
Abstract
Proprotein convertase subtilisin/Kexin type 9 (PCSK9) and pyroptosis both play important roles in myocardial infarction. This study was designed to test the hypothesis that PCSK9 regulates pyroptosis in cardiomyocytes during chronic myocardial ischemia. Primary cardiomyocytes were isolated from WT and PCSK9−/− mice. HL-1 cardiomyocytes were used to set up PCSK9-deficient (PCSK9−/−) and PCSK9-upregulated (PCSK9CRISPRa) cardiomyocyte cell line with CRISPR/Cas9 knockout or activation plasmid. Additional studies were performed with chronic myocardial ischemia in WT and PCSK9−/− mice. We observed that PCSK9 initiates mitochondrial DNA (mtDNA) damage, activates NLRP3 inflammasome signaling (NLRP3, ASC, Caspase-1, IL-1β, and IL-18), and subsequently induces Caspase-1-dependent pyroptosis. There was an intense expression of PCSK9 and pyroptosis marker, GSDMD-NT, in the zone bordering the infarct area. PCSK9−/− significantly suppressed expression of NLRP3 inflammasome signaling, GSDMD-NT, and LDH release. Furthermore, serum levels of PCSK9, NLPR3 inflammasome signaling, and pyroptosis (GSDMD and LDH release) were significantly elevated in patients with chronic myocardial ischemia as compared to those in age-matched healthy subjects. Human hearts with recent infarcts also showed high expression of PCSK9 and GSDMD-NT in the border zone similar to that in the infarcted mouse heart. These observations provide compelling evidence for the role of PCSK9 in regulating Caspase-1-dependent pyroptosis via mtDNA damage and may qualify pro-inflammatory cytokines and pyroptosis as potential targets to treat PCSK9-related cardiovascular diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Myocardial infarction, caused by chronic myocardial ischemia and hypoxia, remains a leading cause of disability and mortality in the US and is rapidly becoming a major issue worldwide [9]. A long-time postulate is that programmed cell death and inflammation play key roles in the development of myocardial infarction [2, 28]. Pyroptosis is a novel form of inflammatory programmed cell death characterized by the caspase-1-dependent formation of pores in the plasma membrane that leads to the release of pro-inflammatory cytokines such as interleukin (IL)-1β and IL-18 [15, 19, 26]. Induction of pyroptosis is closely associated with activation of the NOD-like receptor 3 (NLRP3) inflammasome [30, 31]. The NLRP3 inflammasome activates caspase-1 and cleaves gasdermin D (GSDMD) to generate an N-terminal GSDMD fragment (GSDMD-NT) which further induces the formation of membrane pores and subsequent inflammatory responses [10, 22]. Inhibition of the NLRP3 inflammasome signaling pathway in a mouse model with myocardial infarction reduces infarct size and preserves cardiac function via attenuation of cardiomyocyte pyroptosis [27].
Proprotein convertase subtilisin/Kexin type 9 (PCSK9) degrades low-density lipoprotein cholesterol receptors (LDLr) and subsequently raises low-density lipoprotein cholesterol levels [11]. PCSK9 is mainly secreted by the liver, kidney, and small intestine; however, recent studies showed that PCSK9 is also expressed in vascular smooth muscle cells [5], endothelial cells, [5] and macrophages [6, 8] when these cells are exposed to inflammatory stimuli such as lipopolysaccharide (LPS). PCSK9 is considered an inflammatory molecule and is a part of the salutary effect of PCSK9 inhibitors that may be manifested by the downregulation of inflammatory cascade [13]. We recently showed that PCSK9 is upregulated in ischemic heart tissue and determined the development of infarct size and cardiac function [7]. Palee et al. further confirmed that a PCSK9 inhibitor improves cardiac function and reduces infarct size in rats with ischemia/reperfusion injury [7]. Schlüter et al. also found that oxidized low-density lipoprotein (ox-LDL) significantly impaired contractile function via induction of PCSK9 [25].
