, 45]. Further, the hydrogels based on pure CMC and AA have little applicability for hydrophobic drugs, and their poor mechanical strength is a cause of concern [16–18]. The hydrogels presented here have appreciable mechanical strength and less water solubility and hence could be efficiently used for encapsulation of hydrophobic drugs. The swelling degree of the samples with a range of CD concentrations is presented in Fig. 7. In the case of polymeric network without CD, the SR is about 166 % of the initial weight of hydrogel which decrease with increase in CD concentration and is about 117 % for the sample with 0.1 wt% CD. CD possesses hydrophobic cavities resulting in increase in the hydrophobicity of polymeric network and hence decreases degree of hydration. Also, CD causes the gelation of the polymeric network by cross-linking the polymeric chains, so the exposed area potent for polar interactions with water is reduced. The two phenomena intensify with increase in CD concentration, so at higher wt% of CD due to added hydrophobicity and extensive cross-linking, majority of the polar groups (OH, COOH) of the polymeric network are engaged in bonding both leading to a decrease in the quantity of water absorbed at equilibrium.

Variation of swelling ratio (SR %) for a range of CD concentrations
Drug encapsulation capacity and release kinetics of hydrogels towards IBU
The drug encapsulated within the hydrogel gets stabilized by hydrogen bonding between carboxyl groups of drug molecules and hydroxyl groups of the polymer network, polar interactions of the drug molecules with the ionized groups of the polymer chain and hydrophobic interaction between the respective hydrophobic moieties of the drug and hydrogel. Table 1 shows the data of drug encapsulation capacity (EC) and drug loading capacity (LC) of the studied hydrogels. Since cross-linking between the polymeric chains is unfavourable for hydrogen bonding and polar interactions between the drug molecules and polymer network, so, the polymer network without CD has also an appreciable encapsulation capacity. EC and LC of the hydrogels increase first with increasing the CD concentration that reaches maximum at 0.03 wt% and then remains almost constant with further increase in CD concentration. Two antagonistic phenomena seem to interplay with increase in CD concentration, (a) increase in the number of hydrophobic cavities within the hydrogel which supports the drug encapsulation and (b) increase in the potency of polymer-polymer interactions by cross-linking with CD which decreases the drug-polymer interactions. The hydrogels with lower CD concentrations provide less hydrophobic cavities for drug encapsulation, but at the same time, the lesser cross-linking is favourable for drug-polymer interactions. Since the drug is non-polar [29], the extended hydrophobic cavities with increase in CD concentration seem to have an edge explaining increase of EC and LC with increase in CD concentration. At higher CD concentrations, although there is more room for hydrophobic interactions but the hydrogel is extensively cross-linked and the unbalanced charges are satisfied, thus, a more rigid solid-like structure is obtained which has less appeal for the drug explaining the constancy of EC and LC at higher CD percentage concentrations.
For drug release kinetics, the samples with CD concentration equal to 0.04 wt% were investigated, and burst release was observed. The release profile of IBU from the hydrogel is shown in Fig. 8a. Several models were applied to the experimental data to represent the drug dissolution profile. Zero-order kinetics model is applicable to the pharmaceutical dosage forms that does not disaggregate and releases the drug slowly so that no equilibrium conditions are obtained. According to zero order kinetics [46]:
w t is the amount of drug in the pharmaceutical dosage form at time ‘t’, w o is the initial amount of drug in the pharmaceutical dosage form, K is zero order rate constant and K 0 is the apparent dissolution rate constant or zero order release constant. A regression coefficient (r 2) of 0.90 along with a value of K 0 = 0.00199 min−1 was obtained when zero-order kinetics model was applied for this study. A faster release rate was observed for the first 10 min during which 58 % of the IBU, which was not tightly bound by the polymer chains, was released. However, the subsequent slower release behaviour was probably due to the IBU molecules that were tightly cross-linked with the polymer chains via intermolecular forces, including hydrophobic interaction, electrostatic attraction and hydrogen bonding.

