
Abstract
Intramyocardial hemorrhage is an independent predictor of adverse outcomes in ST-segment elevation myocardial infarction (STEMI). Iron deposition resulting from ischemia–reperfusion injury (I/R) is pro-inflammatory and has been associated with adverse remodeling. The role of iron chelation in hemorrhagic acute myocardial infarction (AMI) has never been explored. The purpose of this study was to investigate the cardioprotection offered by the iron-chelating agent deferiprone (DFP) in a porcine AMI model by evaluating hemorrhage neutralization and subsequent cardiac remodeling. Two groups of animals underwent a reperfused AMI procedure: control and DFP treated (N = 7 each). A comprehensive MRI examination was performed in healthy state and up to week 4 post-AMI, followed by histological assessment. Infarct size was not significantly different between the two groups; however, the DFP group demonstrated earlier resolution of hemorrhage (by T2* imaging) and edema (by T2 imaging). Additionally, ventricular enlargement and myocardial hypertrophy (wall thickness and mass) were significantly smaller with DFP, suggesting reduced adverse remodeling, compared to control. The histologic results were consistent with the MRI findings. To date, there is no effective targeted therapy for reperfusion hemorrhage. Our proof-of-concept study is the first to identify hemorrhage-derived iron as a therapeutic target in I/R and exploit the cardioprotective properties of an iron-chelating drug candidate in the setting of AMI. Iron chelation could potentially serve as an adjunctive therapy in hemorrhagic AMI.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Background
Acute myocardial infarction (AMI) is a major contributor to morbidity and mortality worldwide [77, 80]. In the United States, about 800,000 people experience AMI [8] and about 25% of post-AMI survivors succumb to heart failure (HF), a condition with a 5-year mortality rate of ~ 50% [14, 81]. Although modern medical management has improved the prognosis for AMI patients, chronic adverse remodeling and poor outcomes remain unresolved [47]. While post-AMI reperfusion promotes myocardial salvage, ‘ischemia–reperfusion injury’ (I/R) is often an adverse consequence of tissue reoxygenation following acute myocardial infarction (AMI) [43, 46, 88]. I/R has been associated with additional myocardial damage beyond the effects of the initial ischemia, possibly via several major pathways that promote cell membrane disruption and mitochondrial death, reactive oxygen species (ROS), apoptosis and lipid peroxidation [52, 53, 67, 82]. As part of the I/R-related injury, a phenomenon called ‘no-reflow’, classically observed as a region of microvascular obstruction (MVO) on MRI, has been correlated with poor patient outcomes [28, 45, 86]. Importantly, intramyocardial hemorrhage (IMH), associated with MVO as a result of lethal I/R, is now identified as an independent predictor of adverse outcomes, including mortality [19, 32, 34, 66].
In ST-segment elevation myocardial infarction (STEMI) patients, the presentation of MVO can be as high as 50% [7, 25, 32, 66], while IMH can be found in 50–75% of MVO cases [19, 25, 32, 51, 75]. This is clinically significant, since these patients often experience major adverse cardiovascular events. Although initially thought to represent coincidental biomarkers of infarct severity, recent work suggests that iron deposition in infarcted tissue is indeed a cause for adverse remodeling [17, 19, 41, 56, 57]. Carrick et al. [19] have demonstrated that the predictive power of IMH is greater than MVO in terms of adverse remodeling and N-terminal pro-brain natriuretic peptide (surrogate outcomes), as well as cardiovascular death and heart failure (HF) following discharge (health outcomes). Recently, we have conclusively demonstrated that IMH by itself is pro-inflammatory (attracts macrophages), can aggravate edema development and could contribute to MVO formation in the presence of ischemia [41]. There is further evidence that in human hearts post-AMI, residual (chronic) iron deposition as a consequence of hemorrhage is associated with greater inflammation and adverse remodeling [12, 17, 56]. These observations from multiple investigators strongly support the concept that hemorrhage is not simply a bystander, but an active driver of maladaptive reparative processes that follow mechanical reperfusion in AMI.
The current standard-of-care post-AMI, including anti-platelet agents, statins, ACE inhibitors and β-blockers, has shown promise in limiting infarct size and adverse remodeling [60, 87]. However, in the context of I/R, management of patients with hemorrhagic infarction is currently unknown; it is unclear whether hemorrhage is simply a marker of severity of I/R or whether it offers a novel therapeutic target. Routine clinical therapeutics are not targeted against hemorrhage, as evidenced by the chronic presence or persistent nature of IMH [17, 57]. Thus, to date, IMH remains a challenge for clinicians and there is no effective therapy to address this pathophysiological entity in I/R.
Following AMI, hemorrhage in myocardial tissue undergoes a dynamic transformational process over time as a result of the degradation of hemoglobin into downstream iron by-products, culminating in the formation of extracellular ferritin and hemosiderin (ferric iron); this has been demonstrated in both brain and heart studies [10, 42, 57]. The final form of iron deposition with IMH is very similar to that found in the liver and heart of patients with iron overload syndromes, thalassemia and sickle cell disease [18, 36, 38, 84], where iron chelators like deferoxamine (DFO) and deferiprone (DFP) can selectively bind to labile iron, render it chemically inert and allow for its safe excretion [44, 69]. The role of iron chelation in hemorrhagic AMI, where it would be most suited, has never been evaluated. We hypothesized that sustained treatment with the iron chelator DFP, until iron was cleared from the infarct zone, would blunt post-operative edema, concentric hypertrophy and eccentric remodeling. Our study is the first to investigate this approach and offers promising evidence that the iron chelator DFP can offer cardioprotection by mobilizing hemorrhagic iron by-products. We utilized quantitative MRI techniques, including cardiac function, viability and edema- and hemorrhage-sensitive relaxation parameters—T2 and T2* [40, 48, 57, 70] to assess tissue healing and to evaluate therapeutic efficacy in a porcine model of AMI [13].
Methods
Preclinical protocol
All protocols were approved by the Animal Care Committee of Sunnybrook Research Institute and the study employed non-diseased female Yorkshire pigs (20–25 kg, Caughell Farms, ON) as the model for AMI, as described previously [39,40,41,42]. For imaging and cardiac interventions, animals were sedated under an anesthetic combination of atropine (0.05 mg/kg), ketamine (30 mg/kg) and isoflurane (1–5%) and supported by mechanical ventilation. Complete coronary occlusion was achieved for 90 min distal to the second diagonal branch of the left anterior descending (LAD) artery using a percutaneous balloon dilation catheter (Sprinter OTW, Medtronic, MN), followed by reperfusion. This model has been well validated to consistently create transmural infarction with MVO and hemorrhage [40, 42]. Cardiac interventions were guided by X-ray fluoroscopy (Veradius C Arm, Philips Healthcare) and intra-procedure events were stabilized using antiarrhythmics (amiodarone and lidocaine drip) and defibrillation as required. A comprehensive MRI examination was performed in the healthy state and up to 4 weeks post-AMI, prior to animal sacrifice.
Animal groups
Animals were divided into two groups—untreated controls (N = 7) and DFP treated (N = 7). Oral DFP (Ferriprox®, ApoPharma Inc., Toronto) is readily available in tablet form and is routinely administered to patients with transfusional iron overload. DFP was fed to pigs 1–2 h before the LAD occlusion (pre-loading) and oral treatment was continued daily for 4 weeks until sacrifice. The dose selected, 50 mg/kg, was administered twice daily, with ca. 7 h between doses (total dose 100 mg/kg/day) [83]; this is the maximum dose used in patients with transfusional iron overload [9]. For ease of consumption, the DFP tablets were crushed and mixed with jelly cubes prior to the feed; the control infarcted group was fed with only jelly cubes.
Pharmacokinetic analysis of deferiprone
The pharmacokinetic (PK) analysis of DFP was performed in healthy pigs (N = 3) to assess the systemic availability of the drug following oral administration. Serial blood samples (2–3 mL) were obtained after a dose of 50 mg/kg given orally (Ferriprox® 500 mg tablets, ApoPharma Inc., Toronto): at pre-dose (0 min); at 5, 15, 30 min; and 1, 2, 4, 7 and 12 h after dosing. Blood samples were collected in sodium citrate (3.2%) tubes and immediately centrifuged at 1500g for 10 min at 4 °C. The plasma (1.0 mL) was isolated and stored at − 80 °C for further analysis.
DFP concentration in pig plasma samples was measured by a validated HPLC method. Samples were deproteinized by mixing plasma with equal volume of 0.4 M HClO4, followed by centrifugation at 35,000×g for 15 min. DFP and DFP-glucuronide in the supernatants were resolved on a ZORBAX Eclipse XDB C18 column using a gradient between mobile phase A (10 mM KH2PO4, 4 mM EDTA, 2 mM sodium heptanesulfonate, pH 2.80) and B (100% methanol). Retention times for DFP and DFP-glucuronide were 13.4 min (278 nm) and 8.6 min (264 nm), respectively. Analyte concentration in samples was calculated using calibration curves.
DFP and DFP-glucuronide PK parameters were computed by non-compartmental analysis applying the linear trapezoidal rule with linear interpolation, using Phoenix WinNonlin ver. 6.4 (Certara USA, Inc.). Cmax values were obtained from concentration vs. time plots. Terminal half-life (T1/2) = ln (2)/λz was estimated by linear regression of the time vs. log concentration curve. Area under the curve (AUC) and apparent clearance (CL) were also determined.