These findings support a promising prospective that activation of pyroptosis and the PCSK9–pyroptosis interaction represents one type of myocardial injury in the pathogenesis of myocardial infarction. Although the administration of a PCSK9 inhibitor temporarily improved cardiac function in mice with myocardial ischemia [7], it failed to diminish the death of cardiomyocytes and myocardial infarction continued to deteriorate after the withdrawal of the inhibitor. Thus, to design effective therapeutics for myocardial infarction, it is critical to define the mechanisms of PCSK9-mediated pyroptosis in myocardial infarction and to identify the key molecules mediating PCSK9–pyroptosis interactions. Since PCSK9 is involved in NLRP3 inflammasome activation, we hypothesized that PCSK9 may play a role in the regulation of NLRP3 inflammasome-mediated pyroptosis during chronic myocardial ischemia. This study was designed to test this postulate.
Materials and methods
Animals
C57BL/6 wild-type (WT) and PCSK9−/− mice with the C57BL/6J background were purchased from the Jackson Laboratories (Sacramento, CA) and housed in the animal care facility of our institution. Mice were sacrificed by intraperitoneal injection of pentobarbital (60 mg/kg) and carbon dioxide (CO2) was used for euthanasia of the mice. All experimental procedures were performed per protocols approved by the Institutional Animal Care and Usage Committee and conformed to the guidelines for the Care and Use of Laboratory Animals published by the US National Institutes of Health. To avoid the variable of sex on PCSK9 levels, only male mice approximately 8 weeks of age were used in this study.
Myocardial ischemia protocol
The methodology for the induction of myocardial ischemia has been previously described [7, 14]. In brief, animals were anesthetized with ketamine hydrochloride (60 mg kg−1) and xylazine hydrochloride (8 mg kg−1) intraperitoneally. After endotracheal intubation, mice were mechanically ventilated (tidal volume, 1.2 ml min−1 and respiratory rate, 110 min) and body temperature was maintained between 37.0 °C and 37.5 °C with a heating pad. An electrocardiogram was recorded throughout the procedure. After an equilibration period of 5 min, under aseptic conditions, a left thoracotomy was performed in the 4th intercostal space and the pericardium opened. An 8-0 silk suture was passed around the left coronary artery (LCA) at a point two-thirds of the way between its origin near the pulmonary conus. Occlusion of the LCA caused epicardial cyanosis with regional hypokinesia and expected electrocardiogram changes typical of acute myocardial infarction. Another group of animals underwent the same procedure but without LCA ligation (sham ischemia). The thoracic cavity was closed in layers, using 6-0 sutures, and drained to prevent pneumothorax. Finally, the endotracheal tube was removed and the animals were allowed to recover.
Studies in primary mouse cardiomyocytes
Primary mouse cardiomyocytes were isolated from the hearts of neonatal mice (days 1–3) with Pierce Primary Cardiomyocyte Isolation Kit (Thermo Fisher Scientific). Hypoxic conditions were created by incubating cardiomyocytes in a Controlled Atmosphere Chamber (PASS USA, Lansing, MI) with an atmosphere of 5% CO2/95% N2 at 37 °C. Cells plated in 6-well plates and cultured to 80–90% confluence were transferred to the chamber and incubated in an oxygen-depleted medium for 0.5–7 days. In previous studies, primary cardiomyocytes were exposed to hypoxia for 0.5–2 days in the presence or absence of recombinant mouse PCSK9 protein (0.5, 1 and 2 µg ml−1, Life Technologies, Grand Island, NY).
Assessment of cardiomyocyte metabolic activity
Cell metabolic activity was assessed using an MTT assay kit (ATCC) according to the manufacturer's instructions. Absorbance at 570 nm was measured using a microplate reader.
ELISA for PCSK9, Caspase-1, IL-1β, IL-18, and GSDMD
PCSK9 secretion was measured by Mouse or Human PCSK9 ELISA kit (MBL International, Nagoya, Japan). Caspase-1 secretion was measured by Human Caspase-1 ELISA Kit (Abcam). IL-1β secretion was measured by Human or Mouse IL-1β ELISA Kit (Thermo Fisher Scientific). IL-1β secretion was measured by Human or Mouse IL-1β ELISA Kit (Abcam). GSDMD secretion was measured by Human or Mouse Gasdermin D ELISA Kit (Abcam).
LDH release assay
LDL release in cells or serum was assessed using LDH Assay Kit (Abcam) according to the manufacturer's instructions.