a Ibuprofen release profiles of the hydrogel samples measured in phosphate-buffered solution (pH 7.4) at 37 °C (inset Higuchi model fit to the experimental data), florescence spectra of IBU in b various solvents, c in different concentrations of hydrogel and in d variation of K d with weight of the hydrogel
First-order kinetic model usually gives good results for water-soluble drugs encapsulated in porous matrix where the release of the drug is proportional to the amount of drug remaining in the interior of matrix. According to the model [46]
where Q t is the amount of drug released in time ‘t’, Q 0 is the initial amount of drug in the solution and K 1 is the first-order release constant. This model shows poor applicability to the studied system with r 2 of 0.87 and the value of K 1 = 0.00283 min−1 as IBU is non-polar and almost insoluble in water.
Higuchi model [47] describes drug release as a diffusion process based on the Ficks law and is used to describe the drug dissolution from several types of modified release pharmaceutical dosage forms. As per the model:
where K H is the Higuchi dissolution constant and F t is the amount of drug released in time t per unit area. A good correlation was obtained between Higuchi model and experimental data with r 2 = 0.95 and the value of K H = 0.0011 mg mm−1 min−3/2. Since Higuchi model is used to describe the drug dissolution from the hydrogels [47], it also shows fair applicability domain in the present study (Fig. 8a, inset). The reason being that many of the hypotheses which form the basis of the model are being satisfied like the initial drug concentration of the drug in the polymeric hydrogel is much higher than its solubility in the releases medium, drug particles are smaller than delivery system thickness, hydrogel dissolution is negligible in the release medium and drug diffusivity is constant. However, some discrepancies may be due to some over simplified assumptions of the model like the drug diffusion takes place only in one dimension and also the hydrogel swelling is not negligible in present study, so the diffusion rate increases when the polymeric network becomes loose and filled with water.
Partitioning of IBU
The polarity of the surrounding solvent medium affects the absorption and emission of the fluorophores. The fluorescence spectra of IBU in a number of solvents with different polarities are given in Fig. 8b. The change in polarity around the fluorophore corresponds to the shift in the emission wavelength maximum (Δλ max) which can be quantified experimentally. IBU shows negative solvatochromism which reflects a decrease of the ground-state dipole moment of the drug upon excitation. The increase in electronic transition energy upon increasing solvent polarity results from the solute solvent interactions. The main reason for the negative solvatochromism is the hydrogen-bonding interaction between the polar solvents and IBU which stabilizes the drug in its ground state. Further, increase in the quantum yield by increasing the hydrophobicity of the solvent indicates that for polar solvents, a very efficient non-radiative decay channel opens which is related to the charge transfer nature of the emitting state and involves a further increase in the excited solute dipolarity during the decay, thus resulting in decrease in quantum yield for such solvent systems [48–50]. Figure 8c gives the change in emission spectrum of the drug when the hydrogel (CD = 0.04 wt%) is added to the medium. With increase in the concentration of the hydrogel in the medium, the intensity of the peak at 289 nm corresponding to the IBU occupying the polar region decreases, while a new peak appears at 370 nm indicating that the drug migrates to non-polar region of the hydrogel.
The spectrum clearly shows the partitioning of the drug that occurs between the regions of different polarities. If C 1 is the concentration of the drug in the polar zone and C 2 is its concentration in the non-polar region, then the partition coefficient, K d , is given as
The variation of K d with the weight of added hydrogel is given in Fig. 8d. The plot anticipates that there occurs drift of the encapsulated drug more towards the hydrophobic sites of the hydrogel with increase in hydrogel concentration. Since the drug is non-polar, it shows more tendency to get adhered to the non-polar regions of the hydrogel once the sites are made available. Thus, partitioning exhibits more inclination towards the hydrophobic areas of hydrogel as evident from the Fig. 8c.