MRI protocol
MRI was performed on a 3 T whole-body scanner (MR 750, GE Healthcare) at baseline (healthy state) and at day 1, week 1 and week 4 post-AMI. A steady-state free-precession (SSFP) sequence in cine mode was utilized for cardiac function assessment: 12–15 short-axis slices, TR/TE = 4/1.7 ms, flip angle = 45°, FOV = 24 × 21.6 cm, matrix = 224 × 160, slice thickness = 5 mm (no gap), 8 views/segment, 20 phases/slice, in-plane resolution ~ 1 mm. Hemorrhage was identified with a multi-echo gradient echo T2* acquisition: 8 echoes (1.4–15.8 ms), TR = 16.8 ms, flip angle = 30°, matrix = 128 × 128, 10 short-axis slices, in-plane resolution ~ 1.9 mm. Edema quantification was performed using a previously validated T2-prepared spiral imaging sequence [40, 41]; 10 spirals (12.3 ms, 3072 points), TE = 2.9, 24.3, 45.6, 88.2 and 184.2 ms, in-plane resolution ~ 1 mm. Late gadolinium enhancement (LGE) imaging was initiated at 8–10 min post-injection of contrast agent gadolinium–DTPA (0.2 mmol/kg; Magnevist, Bayer Healthcare). A T1-weighted inversion recovery gradient echo sequence (IR-GRE) sequence was utilized for infarct and MVO delineation: TR/TE = 4.1/1.9 ms, flip angle = 15°, matrix = 224 × 192, 2RR intervals, 12–15 short-axis slices, in-plane resolution ~ 1 mm, inversion time was adjusted to null the myocardium (250–310 ms).
MRI data analysis
All MRI data were analyzed using CVI42 software (Circle Cardiovascular Imaging, Calgary). Cardiac function parameters evaluated were: ejection fraction (EF), end-diastolic volume (EDV), end-systolic volume (ESV), end-diastolic mass and regional end-diastolic wall thickness (EDWT). Infarct size was quantified from the LGE images using the full-width half-maximum thresholding technique [2] and regions of hypoenhancement within the hyperenhanced infarct were manually identified as MVO. Pixel-wise myocardial T2 and T2* maps were generated by fitting with an exponential function. Hemorrhage size was computed from short-axis T2* maps using the threshold criteria of two standard deviations below the remote myocardial T2* measurement (care was taken to avoid artifactual regions at the heart–lung interface) [56]. All volumes were represented as a percentage of the LV volume. For regional tissue characteristics, mean T2 and EDWT were noted based on AHA 17-segment model [20]: a) infarct zone consisting of segments 7–8 (mid-ventricular) and 13–14 (apical) corresponding to the LAD territory at the level of the occlusion and b) remote myocardial territories consisting of segments 4–6 on the lateral side. For T2* regional analysis, only mid-ventricular segments 7–8 were included since the apex is highly prone to susceptibility artifacts on a 3 T scanner.
Histology
At week 4, all animals were euthanized, and hearts were resected. Fixed hearts were sliced at approximately 1 cm intervals along the transverse axis. Slices were processed, embedded in paraffin, and sectioned at 5 µm thickness for histological slides using standard techniques. Large 2 × 3 histology slides were used to accommodate the entire section on one slide. Masson’s Trichrome and Prussian blue stains, as well as the Picrosirius Red and Nuclear Fast Red counterstains were done as per standard staining protocols (available from IHC World: www.ihcworld.com). Antibodies were used following standard staining techniques. Briefly, slides were deparaffinized and hydrated through an ethanol series. Endogenous peroxidase activity was quenched in a 0.3% hydrogen peroxide solution in methanol, and slides were blocked using normal goat serum (Jackson ImmunoResearch Labs, SP15-100). The Mac387 antibody (mouse anti-Calprotectin, clone Mac387, Thermo Fisher, cat# Ma5-12213) was used at 1:100 after trypsin antigen retrieval (Trypsin Enzymatic Antigen Retrieval Solution, Abcam, ab970). A biotin-labeled goat anti-mouse secondary antibody (Jackson ImmunoResearch Labs, 115-065-003) was used at 1:500. Antibody labeling was amplified using the Vectastain Elite ABC HRP RTU Reagent (Vector Labs, #PK-7100), and developed with the ImmPACT SG Peroxidase Substrate (Vector Labs, #SK-4705). After counterstaining with Picrosirius Red to demarcate scar, all slides were dehydrated through an ethanol series, washed in xylenes, and mounted in Permount (Fisher, SP15-100).
Histological quantification was done as follows: slides were digitized at 20× with a TissueScope LE191 scanner (Huron Digital Pathology). Images were processed using custom MATLAB scripts (MathWorks Inc.), and for all images, the right ventricle was manually excluded. Scar size and edema content were calculated using slides stained for Masson’s trichrome. Scar tissue (blue collagen staining), edema (non-vascular white space within the bounds of stained tissue) and total tissue areas in the LV were calculated using HSV color segmentation (modified from available code [4]). To measure iron content, images of the LV stained for Prussian blue were broken into high-resolution panels (~ 400–900 panels per heart). Calcified regions (which also appear blueish-purple) were manually removed, and blue punctae were segmented and individually quantified on every panel using a previously described method [39]. To assess macrophage infiltration, images of the LV stained with the Mac387 antibody were similarly broken into high-resolution panels, and five panels each from the scar and remote myocardium of each heart were randomly selected. Mac387 + cells were manually identified and tallied using Sedeen virtual slide viewer software (Pathcore, Toronto).
Statistics
All MRI parameters were expressed as mean ± standard deviation (SD). To account for longitudinal data (baseline to week 4 post-AMI) and multiple groups, a repeated measures two-way analysis of variance (ANOVA) statistics with multiple comparisons was carried out using the Sidak correction factor (GraphPad Prism®, La Jolla, CA). Within group and across group comparisons were tested and a p value < 0.05 was used to determine statistical significance. For the histological analysis, significance between two groups was tested using an unpaired T test. Multiple groups were compared using one-way ANOVA, followed by a Tukey’s multiple comparisons post-test; significance level was set at 0.05.
Results
Interventional procedure
Four animals died during the acute infarct procedure, two in each group, resulting in a 28% mortality rate associated with the ischemic event. These mortality rates are consistent with our earlier experience with this porcine AMI model [41]. The remaining animals (N = 5 per group) all survived the procedure and were available for subsequent serial MRI and histological analysis after 4 weeks of treatment.
DFP pharmacokinetic analysis
For the PK analysis, an oral dose of 50 mg/kg DFP resulted in a plasma DFP Cmax of 20.3 ± 1.2 µM and AUC 55.4 ± 4.9 h*µM. DFP was biotransformed into its metabolite DFP-glucuronide; the Cmax and AUC for DFP-glucuronide were 147 ± 32.1 µM and 370 ± 51.0 h*µM, respectively (glucuronide:parent ratio 6.7). See Table 1 and Fig. 1.

Serum pharmacokinetic profile of orally administered deferiprone (50 mg/kg) in three healthy animals. Error bars show standard error of mean
Infarction and MVO
Figure 2 demonstrates representative images from the control and DFP groups at day 1 and week 1 post-AMI. All animals in both groups showed transmural infarction with hemorrhage and MVO at day 1. T2* images indicated that hemorrhage was completely resolved in the DFP group at week 1, but not in the control group. Infarct and MVO sizes were comparable between the two groups at all time points, and MVO was significantly resolved by week 1 in both groups (control: p < 0.003; DFP: p = 0.001) compared to day 1 (Fig. 3).

Short-axis images from representative animals subjected to 90 min LAD occlusion without (top panel) and with (bottom panel) treatment of iron chelator deferiprone (DFP). In the DFP group, hemorrhage, as indicated by T2* image (red arrows), was observed only on day 1 but resolved by week 1, unlike the untreated group where hemorrhage persisted even at week 1. The iron neutralizing capacity of DFP is apparent from this example. In both groups, microvascular obstruction (MVO) was seen on day 1 that was partially resolved by week 1. IS infarct segment, HEM hemorrhage, MVO microvascular obstruction

Plots show the time course of a infarct and b MVO size in the control and DFP groups. A significant reduction in infarct size was noted at week 4 and in MVO size at weeks 1 and 4, compared to day 1 in both animal groups. Differences were not significant between groups at any given time point. †p < 0.0001, *p < 0.005 compared to day 1
T2* and hemorrhage
Figure 4a shows the LGE and T2* map in a representative animal indicating the presence and extent of MVO and hemorrhage at day 1 post-AMI. The cumulative time course of T2* in the infarct zone demonstrated a low value at day 1 in both animal groups (p < 0.05), indicative of IMH. However, at week 1, T2* had normalized to baseline levels (complete hemorrhage resolution) in the DFP group, but not in the control group (Fig. 4b) with significantly different T2* values between the groups (p = 0.035); at week 1, only one DFP-treated animal showed a low T2* value (23 ms vs. 32 ms remote) in the infarct segment. At week 4, T2* remained non-significantly below baseline values (p = 0.09) in the control group, suggesting persistence of hemorrhagic by-products; in contrast, hemorrhage resolution was complete in the DFP group earlier at week 1. T2* in the remote region was not significantly different both within groups (compared to baseline) and across groups for a given time point. IMH extent (volume) was significantly reduced at week 1 compared to day 1 in the DFP group (p = 0.038), in contrast to the control group, in which IMH remained elevated (Fig. 4c). By week 4, hemorrhage volume was significantly reduced in both groups (p < 0.01) in comparison to that at day 1.