PCSK9-deficient (PCSK9−/−) and PCSK9-upregulated (PCSK9CRISPRa) HL-1 cardiomyocytes
HL-1 cardiomyocytes were transfected with PCSK9 CRISPR/Cas9 knockout plasmid (Santa Cruz, TX) and PCSK9 homology- directed repair plasmid (puromycin resistance gene and red fluorescent protein sequence). Transfection was confirmed visually and cells were selected with puromycin for 4–6 days. Single clones were isolated and analyzed to exclude off-target cells. PCSK9 CRISPR Activation Plasmid (Santa Cruz) is a synergistic activation mediator transcription activation system designed to specifically upregulate the expression of PCSK9. CRISPR Activation Plasmid Transient Transfection of HL-1 cardiomyocytes was performed with UltraCruz Transfection Reagent (Santa Cruz).
Western blot
The border zone between the ischemic and remote healthy area was carefully dissected under the microscope. Protein from the hearts and cardiomyocytes were purified with RIPA Lysis Buffer System (Santa Cruz, CA) and loaded onto 12% Mini-PROTEAN® TGX™ Precast Gel (Bio-rad, CA) for electrophoresis. The size-separated proteins were then transferred to Hybond ECL Nitrocellulose Membranes (GE Healthcare, NJ). After blocking with 5% BSA buffer for 1 h, the membranes were incubated with primary antibody at 1:1000 dilution overnight at 4 °C. After washing with PBS containing 0.1% Tween-20, membranes were incubated with a secondary antibody for 1 h, and signals were detected with Pierce ECL Western Blotting Substrate (Thermo Scientific, IL). Intensity quantification of the bands was obtained with Image J software and normalized to β-actin.
Antibodies directed at PCSK9 (ab31762), GSDMD-NT (ab215203), NLRP3 (ab263899), and IL-18 (ab243091, ab207323) were purchased from Abcam (San Francisco, CA). Antibodies directed at ASC (67824), Caspase-1 (24232), and IL-1β (12242) were purchased from Cell signaling (Danvers, MA).
Small interfering RNA (siRNA) transfection
The siRNA duplexes corresponding to NLRP3, ASC, Caspase-1, IL-1β, and IL-18 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). To knockdown gene expression, HL-1 cardiomyocytes were transfected with 100 nM of each siRNA for 48 h with Lipofectamine 3000 reagent kit (Thermo Fisher Scientific, Waltham, MA). As a control, cells were transfected with sequence scrambled siRNA control (Santa Cruz). Cell lysates were utilized for ELISA or western blot analysis to verify the efficacy of protein knockdown.
Real-time quantitative PCR
Total RNA was reverse transcribed at 42 °C with SuperScript II (Life Technologies). Gene expression was measured using the SYBR Green PCR core reagents (Applied Biosystems). Relative mRNA levels for each sample were quantified based on Ct (the amplification cycle threshold), normalized to GAPDH as an endogenous mRNA standard, and expressed relative to the sham ischemia (set to a value of 1). Primers used were as follows: PCSK9 forward primer (5′-TTGCAGCAGCTGGGAACTT-3′), PCSK9 reverse primer (5′-CCGACTGATGACCTCTGGA-3′), GAPDH forward primer (5′-GGGTCTTTGCAGCGTATGG-3′), and GAPDH reverse primer (5′-ACCTCCTGTTTCTGGGGACT-3′).
Measurement of cellular mitochondrial ROS by flow cytometry
Cellular total ROS generation was measured with a DCFDA Cellular ROS Detection Assay Kit (Abcam). DCFDA (2′,7′ –dichlorofluorescein diacetate) is a fluorogenic dye that measures hydroxyl, peroxyl, and other ROS activity within the cell. Cells were stained with 20 µM DCFDA for 30 min at 37 °C. The fluorescence of DCFDA was measured on the FL-1 channel with the excitation wavelength at 485 nm and the emission wavelength at 535 nm (FACS Vantage SE, Becton Dickinson). Cellular superoxide generation was measured with a Superoxide Detection Kit (Enzo Life Sciences, NY) on the FL-2 channel with the excitation wavelength at 550 nm and the emission wavelength at 620 nm. Data points were recorded with Flowing Software 2.51 and labeled “M2 percentage fluorescence variation” to indicate the percentage of cells with enhanced mitochondrial ROS production.
H&E staining
To prepare the heart sections for staining, they were deparaffinized in xylene and rehydrated through gradient ethanol. Then, the heart sections were stained using a H&E Staining Kit (Abcam) as per manufacturer’s instructions.