Conclusions
Multi-component thermoreversible hydrogel of natural and synthesized polymers was formulated by semi-IPN methodology and characterized by FTIR, SEM and Rheology. The mechanical properties of the synthesized hydrogel were manipulated by adjusting the concentration of CD. The gelation temperature of hydrogel was found to be a function of CD wt% in polymeric network so that gel can be made workable in broad range of temperatures. Gelation time was found to vary inversely with the CD concentration at a constant temperature due to increase in the cross-linking density. The gelation time and the gelation temperature of the hydrogel synthesized with 0.04 wt% CD could be envisaged to be optimal for in vivo applications. Tensile strength of hydrogel increases with increase in CD concentration in addition to extension in the LVR region. EC and LC of the hydrogels towards ibuprofen increase first with increasing the CD concentration that reaches maximum and then remains almost constant with further increase in CD concentration. The drug release ability of the hydrogel showed that the drug is released very fast in the first 10 min but slows down thereafter. The release kinetics was very well explained in light of various drug release models. The partitioning of the model drug between the polar and hydrophobic regions of the hydrogel was studied as function of hydrogel concentration. In conclusion, the synthesized hydrogel has the potential to be explored further for varied application in various fields.
References
Hatice KC (2005) Synthesis of persulfate containing poly (N-vinyl-2-pyrrolidone) (PVP) hydrogels in aqueous solutions by g-induced radiation. Radiat Phys Chem 72:703–710
Krishna Rao KSV, Naidu BVK, Subha MCS, Sairam M, Aminabhavi TM (2006) Novel chitosan-based pH-sensitive interpenetrating network microgels for the controlled release of cefadroxil. Carbohydr Polym 66:333–344
Myung D, Waters D, Wiseman M, Duhamel PE, Noolandi J, Ta CN (2008) Progress in the development of interpenetrating polymer network hydrogels. Polym Adv Technol 19:647–657
Kiatkamjornwong S, Mongkolsawat K, Sonsuk M (2002) Synthesis and property characterization of cassava starch grafted poly [acrylamide-co- (maleic acid)] superabsorbent via γ-irradiation. Polymer 43:3915–3924
Opdahl A, KimSH KTS, Marmo C, Somorjai GA (2003) Surface mechanical properties of pHEMA contact lenses: viscoelastic and adhesive property changes on exposure to controlled humidity. J Biomed Mater Res A 67:350–356
Yang X, Zhu Z, Liu Q, Chen X, Ma M (2008) Effects of PVA, agar contents, and irradiation doses on properties of PVA/ws-chitosan/glycerol hydrogels made by γ-irradiation followed by freeze-thawing. Radiat Phys Chem 77:954–960
Qiu Y, Park K (2001) Environment-sensitive hydrogels for drug delivery. Adv Drug Deliv Rev 53(3):321–339
Lee KY, Mooney DJ (2001) Hydrogels for tissue engineering. Chem Rev 101(7):1869–1879
Jones A, Vaughan D (2005) Hydrogel dressings in the management of a variety of wound types: a review. J Orthop Nurs 9(Supplement 1):S1–S11
Bharali JD, Shaoo SK, Mozumdar S, Maitra A (2003) Cross-linked polyvinylpyrrolidone nanoparticles: a potential carrier for hydrophilic drugs. J Colloid Interface Sci 258:415–423
Vasir JK, Tambwekar K, Garg S (2003) Bioadhesive microspheres as a controlled drug delivery system. Int J Pharm 255:13–32
Chirila TV, Rakoczy PE, Garrett KL, Lou XCostanble IJ (2002) The use of synthetic polymers for delivery of therapeutic antisense oligodeoxynucleotides. Biomaterials 23:321–342