a LGE and T2* images from a representative animal in the DFP group at day 1 post-AMI. Low T2* values indicate the presence of hemorrhage within the infarct core or region of MVO (arrows). The region of intramyocardial hemorrhage (IMH) is shown as a color overlay on T2*-weighted images with TE = 12.6 ms. White arrows identify susceptibility artifacts at the heart–lung interface, which were manually excluded. b Plot shows the evolution of T2* in the infarct zone of animals in the control and DFP groups. Observations at week 1 noted that T2* recovered to baseline values in the DFP group faster than the control group. c Evolution of hemorrhage extent indicated that the size of hemorrhagic core at week 1 was significantly reduced in the DFP group compared to day 1, but not in the control group. *p < 0.05, **p < 0.005, ***p < 0.0005 compared with baseline; §p < 0.05 compared to other group at the same time point; †p < 0.05, ††p < 0.01 compared with day 1
T2 and edema
Figure 5a shows the LGE and T2 map in a representative animal identifying infarction and edema, respectively, at week 1 post-AMI. Infarct T2 was significantly elevated at week 1 in both groups (p < 0.0001), indicative of ongoing inflammatory processes. In the infarct zone (Fig. 5b) of a DFP-treated pig, T2 was significantly reduced by week 4 compared to week 1 (p = 0.004), suggesting edema resolution. However, in the control group, T2 remained elevated at week 4 compared to week 1 (p = NS), indicative of persistent edema. In addition, at week 4, infarct T2 was significantly lower in the DFP group compared to the control group (p = 0.032), suggestive of greater resolution of inflammation with DFP treatment. In the remote myocardium (Fig. 5c), T2 was significantly elevated at week 1 in the control group compared to baseline (p = 0.038) suggestive of either a global inflammatory response or a possible hyperemic response (increased blood flow) reflected by the increased T2 blood oxygenation level-dependent (BOLD) effect [40, 42]. A previous study has demonstrated that blood flow in the remote myocardium can be significantly elevated in the first 2 weeks following experimental AMI, which is restored to normal values thereafter [65]. In contrast, remote T2 remained unaltered throughout the healing process in the DFP group.

a LGE and T2 images from a representative animal in the control group at week 1 post-AMI. The elevated T2 signal indicates the presence of edema within the infarct zone (arrows); color overlay shown on T2-weighted images with TE = 88 ms. b Plot shows the evolution of T2 in the infarct zone of animals in the control and DFP groups. Infarct T2 was significantly elevated post-AMI in both groups indicative of edematous development. With DFP, inflammation or edema was significantly reduced by week 4 with T2 values approaching baseline levels. In the untreated group, edema persisted at week 4. c Plot shows the evolution of T2 in the remote myocardium of the control and DFP groups. Remote T2 was significantly elevated at week 1 in the control group compared to baseline suggestive of remote myocardial remodeling. Remote T2 remained unchanged in the DFP group. *p < 0.05, **p < 0.001, ***p < 0.0001 compared with baseline; §p < 0.05 compared with other group at the same time point; †p < 0.05 compared with baseline
Regional wall thickness
At day 1, infarct wall thickness was significantly elevated from baseline (p = 0.0027) in the control group, consistent with edema formation; this was not significant in the DFP-treated group (Fig. 6a). At week 4, infarct wall thickness remained significantly higher in the control group suggesting persistent swelling in comparison to the DFP-treated group (p = 0.034). In the control group, remote zone wall thickness was also significantly elevated throughout the healing process when compared to baseline (Fig. 6b). In contrast, remote zone wall thickness was only significantly increased at week 4 in the DFP group. Overall, there was a trend toward greater remote zone wall thickness in the control group when compared to DFP group at each time point, thus pointing toward a beneficial remote zone remodeling process in DFP-treated animals (p = NS). These variations in wall thickness agreed well with T2 characteristics in the infarct and remote myocardium (Fig. 5).

Plots show the cumulative time course of end-diastolic wall thickness (EDWT) in the a infarcted and b remote myocardial territories. a In the control group, infarct wall thickness was greater than that in the DFP group during the 4-week healing, indicative of reduced edematous swelling with the iron-chelating agent. b In the control group, remote zone wall thickness was significantly elevated at all time points post-AMI and exhibited a trend towards greater EDWT than the DFP group. *p < 0.01, **p < 0.005, ***p < 0.0001 compared with baseline; §p < 0.05 compared to the other group at the same time point
Global cardiac function
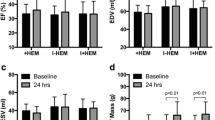
EF was not significantly different between the two groups (Fig. 7a). In the control group, ED mass was significantly elevated throughout the healing process (p < 0.001). In contrast, significance was noted only from week 1 onwards in the DFP group (p < 0.01). ED mass was also significantly greater in the control group compared to DFP, indicative of greater compensatory hypertrophy, at weeks 1 (p = 0.031) and 4 (p = 0.0003) (Fig. 7b). At week 4, EDV was significantly elevated in both the control (56% increase, p < 0.0001) and DFP (23% increase, p = 0.016) groups compared to baseline; however, EDV in the DFP group was significantly lower than control at week 4 (33% less, p = 0.003) (Fig. 7c). Similar to EDV, ESV also demonstrated a greater and earlier elevation in the control group at week 1 (39% increase, p = 0.0015) and week 4 (62% increase, p < 0.0001) compared to baseline, as opposed to the DFP group which showed an elevation only at week 4 (45% increase, p = 0.0016) compared to baseline. These structural changes suggest a beneficial remodeling process with the iron-chelating agent. EF was not found to be significantly different across groups or across time within groups.

Plots show the cumulative time course of a ejection fraction (EF), b end-diastolic (ED) mass, c end-diastolic volume (EDV) and d end-systolic volume (ESV) in the two groups. a EF was not significantly different between the two groups at all time points. b In the DFP group, myocardial enlargement, measured by end-diastolic (ED) mass, was significantly reduced at weeks 1 and 4 compared to the control group. c At week 4, end-diastolic ventricular volume (EDV) was significantly less in the DFP group compared to the control, suggesting alleviated chronic preload stress with DFP. d Differences in ESV were similar to that of EDV. *p < 0.01, **p < 0.001, ***p < 0.0001 compared with baseline; §p < 0.05, §§p < 0.001 compared to the other group at the same time point
Histology
Animals were killed 4 weeks post-MI, and hearts were processed for histological examination of the scar, edema, iron deposition and inflammatory response. Infarct scar was delineated using Masson’s trichrome stained sections (Fig. 8a, b). Scar area (as percent of LV area) showed no significant difference between control and DFP-treated animals (9.03 ± 7.46% for control, 9.37 ± 1.44% for DFP, Fig. 8c), consistent with observations from LGE imaging (Fig. 3a). Edema quantification revealed that DFP treatment led to a trending reduction (p = NS) in edema in the LV as a whole (Fig. 8h) and also when broken down into scar and remote regions (Fig. 8i, j) with greater effect in the scar region. Quantification of blue (ferric iron) punctae observed on Prussian blue stain (Fig. 9a–d) revealed that they are significantly smaller in size (p = 0.0002) and visually less intensely stained (Fig. 9 c–e) in the DFP group. A determination of the distribution of punctae across the tissue, however, showed no change in punctae density with DFP treatment (Fig. 9f), and a count of the total number of punctae present in each entire heart also showed no difference (Fig. 9g). Characterization of the inflammatory response revealed a significant increase in the number of macrophages found in the scar of DFP-treated group compared to that found in the remote myocardium of DFP treated as well as scar and remote regions of the control group (p < 0.0001, Fig. 10b).

Images show Masson’s trichrome staining to analyze scar size and extent of edema. a, b Sample histology images of hearts from saline (a) or DFP (b)-treated animals. Approximate location of panels d–g are demarcated in panel b. Bars = 5 mm. c Histological measurement of scar size showed no difference between the two groups. d–g Zoomed-in areas from the approximate locations demarcated in b showing examples of low (d, f) and high (e, g) levels of edema in both the remote myocardium (d, e) and scar area (f, g). Edema was evidenced by the increased space between tissue fibers. Bars = 100 µm. h–j Quantification of the area of edema within the whole LV (h), only the scar (i), and the non-scar (j) regions of myocardium. Black line is mean, and error bars are standard error of mean. Though none of the comparisons reached statistical significance, there was a trend toward decreased edema with DFP treatment in all tissue regions

Images show histological evaluation of iron within the tissue via Prussian blue staining. a–d Sample images of staining in the remote myocardium (a, b) or scar (c, d) regions of animals treated with saline (a, c) or DFP (b, d). Blue punctae, indicating the presence of iron, was rarely present in the remote myocardium. Within the scar, punctae in the DFP-treated group generally appeared smaller and less prominent than those in the control group. Bars = 50 µm. e–g Quantification of Prussian blue punctae. Black line shows mean, and error bars are standard error of mean. e Punctae across panels were significantly smaller in DFP-treated animals (p = 0.0002). f The density of punctae throughout the tissue panels did not change with DFP treatment. g The total number of punctae present in each heart was equivalent in both groups

a Sample image of Mac387 + cells [red = Picrosirius Red (collagen), black = Mac387 staining (macrophages and monocytes), bar = 50 µm]. b Quantification of Mac387 + cells within either the remote myocardium or scar area. Black line shows mean, and error bars are standard error of mean. There was a significant increase in the number of macrophages present in the scar of DFP-treated hearts. **p < 0.01; ****p < 0.0001
Discussion
This is the first report investigating the cardioprotective potential of the iron-chelating agent deferiprone in the context of hemorrhagic myocardial infarction. Our study demonstrated the following key benefits offered by the iron-chelating agent DFP: (1) earlier resolution of hemorrhage; (2) evidence suggestive of reduced chronic edema; (3) less compensatory hypertrophy; and (4) prevention of LV dilatation (signature of negative remodeling). Overall, our findings suggest that an iron chelation strategy employed early in the acute phase of I/R can mitigate hemorrhagic iron deposition and alleviate chronic adverse remodeling post-AMI.