Immunostaining
The heart sections were immunohistochemically analyzed using a Mouse/Rabbit Specific HRP/3,3′-diaminobenzidine detection immunohistochemistry kit (Abcam, San Francisco, CA) as per manufacturer’s instructions. For immunofluorescent staining [28], the heart sections were stained with different antibodies for 1 h. Next, the sections were washed with PBS 3 times and incubated with FITC or Texas Red‐conjugated secondary antibody (1:1000, v/v, dilution) for 30 min at room temperature. After several washes in PBS and deionized water, the sections were covered with ProlongH Gold antifade reagent with 4-diamidino‐2‐phenylindole (DAPI; Life Technologies, Carlsbad, CA).
Human study
This study was conducted at the First Affiliated Hospital of Xinxiang Medical University. Ethical research committee approval was obtained at each contributing site and all data were combined. Eligible patients were between the ages of 55 and 70 years and had received a confirmed diagnosis of chronic myocardial ischemia. Written informed consent was obtained from all patients. The diagnostic criteria are based on published consensus guidelines. A diagnosis of chronic myocardial ischemia was established with the history, electrocardiogram, and laboratory test for creatine kinase, troponin-I or -T, and myoglobin. A group of healthy subjects without known disease served as the control.
Human ischemic heart blocks were obtained from the Department of Pathology archives from the First Affiliated Hospital of Xinxiang Medical University. Patients were approximately 65–80 years (n = 5) and all had documented myocardial infarction in the preceding week. The histological sections revealed regions of myocardial necrosis and non-infarcted healthy regions that were interspersed. The protocol for the collection of archived tissue sections was approved on an exempt basis by the Institutional Review Board and conformed to the Helsinki declaration. Multiple sections of the hearts were immunostained for PCSK9, Caspase-1, IL-1β, and GSDMD-NT.
Statistical analysis
The data was represented as mean ± SD. The significant difference between the two groups was tested using an unpaired t-test. Multiple comparisons were analyzed using a one-way ANOVA followed by a Tukey post hoc comparisons test. Spearman correlation coefficients (r2) were used to assess the relationship between variables. All analyses were performed with GraphPad Prism version 7.00 (GraphPad Software, CA). A p value of < 0.05 was considered statistically significant.
Results
PCSK9 regulates mtDNA damage in cardiomyocytes during hypoxia
Myocardial hypoxia is a major factor in the pathology of cardiac ischemia and myocardial infarction. We first studied the effects of PCSK9 on the viability of cardiomyocytes during hypoxia [14, 20]. As shown in Fig. 1a, cell viability decreased slightly as the period of hypoxia increased from 12 to 48 h but it declined rapidly thereafter. Therefore, we maintained the duration of hypoxia from 12 to 48 h in subsequent experiments. Human recombinant PCSK9 (hrPCSK9) slightly inhibited cell viability at high concentrations in normoxia (Fig. 1b). Interestingly, in hypoxia, hrPCSK9 treatment showed that hrPCSK9 significantly decreased cell viability in a dose-dependent manner (Fig. 1b), while PCSK9 deficiency (PCSK9−/−) blocked hypoxia-induced cell death (Fig. 1c). Next, we investigated the role of hypoxia in regulating PCSK9 expression. Our data showed that hypoxia-induced PCSK9 expression in both mRNA and protein levels (Fig. 1d, e).