Gulyuz U, Okay O (2013) Self-healing polyacrylic acid hydrogels. Soft Matter 9:10287
Nguyena MK, Alsberg E (2014) Bioactive factor delivery strategies from engineered polymer hydrogels for therapeutic medicine. Prog Polym Sci 39:1235–1265
Sahiner N (2013) Soft and flexible hydrogel templates of different sizes andvarious functionalities for metal nanoparticle preparationand their use in catalysis. Prog Polym Sci 38:1329–1356
Wang W, WangQ WA (2011) pH-Responsive Carboxymethylcellulose-g Poly(sodium acrylate)/Polyvinylpyrrolidone Semi-IPN Hydrogels with Enhanced Responsive and Swelling Properties. Macromol Res 19:57–65
Peng X-W, Ren J-L, Lin-XinZhong FP, Sun R-C (2011) Xylan-rich Hemicelluloses-graft-Acrylic Acid Ionic Hydrogels with Rapid Responses to pH, Salt, and Organic Solvents. J Agric Food Chem 59:8208–8215
Omidian H, Park K (2012) Fundamentals and Applications of Controlled Release Drug Delivery. Adv Deliv Sci Technol 75:105
Guo JH, Skinner GW, Harcum WW, Barnum PE (1998) Pharmaceutical applications of naturally occurring water-soluble polymers. Pharm Sci Technol Today 1:254
Omidian H, et al (2005) Hydrogels having enhanced elasticity and mechanical strengthproperties in US patent, 6,960,617
Omidian H, Rocca JG (2006) Formation of strong superporous hydrogels in US patent, 7, 056,957
Omidian H, Rocca JG, Park K (2006) Elastic, superporous hydrogel hybrids of polyacrylamide and sodium alginate. Macromol Biosci 6:703–710
Dhar N, Akhlaghi SP, Tam KC (2012) Biodegradable and biocompatible polyampholyte microgels derived from chitosan, carboxymethyl cellulose and modified methyl cellulose. Carbohydr Polym 87:101–109
Manakker F, Vermonden T, Nostrum CF, Hennink WE (2009) Cyclodextrin-based polymeric materials: synthesis, properties, and pharmaceutical /biomedical applications. Biomacromolecules 10:3157
Jiang GB, Quan D, Liao K, Wang H (2006) Novel Polymer Micelles Prepared from Chitosan Grafted Hydrophobic Palmitoyl Groups for Drug Delivery. Mol Pharm 3:152–160
Liu TY, Lin YL (2010) Novel pH-sensitive chitosan-based hydrogel for encapsulating poorly water-soluble drugs. Acta Biomater 6:1423–1429
Wang J, Somasundaran P (2005) Adsorption and conformation of carboxymethyl cellulose at solid–liquid interfaces using spectroscopic, AFM and allied techniques. J Colloid Interface Sci 291:75–83
Gajare P, Patil C, Kalyane N, Pore Y (2009) Effect 0f Hydrophilic Polymers on Pioglitazone Complexation with Hydroxypropyl-β-Cyclodextrin. Dig J Nanomater Biostruct 4:891–897
Kulbida A, Ramos MN, Rasanen M, Nieminen J, Schrems O, Fausto R (1995) Rotational Isomerism in Acrylic Acid- A Combined Matrix-isolated IR, Raman and ab initio Molecular Orbital Study. J Chem Soc Faraday Trans 91:1571–1585
Oxley J, Smith J, Brady J, Dubnikova F, Kosloff R, Zeiri L, Zeiri Y (2008) Raman and Infrared Fingerprint Spectroscopy of Peroxide-Based Explosives. Appl Spectrosc 62:906
McMurry J (2007) Organic chemistry with biological approach. Cengage Learning. Inc. ISBN-13: 978-0-495-39144-9
Aouada FA, de Moura MR, Rubira AF, Muniz EC, Fernandes PRG, Mukai H, da Silveira ACF, Itri R (2006) Birefringent hydrogels based on PAAm and lyotropic liquid crystal: optical, morphological and hydrophilic characterization. Eur Polym J42:2781–2790
Yang X, Liu Q, Chen X, Yu F, Zhu Z (2008) Investigation of PVA/ws-chitosan hydrogels prepared by combined c-irradiation and freeze-thawing. Carbohydr Polym 73:401–408