The current standard-of-care post-AMI has shown promise in limiting infarct size and adverse remodeling [60, 87]; however, even with modern pharmacotherapies, morbidity and mortality remain high [77]. Treatment strategies for IMH patients is currently unavailable. It has particularly been noted that patients with IMH are prone to exhibit adverse remodeling and progress toward HF with poor long-term outcomes, including mortality [12, 17, 19, 25, 32]. Adjunctive protective pathways are being investigated to improve long-term outcomes of AMI, but these have not taken IMH into consideration. For example, although anti-inflammatory strategies have demonstrated benefit in animal models [63], recent clinical trials (involving anti-CD18, anti-CD11, anti-C5) have been negative [5, 6, 26]. These failures could be attributed to the pleiotropic nature of cytokines [30] and the fact that restricting inflammatory processes during healing may impair their beneficial effects. It is likely that it would be essential to first eliminate the “suspected source” (iron in this case) of the amplified inflammatory response rather than address its consequences afterward with non-specific anti-inflammatory agents. In this regard, our study design was specific in targeting iron species arising from the degradation by-products of hemorrhage, which are known to be a source of an inflammatory response [10, 12, 42, 56]. Thus, our findings are suggestive that neutralization of tissue iron deposits following hemorrhagic infarction by iron chelation can mitigate downstream adverse consequences (inflammation and remodeling).
Greater initial ischemic insult durations have been associated with greater degrees of myocardial hemorrhage post-AMI, and reperfusion appears to be a prerequisite [35, 49]. Recent studies have demonstrated that hemorrhage is associated with larger infarctions, reduced salvage, greater MVO and lower ejection fraction; it was also the strongest independent predictor of adverse left ventricular (LV) remodeling with an increased risk of arrhythmia, and predictive power even greater than MVO [19, 66]. Hemorrhage may also lead to stimulation of the host’s innate immune system [15] and persistent hemorrhage post-AMI is associated with a sustained inflammatory response [12, 16, 57]. Our recent study further confirmed that hemorrhagic by-products, independent of ischemia, are inherently pro-inflammatory in I/R and that greater hemorrhage (lower T2*) is associated with a heightened inflammatory response (higher T2), including macrophage infiltration into myocardial tissue.[41]. While disappearance of iron deposits in tissues is an expected consequence of DFP treatment, it cannot be excluded that DFP may act through mechanisms others than iron removal in post-AMI (unknown currently). Nevertheless, prior observations by various investigators (as mentioned above) strongly point toward the association between hemorrhage and inflammation and adverse outcomes, thereby reinforcing the cause–effect relationship and advancing the rationale for an iron removal strategy [24]. In support of this hypothesis, our findings clearly show that early elimination of iron deposits arising from hemorrhage are associated with a beneficial remodeling process. While we did not note any significant differences in ejection fraction between the two groups, our study was limited to a 4-week observation period, which might be too early to observe an impact on cardiac function. Nevertheless, we did see reduced edema, myocardial mass, ventricular expansion and remote hypertrophy during the healing phase in the presence of the iron chelator.
The use of iron chelation in I/R is not a new concept. Deferoxamine (DFO) has been employed in limited experimental and clinical settings of AMI [3, 21, 23, 58], with the prospect of limiting iron-induced oxidative stress at reperfusion. Canine studies with DFO have demonstrated a reduction in infarct size and oxidative injury in the acute phase of AMI [62, 73], but the same investigators have also reported negative findings. More recently, a small randomized trial by Chan et al. in 60 patients has indicated that adjunctive DFO treatment acutely during primary percutaneous coronary intervention (PPCI) can reduce oxidative stress but not affect infarct size, or improve myocardial salvage or cardiac function at 3 months post-AMI [21]. Apart from which iron chelator was employed, there are important design-related differences between prior studies and our present study. Firstly, it should be noted that all prior preclinical and human studies did not account for the presence of IMH. This is a particularly important point for the human study where no risk stratification was performed to evaluate maximal benefit impacted by the therapy.
Secondly, it is important to emphasize that prior studies (with DFO) have only investigated the effects of a single dose, but not sustained administration of an iron-chelating agent during the entire infarct healing process. A single dose of an iron chelator at reperfusion would be incapable of eliminating ferric iron by-products as these are only formed beyond the first few days of hemorrhagic injury [10, 42]; the PK profile of DFP is in the hours time frame. At day 1, we did not see any significant difference in infarct size between the two groups, suggesting that the single dose prior to reperfusion did not influence hemorrhage or MVO and infarct size. This suggests that a sustained dosing regimen would be necessary to access iron deposited by hemorrhagic transformation and to subsequently affect the infarct healing process. Our study has clearly demonstrated that sustained DFP treatment can resolve hemorrhage faster, alleviate inflammation and mitigate chronic adverse remodeling post-AMI. Although a head-to-head comparison between DFO and DFP is lacking, the small size of DFP (139 Da vs. 560 Da for DFO) [44] make is more ideally suited to the hemorrhagic situation by facilitating better penetration into the core infarct region through the MVO; future studies will need to confirm this.
Recently, the TACT (Trial to Assess Chelation Therapy) trial in post-AMI patients showed a non-significant reduction in adverse cardiovascular events at 6 months following treatment with disodium ethylenediaminetetraacetic acid (EDTA) [59]. Disodium EDTA is known to predominantly bind divalent cations and some trivalent cations such as calcium, magnesium, lead, zinc, iron and aluminum [59]. Thus, EDTA is essentially a broad-spectrum mineral chelator and not specific to the ferric iron species optimal for targeting IMH. Thus, the TACT trial only included agents with non-specific actions and not directly assessed targeted elimination of IMH. We hypothesize that iron species play a critical role in I/R and can potentially serve as a new therapeutic target [41]. Similar to hemorrhagic stroke, to date, there is no available targeted therapy for reperfusion hemorrhage; in this light, iron chelation for IMH appears to be an attractive option.
Oral DFP is readily available in tablet form; however, published PK data on pigs are currently lacking. In humans, the reported average DFP AUC (h*µM) and Cmax (µM) are 660 and 283, respectively, after a 50 mg/kg oral dose [29, 64, 1]. Humans and pigs appear to be similar in that there is extensive biotransformation of DFP to its metabolite DFP-glucuronide, although, DFP exposure in pigs was ca. 12-fold lower than that in humans owing to much higher glucuronidation in pigs. Despite this lower exposure, it is promising that the evidence of efficacy was still obtained in our pig model of AMI at a DFP exposure that is readily achievable in patients. Furthermore, the half-life of DFP in our pig was determined to be 2.11 h, which is very comparable to 1.77 h [29, 64, 1] in humans. The half-life profile confirms that a daily dose separation of 7–8 h should ensure continuity in the action of the iron chelator.
Improvement in EF is routinely considered the primary end point for determining the efficacy and successful translation of new therapeutic options into the clinic for heart failure patients with reduced EF [50]. In our study, we did not observe any significant alterations in EF with DFP administration relative to the control group. A possible reason for this observation could be that our study interrogated the disease progression only over a 4-week period post-MI, which is too early to conclude improvements in global function. A recent study in AMI patients has indicated that the majority of improvement in EF occurs within the 1 month time frame [54]. Thus, in this relatively early phase of MI, it is likely that the hearts are still capable of functionally overcompensating for the lost myocardium, thus not demonstrating any perceivable change in EF across groups. Nevertheless, our findings did demonstrate significantly reduced ventricular volumes in the DFP group (relative to controls), whose elevation is also considered an important marker of progression toward heart failure [55]. To be more conclusive, future studies will need to evaluate functional improvements in the true chronic phase post-AMI (> 6 weeks).
It is important to keep in mind the non-specific nature of the MRI relaxation effects in the presence of multi-factorial tissue pathology. For example, the response and sensitivity of MRI relaxation times T2 and T2* depend on the pathophysiological state of the tissue matrix [22]. Edema, comprising free water, imposes an inefficient medium for T2 relaxation due to short molecular correlation times and results in elevated T2-weighted signal or long T2 relaxation times [31, 33, 42, 89]. On the other hand, the presence of hemorrhage results in the shortening of T2 and T2* relaxation times due to the breakdown products of the heme iron (paramagnetic centers) [10]. Hemorrhagic transformation from deoxyhemoglobin (early acute) to methemoglobin (acute) to ferritin and hemosiderin (subacute, chronic) promotes a progressive increase in T2 and T2* shortening [10]; T2 signal loss is attributed to a diffusion-mediated mechanism, while T2* to a dephasing mechanism as a result of these paramagnetic disturbances. Thus, the degree and extent of both edema and hemorrhage by the T2 parameter might be underestimated in the presence of hemorrhagic iron. Nevertheless, the dynamics of edema evolution by the T2 parameter observed in our study were very similar to that observed in a recent porcine study by Fernández-Jiménez et al. [27]. Using a 40 min I/R model, they noted a T2 elevation of 42% at day 4 and 70% at week 1 (negligible change at day 1), compared to 15% change at day 1 and 38% at week 1 in our study. Differences between the observations could be attributed to the differing pig strains, but trends observed are identical. T2* is also elevated with edema, but to a lesser extent than T2 [40] and is considered a more sensitive indicator or tissue iron concentration [37, 42, 57]. A recent study in a porcine model of AMI by Rossello et al. [78] has shown that T2* is positively correlated with tissue water content and that T2* is influenced by both IMH and edema, very similar to T2. In fact, it was demonstrated that T2’ better represented histological IMH with little association with myocardial water content. These combinational effects of edema and IMH on T2* can help explain why we see a dramatic increase in infarct T2* from day 1 to week 1 in the DFP group relative to control. The identical T2s between the two groups at week 1 suggests that edema may roughly be comparable between the groups at this time point, which offers the strong evidence that the observed T2* elevation does in fact imply faster hemorrhage resolution at week 1 with DFP, with minimal impact from edema.
It should also be noted that the hypointense region within hyperenhanced infarct identified on an LGE image is an indirect measure of no-reflow or microvascular obstruction as a result of the contrast agent being unable to diffuse into the infarct core. However, previous studies have demonstrated that these hypointense regions are spatially correlated with the region of no-reflow as measured by thioflavin-S staining (intact endothelium) [76]. These non-specific responses should be considered when attributing an MRI marker to a given tissue injury feature as they can influence interpretation of response to therapy.
Considering these non-specific MRI effects, we performed detailed histological analyses to quantify features such as infarction, edema and iron and macrophage infiltration. Overall, MRI measures were in agreement with ground truth histological findings at end study (week 4). We noted that although there was no change in the number or density of iron punctae across the groups, the average size of iron clusters was significantly less in the DFP group compared to control, indicating that iron was indeed removed by DFP. This likely means that there were possibly equivalent amounts of iron initially deposited in each group, and treatment with DFP shrank the individual clusters of iron deposits. This is in agreement with observations by T2* MRI in the scar region, where low T2* values at day 2 were comparable between the groups, indicating similar iron levels, and at weeks 1 and 4, T2* was significantly elevated in the DFP group suggesting removal of iron. Furthermore, there was trend toward less global edematous response in the DFP-treated group compared to control similar to that indicated by the T2 response; significance was not achieved per heart given the small sample size. In this light, we speculate that greater degree of wall thickness observed in the remote zone of control group (relative to DFP) could be due to both compensatory hypertrophy and persistent edema, although it is difficult to determine their relative contributions. The macrophage quantification results were not what we would have predicted. The evidence of decreased edema (by both T2 and histology) and decreased wall thickness would suggest that there would be reduced immune response, and therefore fewer macrophages with treatment. However, our antibody cannot distinguish between the many roles that macrophages can have (e.g., reparative vs inflammatory). At the later 4 week time point post-MI, macrophages are more likely of the reparative variety, as the inflammatory ones are generally resolved within the first few days of the initial injury [68]. Thus, this increase in macrophages within the scar of DFP-treated hearts is likely to indicate an increase in the reparative mechanisms that occur post-MI, as a consequence of the DFP treatment. Future molecular studies interrogating the inflammatory state in both the acute and chronic phases of MI are warranted to confirm this.
The consequences of I/R partly negate the benefits offered by reperfusion. This has initiated a new interest in targeting the post-reperfusion components for extending the cardioprotection brought about by reperfusion alone. Thus, I/R remains a therapeutic challenge and novel adjunctive therapies for IMH could offer an incremental benefit resulting in better patient outcomes and a further reduction in mortality.
Limitations
This is a proof-of-concept study and hence we acknowledge several limitations. Animal numbers are small; however, we have shown previously that porcine groups of four to seven animals are sufficient to distinguish tissue response and remodeling processes across animal groups with different injury patterns via MRI [40, 41]. The study design involved pre-loading of DFP, i.e., treatment prior to the onset of ischemia, which is not a realistic clinical scenario. Our study also cannot determine the relative beneficial contribution of iron chelation in curbing iron-induced oxidative stress at the time of reperfusion, compared with the interaction with hemorrhagic iron in the acute/subacute stage. Nevertheless, prior studies by Reddy et al. and Lesnefsky et al. [61, 74] in animals and by Chan et al [21] in AMI patients evaluated DFO as a single dose therapy and found no significant beneficial effect on chronic remodeling. In contrast, our study demonstrated that sustained DFP administration is able to mitigate inflammation and adverse myocardial remodeling. This suggests that cardioprotection may likely arise from the neutralization of iron by-products that form in the acute/subacute stage, rather than of those available at the time of reperfusion. Importantly, this also supports the premise that DFP would have shown efficacy if pre-loading were not part of the study design, i.e., DFP administered post-reperfusion, as would be the case in clinical use, would still have shown a beneficial response. Nevertheless, future preclinical studies focused on human dosing strategies, such as at reperfusion or after, are warranted to demonstrate the efficacy of DFP in a clinically translatable situation.
A potential obstacle is the limited access of DFP to the infarct core, since hemorrhage occurs within the region of MVO (as seen in a 90 min porcine model) [42, 85]. In this study we did not perform DFP assays to examine the penetrability of the drug in the infarct core. However, MVO is also partially resolved by week 1 [42] and hence it might be reasonable to assume better accessibility of the drug to the hemorrhagic core beyond this time point (particularly in the case of sustained treatment as in our study). To bring this into context, the MR contrast agent gadolinium–DTPA, a much larger complex (938 Da) [79], can fill into the MVO region with late enhancement imaging [85]. Hence, it is reasonable that DFP should be able to get to the infarct site; importantly, based on the evolution of hemorrhage, ferric iron begins to appear only after 3–5 days [10, 42], at which time DFP should be most effective. In our study, IMH was still present only in one DFP-treated animal at week 1, as opposed to complete resolution in the rest of the animals in that group. A recent study in procine I/R demonstrated that IMH peaks at day 4 post-AMI, where differences in drug intervention could be expected to be much larger. Although the present study missed this time point, we were nevertheless able to note statistically significant differences in IMH between the control and treatment groups.
Future studies examining DFP assays in tissue specimens will help assess penetrability. The most common side effects of DFP are gastrointestinal upset, vomiting and diarrhea; however, the pigs in our study did not suffer from any of these with the dosing regimen during the 4-week period. The optimal dose level and dosing frequency for the administration of iron chelators in AMI is also not known; future dose-ranging studies will need to be designed to establish these variables. PK analysis was only performed in healthy animals; however, it is uncertain whether the profile would alter or not post-AMI. Future studies would have to address this aspect.
The 4-week end point is not truly chronic to observe remodeling, as inflammation is not completely resolved, and scar is not mature at this phase. This is probably why we did not observe any significant infarct zone thinning at 4 weeks, compared to baseline. Nevertheless, this time point was sufficient to detect significant differences between the two concerned animal groups. Our histological analysis revealed interesting information about the tissue state, but also had some limitations: (a) our animal count was too small to expect statistical significance when comparing histological averages for each whole heart; (b) we were only able to perform staining and analysis in a single mid-ventricular section per heart given technical challenges with tissue storage; (c) we measured the space present between cells and tissue fibers to estimate edema; however, these spaces could be impacted by artifacts resulting from mechanical handling of the tissue; we have been careful to manually exclude such regions from the analysis. Lastly, cardiovascular implications in males and females can be quite different as evident from recent reports [11, 71, 72]; however, ours was a proof-of-concept study that utilized only female pigs and hence cannot assess sex-related responses to the drug.
Conclusions
Iron deposition as a result of reperfusion hemorrhage presents a serious condition in STEMI patients, contributing to increased inflammation and chronic adverse remodeling. To date, there is no available targeted therapy for reperfusion hemorrhage, which has now been confirmed to be an independent predictor of adverse outcomes in humans. An approach targeting the components of I/R early in the acute phase potentially offers an attractive mechanism to improve long-term outcomes. Our proof-of-concept study is the first to identify hemorrhage-derived iron as a potential therapeutic target in I/R and exploit the cardioprotective properties of an iron-chelating drug candidate in the setting of AMI. We have demonstrated that elimination of hemorrhagic iron can result in faster resolution of inflammation and prevent tissue hypertrophy and ventricular dilatation (early signatures of heart failure), thus promoting a beneficial remodeling process. The identification of this new therapeutic target in the appropriate patient population (large myocardial infarctions that produce IMH) should be considered for future clinical trials.