Moura MJ, Figueiredo MM, Gil MH (2007) Rheological Study of Genipin Cross-Linked Chitosan Hydrogels. Biomacromolecules 8:3823–3829
Hou Q, De Bank PA, Shakesheff KM (2004) Injectable scaffolds for tissue regeneration. J Mater Chem 14:1915–1923
Hafeti A, Amsden B (2002) Biodegradable injectable in situ forming drug delivery systems. J Control Release 80:9–28
Fechine GJM, Barros JAG, Alcantara MR, Catalani LH (2006) Fluorescence polarization and rheological studies of the poly (N-vinyl-2-pyrrolidone) hydrogels produced by UV radiation. Polymer 47:2629–2633
Sharma SC, Shrestha LK, Tsuchiya K, Sakai K, Sakai H, Abe M (2009) Viscoelastic Wormlike Micelles of Long Polyoxyethylene Chain Phytosterol with Lipophilic Nonionic Surfactant in Aqueous Solution. J Phys Chem B 113:3043–3050
BirdRB ARC, Hassager O (1987) Dynamics of polymeric liquids—volume 1: fluid mechanics, 2nd edn. John Wiley, New York
Rubinstein M, Colby RH (2003) Polymer Physics(Oxford University Press)
Anseth KS, Bowman CN, Brannon-Peppas L (1996) Mechanical properties of hydrogels and their experimental determination. Biomaterials 17:1647–1657
Kavanagh GM, Ross-Murphy Simon B (1998) Rheological characterisation of polymer gels. Prog Polym Sci 23:533–562
Ghosh K, Shu XZ, Mou R, Lombardi J, Prestwich GD, Rafailovich MH, Clark RAF (2005) Rheological Characterization of in Situ Cross-Linkable Hyaluronan Hydrogels. Biomacromolecules 6:2857–2865
Kono H (2014) Characterization and properties of carboxymethylcellulose hydrogels crosslinked by polyethyleneglycol. Carbohydr Polym 106:84–93
Nho YC, Park JS, Lim YM (2014) Preparation of Poly(acrylic acid) Hydrogel by Radiation Crosslinking and Its Application for Mucoadhesives. Polymers 6:890–898
Costa P, Lobo JMS (2001) Modeling and comparison of dissolution profiles. Eur J Pharm Sci 13:123–133
Higuchi T (1963) Mechanism of sustained-action medication. Theoretical analysis of rate of release of solid drugs dispersed in solid matrices. J Pharm Sci 52:1145–1149
Cowley JD, Pasha I (1981) The fluorescence of some dipolar NN-dialkyl-4-(dichloro-1,3,5-triazinyl)anilines. Part 2. Temperature and solvent effects on the radiationless decay of an intramolecular charge-transfer excited singlet state. J Chem Soc Perkin Trans 2:918
Cowley JD, Okame E, Todd RST (1991) Triazinylaniline derivatives as fluorescence probes. Part 1. Absorption and fluorescence in organic solvents and in aqueous media in relation to twisted intramolecular charge-transfer state formation, hydrogen bonding, and protic equilibria. J Chem Soc Perkin Trans 2:1495
El-Daly SA, Abdel-Kader MH, Issa RM, El-Sherbini EA (2003) Influence of solvent polarity and medium acidity on the UV_/Vis spectral behavior of 1-methyl-4-[4-amino-styryl] pyridinum iodide. Spectrochim Acta A 59:405–411
Acknowledgments
AAD acknowledges the Department of Science and Technology (DST), Govt. of India, for providing funds under the FIST scheme to the Department of Chemistry, University of Kashmir, for procuring various instruments.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Maswal, M., Chat, O.A. & Dar, A.A. Rheological characterization of multi-component hydrogel based on carboxymethyl cellulose: insight into its encapsulation capacity and release kinetics towards ibuprofen. Colloid Polym Sci 293, 1723–1735 (2015). https://doi.org/10.1007/s00396-015-3545-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00396-015-3545-4