Abbreviations
- AMI:
-
Acute myocardial infarction
- MVO:
-
Microvascular obstruction
- IMH:
-
Intramyocardial hemorrhage
- STEMI:
-
ST-segment elevation myocardial infarction
- LAD:
-
Left anterior descending
- MRI:
-
Magnetic resonance imaging
- LGE:
-
Late gadolinium enhancement
- SSFP:
-
Steady-state free precession
- IR-GRE:
-
Inversion recovery gradient echo sequence
- LV:
-
Left ventricle
- EF:
-
Ejection fraction
- EDV:
-
End-diastolic volume
- ANOVA:
-
Analysis of variance
- I/R:
-
Ischemia–reperfusion injury
- MT:
-
Masson’s trichrome
- PB:
-
Prussian blue
- SD:
-
Standard deviation
- DFP:
-
Deferiprone
- DFO:
-
Desferoxamine
- PPCI:
-
Primary percutaneous coronary intervention
- EDTA:
-
Ethylenediaminetetraacetic acid
- HF:
-
Heart failure
References
Al-Refaie FN, Sheppard LN, Nortey P, Wonke B, Hoffbrand AV (1995) Pharmacokinetics of the oral iron chelator deferiprone (L1) in patients with iron overload. Br J Haematol 89:403–408
Amado LC, Gerber BL, Gupta SN, Rettmann DW, Szarf G, Schock R, Nasir K, Kraitchman DL, Lima JA (2004) Accurate and objective infarct sizing by contrast-enhanced magnetic resonance imaging in a canine myocardial infarction model. J Am Coll Cardiol 44:2383–2389. https://doi.org/10.1016/j.jacc.2004.09.020
Ambrosio G, Zweier JL, Jacobus WE, Weisfeldt ML, Flaherty JT (1987) Improvement of postischemic myocardial function and metabolism induced by administration of deferoxamine at the time of reflow: the role of iron in the pathogenesis of reperfusion injury. Circulation 76:906–915
Analyst I (2015) Detect colored regions in an image via thresholding in HSV color space–SimpleColorDetectionByHue(). In: File Exch.—MATLAB Cent. https://www.mathworks.com/matlabcentral/fileexchange/28512
APEX AMI Investigators, Armstrong PW, Granger CB, Adams PX, Hamm C, Holmes D, O’Neill WW, Todaro TG, Vahanian A, Van de Werf F (2007) Pexelizumab for acute ST-elevation myocardial infarction in patients undergoing primary percutaneous coronary intervention: a randomized controlled trial. JAMA 297:43–51. https://doi.org/10.1001/jama.297.1.43
Baran KW, Nguyen M, McKendall GR, Lambrew CT, Dykstra G, Palmeri ST, Gibbons RJ, Borzak S, Sobel BE, Gourlay SG, Rundle AC, Gibson CM, Barron HV, Limitation of Myocardial Infarction Following Thrombolysis in Acute Myocardial Infarction (LIMIT AMI) Study Group (2001) Double-blind, randomized trial of an anti-CD18 antibody in conjunction with recombinant tissue plasminogen activator for acute myocardial infarction: limitation of myocardial infarction following thrombolysis in acute myocardial infarction (LIMIT AMI) study. Circulation 104:2778–2783
Bekkers SC, Smulders MW, Passos VL, Leiner T, Waltenberger J, Gorgels AP, Schalla S (2010) Clinical implications of microvascular obstruction and intramyocardial haemorrhage in acute myocardial infarction using cardiovascular magnetic resonance imaging. Eur Radiol 20:2572–2578. https://doi.org/10.1007/s00330-010-1849-9
Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, de Ferranti SD, Floyd J, Fornage M, Gillespie C, Isasi CR, Jiménez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Mackey RH, Matsushita K, Mozaffarian D, Mussolino ME, Nasir K, Neumar RW, Palaniappan L, Pandey DK, Thiagarajan RR, Reeves MJ, Ritchey M, Rodriguez CJ, Roth GA, Rosamond WD, Sasson C, Towfighi A, Tsao CW, Turner MB, Virani SS, Voeks JH, Willey JZ, Wilkins JT, Wu JH, Alger HM, Wong SS, Muntner P, American Heart Association Statistics Committee, and Stroke Statistics Subcommittee (2017) Heart disease and stroke statistics-2017 update: a report from the American Heart Association. Circulation. https://doi.org/10.1161/cir.0000000000000485
Berdoukas V, Farmaki K, Carson S, Wood J, Coates T (2012) Treating thalassemia major-related iron overload: the role of deferiprone. J Blood Med 3:119–129. https://doi.org/10.2147/JBM.S27400
Bradley WGJ (1993) MR appearance of hemorrhage in the brain. Radiology 189:15–26
Bucholz EM, Butala NM, Rathore SS, Dreyer RP, Lansky AJ, Krumholz HM (2014) Sex differences in long-term mortality after myocardial infarction: a systematic review. Circulation 130:757–767. https://doi.org/10.1161/circulationaha.114.009480
Bulluck H, Rosmini S, Abdel-Gadir A, White SK, Bhuva AN, Treibel TA, Fontana M, Ramlall M, Hamarneh A, Sirker A, Herrey AS, Manisty C, Yellon DM, Kellman P, Moon JC, Hausenloy DJ (2016) Residual myocardial iron following intramyocardial hemorrhage during the convalescent phase of reperfused ST-segment-elevation myocardial infarction and adverse left ventricular remodeling. Circ Cardiovasc Imaging. https://doi.org/10.1161/circimaging.116.004940
Bøtker HE, Hausenloy D, Andreadou I, Antonucci S, Boengler K, Davidson SM, Deshwal S, Devaux Y, Di Lisa F, Di Sante M, Efentakis P, Femminò S, García-Dorado D, Giricz Z, Ibanez B, Iliodromitis E, Kaludercic N, Kleinbongard P, Neuhäuser M, Ovize M, Pagliaro P, Rahbek-Schmidt M, Ruiz-Meana M, Schlüter K-D, Schulz R, Skyschally A, Wilder C, Yellon DM, Ferdinandy P, Heusch G (2018) Practical guidelines for rigor and reproducibility in preclinical and clinical studies on cardioprotection. Basic Res Cardiol 113:39. https://doi.org/10.1007/s00395-018-0696-8
Cahill TJ, Kharbanda RK (2017) Heart failure after myocardial infarction in the era of primary percutaneous coronary intervention: mechanisms, incidence and identification of patients at risk. World J Cardiol 9:407–415. https://doi.org/10.4330/wjc.v9.i5.407
Cai B, Deitch EA, Ulloa L (2010) Novel insights for systemic inflammation in sepsis and hemorrhage. Mediat Inflamm 2010:642462. https://doi.org/10.1155/2010/642462
Cannan C, Eitel I, Hare J, Kumar A, Friedrich M (2010) Hemorrhage in the myocardium following infarction. JACC Cardiovasc Imaging 3:665–668. https://doi.org/10.1016/j.jcmg.2009.12.019
Carberry J, Carrick D, Haig C, Ahmed N, Mordi I, McEntegart M, Petrie MC, Eteiba H, Hood S, Watkins S, Lindsay M, Davie A, Mahrous A, Ford I, Sattar N, Welsh P, Radjenovic A, Oldroyd KG, Berry C (2017) Persistent iron within the infarct core after ST-segment elevation myocardial infarction: implications for left ventricular remodeling and health outcomes. JACC Cardiovasc Imaging. https://doi.org/10.1016/j.jcmg.2017.08.027
Carpenter J-P, He T, Kirk P, Roughton M, Anderson LJ, de Noronha SV, Baksi AJ, Sheppard MN, Porter JB, Walker JM, Wood JC, Forni G, Catani G, Matta G, Fucharoen S, Fleming A, House M, Black G, Firmin DN, St Pierre TG, Pennell DJ (2014) Calibration of myocardial T2 and T1 against iron concentration. J Cardiovasc Magn Reson 16:62. https://doi.org/10.1186/s12968-014-0062-4
Carrick D, Haig C, Ahmed N, McEntegart M, Petrie MC, Eteiba H, Hood S, Watkins S, Lindsay MM, Davie A, Mahrous A, Mordi I, Rauhalammi S, Sattar N, Welsh P, Radjenovic A, Ford I, Oldroyd KG, Berry C (2016) Myocardial hemorrhage after acute reperfused ST-segment-elevation myocardial infarction: relation to microvascular obstruction and prognostic significance. Circ Cardiovasc Imaging 9:e004148. https://doi.org/10.1161/circimaging.115.004148
Cerqueira MD, Weissman NJ, Dilsizian V, Jacobs AK, Kaul S, Laskey WK, Pennell DJ, Rumberger JA, Ryan T, Verani MS, American Heart Association Writing Group on Myocardial Segmentation, and Registration for Cardiac Imaging (2002) Standardized myocardial segmentation and nomenclature for tomographic imaging of the heart. A statement for healthcare professionals from the Cardiac Imaging Committee of the Council on Clinical Cardiology of the American Heart Association. Circulation 105:539–542
Chan W, Taylor AJ, Ellims AH, Lefkovits L, Wong C, Kingwell BA, Natoli A, Croft KD, Mori T, Kaye DM, Dart AM, Duffy SJ (2012) Effect of iron chelation on myocardial infarct size and oxidative stress in ST-elevation-myocardial infarction. Circ Cardiovasc Interv 5:270–278. https://doi.org/10.1161/circinterventions.111.966226
Cheng H-LM, Stikov N, Ghugre NR, Wright GA (2012) Practical medical applications of quantitative MR relaxometry. J Magn Reson Imaging 36:805–824. https://doi.org/10.1002/jmri.23718
Dendorfer A, Heidbreder M, Hellwig-Burgel T, Johren O, Qadri F, Dominiak P (2005) Deferoxamine induces prolonged cardiac preconditioning via accumulation of oxygen radicals. Free Radic Biol Med 38:117–124. https://doi.org/10.1016/j.freeradbiomed.2004.10.015
Dharmakumar R (2016) “Rusty hearts”: is it time to rethink iron chelation therapies in post-myocardial-infarction setting? Circ Cardiovasc Imaging. https://doi.org/10.1161/circimaging.116.005541
Eitel I, Kubusch K, Strohm O, Desch S, Mikami Y, de Waha S, Gutberlet M, Schuler G, Friedrich MG, Thiele H (2011) Prognostic value and determinants of a hypointense infarct core in T2-weighted cardiac magnetic resonance in acute reperfused ST-elevation-myocardial infarction. Circ Cardiovasc Imaging 4:354–362. https://doi.org/10.1161/circimaging.110.960500
Faxon DP, Gibbons RJ, Chronos NAF, Gurbel PA, Sheehan F, Investigators HALT-MI (2002) The effect of blockade of the CD11/CD18 integrin receptor on infarct size in patients with acute myocardial infarction treated with direct angioplasty: the results of the HALT-MI study. J Am Coll Cardiol 40:1199–1204
Fernández-Jiménez R, Galán-Arriola C, Sánchez-González J, Agüero J, López-Martín GJ, Gomez-Talavera S, Garcia-Prieto J, Benn A, Molina-Iracheta A, Barreiro-Pérez M, Martin-García A, García-Lunar I, Pizarro G, Sanz J, Sánchez PL, Fuster V, Ibanez B (2017) Effect of ischemia duration and protective interventions on the temporal dynamics of tissue composition after myocardial infarction. Circ Res 121:439–450. https://doi.org/10.1161/circresaha.117.310901
Fishbein MC, Y-Rit J, Lando U, Kanmatsuse K, Mercier JC, Ganz W (1980) The relationship of vascular injury and myocardial hemorrhage to necrosis after reperfusion. Circulation 62:1274–1279
Fradette C, Pichette V, Sicard É, Stilman A, Jayashankar S, Tsang YC, Spino M, Tricta F (2016) Effects of renal impairment on the pharmacokinetics of orally administered deferiprone. Br J Clin Pharmacol 82:994–1001. https://doi.org/10.1111/bcp.13037
Frangogiannis NG, Smith CW, Entman ML (2002) The inflammatory response in myocardial infarction. Cardiovasc Res 53:31–47
Friedrich MG, Kim HW, Kim RJ (2011) T2-weighted imaging to assess post-infarct myocardium at risk. JACC Cardiovasc Imaging 4:1014–1021. https://doi.org/10.1016/j.jcmg.2011.07.005
Ganame J, Messalli G, Dymarkowski S, Rademakers FE, Desmet W, Van de Werf F, Bogaert J (2009) Impact of myocardial haemorrhage on left ventricular function and remodelling in patients with reperfused acute myocardial infarction. Eur Heart J 30:1440–1449. https://doi.org/10.1093/eurheartj/ehp093
Garcia-Dorado D, Oliveras J, Gili J, Sanz E, Perez-Villa F, Barrabes J, Carreras MJ, Solares J, Soler-Soler J (1993) Analysis of myocardial oedema by magnetic resonance imaging early after coronary artery occlusion with or without reperfusion. Cardiovasc Res 27:1462–1469
Garcia-Dorado D, Ruiz-Meana M, Piper HM (2009) Lethal reperfusion injury in acute myocardial infarction: facts and unresolved issues. Cardiovasc Res 83:165–168. https://doi.org/10.1093/cvr/cvp185
Garcia-Dorado D, Theroux P, Solares J, Alonso J, Fernandez-Aviles F, Elizaga J, Soriano J, Botas J, Munoz R (1990) Determinants of hemorrhagic infarcts: histologic observations from experiments involving coronary occlusion, coronary reperfusion, and reocclusion. Am J Pathol 137:301–311
Ghugre NR, Coates TD, Nelson MD, Wood JC (2005) Mechanisms of tissue-iron relaxivity: nuclear magnetic resonance studies of human liver biopsy specimens. Magn Reson Med 54:1185–1193. https://doi.org/10.1002/mrm.20697
Ghugre NR, Enriquez CM, Gonzalez I, Nelson MDJ, Coates TD, Wood JC (2006) MRI detects myocardial iron in the human heart. Magn Reson Med 56:681–686. https://doi.org/10.1002/mrm.20981
Ghugre NR, Gonzalez-Gomez I, Butensky E, Noetzli L, Fischer R, Williams R, Harmatz P, Coates TD, Wood JC (2009) Patterns of hepatic iron distribution in patients with chronically transfused thalassemia and sickle cell disease. Am J Hematol 84:480–483. https://doi.org/10.1002/ajh.21456
Ghugre NR, Gonzalez-Gomez I, Shimada H, Coates TD, Wood JC (2010) Quantitative analysis and modelling of hepatic iron stores using stereology and spatial statistics. J Microsc 238:265–274. https://doi.org/10.1111/j.1365-2818.2009.03355.x
Ghugre NR, Pop M, Barry J, Connelly KA, Wright GA (2013) Quantitative magnetic resonance imaging can distinguish remodeling mechanisms after acute myocardial infarction based on the severity of ischemic insult. Magn Reson Med 70:1095–1105. https://doi.org/10.1002/mrm.24531
Ghugre NR, Pop M, Thomas R, Newbigging S, Qi X, Barry J, Strauss BH, Wright GA (2017) Hemorrhage promotes inflammation and myocardial damage following acute myocardial infarction: insights from a novel preclinical model and cardiovascular magnetic resonance. J Cardiovasc Magn Reson 19:50. https://doi.org/10.1186/s12968-017-0361-7
Ghugre NR, Ramanan V, Pop M, Yang Y, Barry J, Qiang B, Connelly KA, Dick AJ, Wright GA (2011) Quantitative tracking of edema, hemorrhage, and microvascular obstruction in subacute myocardial infarction in a porcine model by MRI. Magn Reson Med 66:1129–1141. https://doi.org/10.1002/mrm.22855
Hausenloy DJ, Botker HE, Engstrom T, Erlinge D, Heusch G, Ibanez B, Kloner RA, Ovize M, Yellon DM, Garcia-Dorado D (2017) Targeting reperfusion injury in patients with ST-segment elevation myocardial infarction: trials and tribulations. Eur Heart J 38:935–941. https://doi.org/10.1093/eurheartj/ehw145
Hershko C, Link G, Konijn AM, Cabantchik ZI (2005) Objectives and mechanism of iron chelation therapy. Ann N Acad Sci 1054:124–135. https://doi.org/10.1196/annals.1345.015
Heusch G (2016) The coronary circulation as a target of cardioprotection. Circ Res 118:1643–1658. https://doi.org/10.1161/circresaha.116.308640
Heusch G, Gersh BJ (2017) The pathophysiology of acute myocardial infarction and strategies of protection beyond reperfusion: a continual challenge. Eur Heart J 38:774–784. https://doi.org/10.1093/eurheartj/ehw224
Heusch G, Libby P, Gersh B, Yellon D, Böhm M, Lopaschuk G, Opie L (2014) Cardiovascular remodelling in coronary artery disease and heart failure. Lancet Lond Engl 383:1933–1943. https://doi.org/10.1016/S0140-6736(14)60107-0
Heusch P, Nensa F, Heusch G (2015) Is MRI really the gold standard for the quantification of salvage from myocardial infarction? Circ Res 117:222–224. https://doi.org/10.1161/circresaha.117.306929
Higginson LA, Beanlands DS, Nair RC, Temple V, Sheldrick K (1983) The time course and characterization of myocardial hemorrhage after coronary reperfusion in the anesthetized dog. Circulation 67:1024–1031
Hinder M, Yi BA, Langenickel TH (2018) Developing drugs for heart failure with reduced ejection fraction: what have we learned from clinical trials? Clin Pharmacol Ther 103:802–814. https://doi.org/10.1002/cpt.1010
Husser O, Monmeneu JV, Sanchis J, Nunez J, Lopez-Lereu MP, Bonanad C, Chaustre F, Gomez C, Bosch MJ, Hinarejos R, Chorro FJ, Riegger GAJ, Llacer A, Bodi V (2013) Cardiovascular magnetic resonance-derived intramyocardial hemorrhage after STEMI: Influence on long-term prognosis, adverse left ventricular remodeling and relationship with microvascular obstruction. Int J Cardiol 167:2047–2054. https://doi.org/10.1016/j.ijcard.2012.05.055
Jaffe R, Charron T, Puley G, Dick A, Strauss BH (2008) Microvascular obstruction and the no-reflow phenomenon after percutaneous coronary intervention. Circulation 117:3152–3156. https://doi.org/10.1161/circulationaha.107.742312
Jaffe R, Dick A, Strauss BH (2010) Prevention and treatment of microvascular obstruction-related myocardial injury and coronary no-reflow following percutaneous coronary intervention: a systematic approach. JACC Cardiovasc Interv 3:695–704. https://doi.org/10.1016/j.jcin.2010.05.004
Johanna S, Josephine M, Nils W, Mahbubul A, Viveka F-K (2014) Evolution of left ventricular ejection fraction after acute myocardial infarction. Circulation 130:743–748. https://doi.org/10.1161/CIRCULATIONAHA.114.009924
Jugdutt BI (2003) Ventricular remodeling after infarction and the extracellular collagen matrix: when is enough enough? Circulation 108:1395–1403. https://doi.org/10.1161/01.CIR.0000085658.98621.49
Kali A, Cokic I, Tang R, Dohnalkova A, Kovarik L, Yang H-J, Kumar A, Prato FS, Wood JC, Underhill D, Marbán E, Dharmakumar R (2016) Persistent microvascular obstruction after myocardial infarction culminates in the confluence of ferric iron oxide crystals, proinflammatory burden, and adverse remodeling. Circ Cardiovasc Imaging. https://doi.org/10.1161/circimaging.115.004996
Kali A, Kumar A, Cokic I, Tang RLQ, Tsaftaris SA, Friedrich MG, Dharmakumar R (2013) Chronic manifestation of postreperfusion intramyocardial hemorrhage as regional iron deposition: a cardiovascular magnetic resonance study with ex vivo validation. Circ Cardiovasc Imaging 6:218–228. https://doi.org/10.1161/CIRCIMAGING.112.000133
Kobayashi S, Tadokoro H, Wakida Y, Kar S, Nordlander R, Haendchen RV, Corday E (1991) Coronary venous retroinfusion of deferoxamine reduces infarct size in pigs. J Am Coll Cardiol 18:621–627
Lamas GA, Goertz C, Boineau R, Mark DB, Rozema T, Nahin RL, Lindblad L, Lewis EF, Drisko J, Lee KL, Investigators TACT (2013) Effect of disodium EDTA chelation regimen on cardiovascular events in patients with previous myocardial infarction: the TACT randomized trial. JAMA 309:1241–1250. https://doi.org/10.1001/jama.2013.2107
Landmesser U, Wollert KC, Drexler H (2009) Potential novel pharmacological therapies for myocardial remodelling. Cardiovasc Res 81:519–527. https://doi.org/10.1093/cvr/cvn317
Lesnefsky EJ, Hedlund BE, Hallaway PE, Horwitz LD (1990) High-dose iron-chelator therapy during reperfusion with deferoxamine-hydroxyethyl starch conjugate fails to reduce canine infarct size. J Cardiovasc Pharmacol 16:523–528
Lesnefsky EJ, Repine JE, Horwitz LD (1990) Deferoxamine pretreatment reduces canine infarct size and oxidative injury. J Pharmacol Exp Ther 253:1103–1109
Libby P, Maroko PR, Bloor CM, Sobel BE, Braunwald E (1973) Reduction of experimental myocardial infarct size by corticosteroid administration. J Clin Invest 52:599–607. https://doi.org/10.1172/JCI107221
Limenta LMG, Jirasomprasert T, Tankanitlert J, Svasti S, Wilairat P, Chantharaksri U, Fucharoen S, Morales NP (2008) UGT1A6 genotype-related pharmacokinetics of deferiprone (L1) in healthy volunteers. Br J Clin Pharmacol 65:908–916. https://doi.org/10.1111/j.1365-2125.2008.03103.x
MacLean MJ, Biro GP (1992) Time course of myocardial bloodflow changes during healing of myocardial infarct in pigs. Can J Cardiol 8:749–755
Mather AN, Fairbairn TA, Ball SG, Greenwood JP, Plein S (2011) Reperfusion haemorrhage as determined by cardiovascular MRI is a predictor of adverse left ventricular remodelling and markers of late arrhythmic risk. Heart 97:453–459. https://doi.org/10.1136/hrt.2010.202028
Murphy E, Steenbergen C (2008) Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol Rev 88:581–609. https://doi.org/10.1152/physrev.00024.2007
Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo J-L, Libby P, Weissleder R, Pittet MJ (2007) The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med 204:3037–3047. https://doi.org/10.1084/jem.20070885
Olivieri NF, Brittenham GM (1997) Iron-chelating therapy and the treatment of thalassemia. Blood 89:739–761
O’Regan DP, Ahmed R, Karunanithy N, Neuwirth C, Tan Y, Durighel G, Hajnal JV, Nadra I, Corbett SJ, Cook SA (2009) Reperfusion hemorrhage following acute myocardial infarction: assessment with T2* mapping and effect on measuring the area at risk. Radiology 250:916–922. https://doi.org/10.1148/radiol.2503081154
Pepe A, Gamberini MR, Missere M, Pistoia L, Mangione M, Cuccia L, Spasiano A, Maffei S, Cadeddu C, Midiri M, Borgna C, Meloni A, Working Group of Gender Medicine of Italian Society of Cardiology (2018) Gender differences in the development of cardiac complications: a multicentre study in a large cohort of thalassaemia major patients to optimize the timing of cardiac follow-up. Br J Haematol 180:879–888. https://doi.org/10.1111/bjh.15125
Potts J, Sirker A, Martinez SC, Gulati M, Alasnag M, Rashid M, Kwok CS, Ensor J, Burke DL, Riley RD, Holmvang L, Mamas MA (2018) Persistent sex disparities in clinical outcomes with percutaneous coronary intervention: Insights from 6.6 million PCI procedures in the United States. PLoS One 13:e0203325. https://doi.org/10.1371/journal.pone.0203325
Reddy BR, Kloner RA, Przyklenk K (1989) Early treatment with deferoxamine limits myocardial ischemic/reperfusion injury. Free Radic Biol Med 7:45–52
Reddy BR, Wynne J, Kloner RA, Przyklenk K (1991) Pretreatment with the iron chelator desferrioxamine fails to provide sustained protection against myocardial ischaemia-reperfusion injury. Cardiovasc Res 25:711–718
Robbers LFHJ, Eerenberg ES, Teunissen PFA, Jansen MF, Hollander MR, Horrevoets AJG, Knaapen P, Nijveldt R, Heymans MW, Levi MM, van Rossum AC, Niessen HWM, Marcu CB, Beek AM, van Royen N (2013) Magnetic resonance imaging-defined areas of microvascular obstruction after acute myocardial infarction represent microvascular destruction and haemorrhage. Eur Heart J 34:2346–2353. https://doi.org/10.1093/eurheartj/eht100
Rochitte CE, Lima JA, Bluemke DA, Reeder SB, McVeigh ER, Furuta T, Becker LC, Melin JA (1998) Magnitude and time course of microvascular obstruction and tissue injury after acute myocardial infarction. Circulation 98:1006–1014. https://doi.org/10.1161/01.cir.98.10.1006
Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Makuc DM, Marcus GM, Marelli A, Matchar DB, Moy CS, Mozaffarian D, Mussolino ME, Nichol G, Paynter NP, Soliman EZ, Sorlie PD, Sotoodehnia N, Turan TN, Virani SS, Wong ND, Woo D, Turner MB (2012) Heart disease and stroke statistics–2012 update: a report from the American Heart Association. Circulation 125:e2–e220. https://doi.org/10.1161/CIR.0b013e31823ac046
Rossello X, Lopez-Ayala P, Fernández-Jiménez R, Oliver E, Galán-Arriola C, de Molina-Iracheta A, Agüero J, López GJ, Lobo-Gonzalez M, Vílchez-Tschischke JP, Fuster V, Sánchez-González J, Ibanez B (2019) R2 prime (R2’) magnetic resonance imaging for post-myocardial infarction intramyocardial haemorrhage quantification. Eur Heart J Cardiovasc Imaging. https://doi.org/10.1093/ehjci/jez306
Sampson JH, Brady M, Raghavan R, Mehta AI, Friedman AH, Reardon DA, Petry NA, Barboriak DP, Wong TZ, Zalutsky MR, Lally-Goss D, Bigner DD (2011) Colocalization of gadolinium-diethylene triamine pentaacetic acid with high-molecular-weight molecules after intracerebral convection-enhanced delivery in humans. Neurosurgery 69:668–676. https://doi.org/10.1227/NEU.0b013e3182181ba8
Statistics Canada (2011c, October) Mortality, summary list of causes 2008
Terzic A, Behfar A (2016) Stem cell therapy for heart failure: ensuring regenerative proficiency. Trends Cardiovasc Med 26:395–404. https://doi.org/10.1016/j.tcm.2016.01.003
Vander Heide RS, Steenbergen C (2013) Cardioprotection and myocardial reperfusion: pitfalls to clinical application. Circ Res 113:464–477. https://doi.org/10.1161/CIRCRESAHA.113.300765
Wood JC, Aguilar M, Otto-Duessel M, Nick H, Nelson MD, Moats R (2008) Influence of iron chelation on R1 and R2 calibration curves in gerbil liver and heart. Magn Reson Med 60:82–89. https://doi.org/10.1002/mrm.21660
Wood JC, Enriquez C, Ghugre N, Otto-Duessel M, Aguilar M, Nelson MD, Moats R, Coates TD (2005) Physiology and pathophysiology of iron cardiomyopathy in thalassemia. Ann N Acad Sci 1054:386–395. https://doi.org/10.1196/annals.1345.047
Wu KC (2012) CMR of microvascular obstruction and hemorrhage in myocardial infarction. J Cardiovasc Magn Reson 14:68. https://doi.org/10.1186/1532-429X-14-68
Wu KC, Zerhouni EA, Judd RM, Lugo-Olivieri CH, Barouch LA, Schulman SP, Blumenthal RS, Lima JA (1998) Prognostic significance of microvascular obstruction by magnetic resonance imaging in patients with acute myocardial infarction. Circulation 97:765–772
Ye Y, Perez-Polo JR, Birnbaum Y (2010) Protecting against ischemia-reperfusion injury: antiplatelet drugs, statins, and their potential interactions. Ann N Acad Sci 1207:76–82. https://doi.org/10.1111/j.1749-6632.2010.05725.x
Yellon DM, Hausenloy DJ (2007) Myocardial reperfusion injury. N Engl J Med 357:1121–1135. https://doi.org/10.1056/NEJMra071667
Zia MI, Ghugre NR, Connelly KA, Strauss BH, Sparkes JD, Dick AJ, Wright GA (2012) Characterizing myocardial edema and hemorrhage using quantitative T2 and T2* mapping at multiple time intervals post ST-segment elevation myocardial infarction. Circ Cardiovasc Imaging 5:566–572. https://doi.org/10.1161/CIRCIMAGING.112.973222
Acknowledgements
We would like to thank ApoPharma Inc. for supplying us with deferiprone (Ferriprox®) for use in this study.
Funding
The study was funded by the Ontario Research Fund—Research Excellence (ORF-RE7-21). We also acknowledge support from the Sunnybrook Research Institute Summer Student Program. The pharmacokinetic analysis of deferiprone was funded by ApoPharma Inc.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
A joint patent application between Sunnybrook Research Institute, Childrens Hospital Los Angeles and ApoPharma Inc. has been filed related to the methods of treatment with deferiprone in ischemia–reperfusion injury. NG, GW, JW, JC and MS have been listed as inventors on the patent application.
Ethics approval
Animal procedures were conducted in accordance with protocols approved by the Animal Care Committee of Sunnybrook Research Institute.
Rights and permissions
About this article

Cite this article
Behrouzi, B., Weyers, J.J., Qi, X. et al. Action of iron chelator on intramyocardial hemorrhage and cardiac remodeling following acute myocardial infarction. Basic Res Cardiol 115, 24 (2020). https://doi.org/10.1007/s00395-020-0782-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00395-020-0782-6