PCSK9 regulates mtDNA damage in cardiomyocytes during hypoxia. a Cell viability in primary cardiomyocytes during hypoxia. b hrPCSK9 decreases cell viability in primary cardiomyocytes after exposure to normoxia or hypoxia for 24 h. c PCSK9−/− enhances cell viability in primary cardiomyocytes after exposure to to normoxia or hypoxia for 48 h. d and e Hypoxia induces PCSK9 expression at both the mRNA level and the protein level in primary cardiomyocytes. f Hypoxia induces mtDNA damage in primary cardiomyocytes. g Effects of MitoTEMPOL pretreatment (5 μM for 2 h) and PCSK9−/− on hypoxia-mediated mtDNA damage in primary cardiomyocytes after exposure to normoxia or hypoxia for 12 h. h hrPCSK9 induces mtDNA damage in primary cardiomyocytes. i PCSK9CRISPRa induces mtDNA damage in HL-1 cardiomyocytes. MitoTEMPOL was pretreated at 5 μM for 2 h. h, i Cardiomyocytes were exposed to normoxia or hypoxia for 12 h. The data points represent five biological replicates (n = 5) from two independent experiments shown as mean ± SD. The differences between groups were examined using an unpaired t test. Multiple comparisons were analyzed using a one-way ANOVA followed by a Tukey post hoc comparisons test. *p < 0.05, **p < 0.01, *** p < 0.001, ****p < 0.0001 vs. control or as indicated
Mitochondrial DNA (mtDNA) damage activates NLRP3 inflammasome activation which is a crucial factor in mediating inflammatory responses and pyroptosis during myocardial ischemia. In this experiment, hypoxia time-dependently increased mtDNA damage as expected (Fig. 1f). Hypoxia-mediated mtDNA damage was lower in mice pretreated with a mitochondria-targeted antioxidant (MitoTEMPOL) and in PCSK9−/− cardiomyocytes (Fig. 1g). Further analysis showed that treatment with hrPCSK9 alone also induced mtDNA damage (Fig. 1h), while pretreatment with MitoTEMPOL inhibited mtDNA damage mediated by upregulation of PCSK9 (PCSK9CRISPRa) (Fig. 1i). This observation indicates that PCSK9 plays a key role in regulating mtDNA damage in cardiomyocytes.
PCSK9 regulates pyroptosis via mtDNA damage in HL-1 cardiomyocytes
Excess production of mitochondrial reactive oxygen species (mtROS) and mtDNA damage are key triggers in the activation of pyroptosis [29]. It remains unclear how PCSK9 relates to these forms of mitochondrial dysfunction and we do not yet know how it affects the activation of GSDMD (cleavage to GSDMD-NT) and release of LDH, two critical mediators of pyroptosis. After we confirmed the role of PCSK9 in regulating mtDNA damage, we further investigated the relationship between PCSK9 and the production of mtROS in cardiomyocytes during hypoxia. Our data showed that mtROS production was greater during hypoxia than normoxia. In the condition of hypoxia, mtROS production increased with the addition of hrPCSK9 (4 µg ml−1) and in PCSK9CRISPRa HL-1 cardiomyocytes but was inhibited in PCSK9−/− HL-1 cardiomyocytes (Fig. 2a). Thus, we postulated that PCSK9 may play a role in regulating pyroptosis via mtROS-mediated mtDNA damage. As shown in Fig. 2b, c, GSDMD-NT levels and LDH release increased with hrPCSK9 treatment or PCSK9CRISPRa genetic background but they decreased with PCSK9−/− background. Furthermore, pretreatment with MitoTEMPOL inhibited the effects of hrPCSK9 or PCSK9CRISPRa on GSDMD-NT levels and LDH release (Fig. 2d, e). These results indicate that mtDNA damage is involved in PCSK9-mediated pyroptosis.

PCSK9 regulates pyroptosis via mtDNA damage in HL-1 cardiomyocytes. a PCSK9 regulates mtROS production. b and c Effect of PCSK9 on GSDMD-NT expression and LDH release. a–c HL-1 cardiomyocytes were exposed to normoxia or hypoxia for 48 h. d and e Pretreatment with MitoTEMPOL (5 μM for 2 h) inhibits GSDMD-NT levels and LDH release. d, e HL-1 cardiomyocytes were exposed to normoxia or hypoxia for 12 h. Data points represent five biological replicates (n = 5) from two independent experiments shown as mean ± SD. The differences between groups were tested using an unpaired t test. Multiple comparisons were analyzed using a one-way ANOVA followed by a Tukey post hoc comparisons test. *p < 0.05, **p < 0.01, ***p < 0.001 vs. control or as indicated
PCSK9 regulates pyroptosis via NLRP3 inflammasome signaling in HL-1 cardiomyocytes
We previously showed that hypoxia highly induced expression of PCSK9 in primary cardiomyocytes [7]. Other studies have shown that the NLRP3 inflammasome and pyroptosis were activated by hypoxic conditions [4]. Therefore, we postulated a link between PCSK9, NLRP3 inflammasome signaling, and pyroptosis in cardiomyocytes during hypoxia. Our data showed that NLRP3 inflammasome signaling (NLRP3, ASC, cleaved caspase-1 P20, IL-1β, and IL-18) increased with hrPCSK9 pretreatment (4 µg ml−1) and PCSK9CRISPRa. However, NLRP3 inflammasome signaling decreased with PCSK9−/− (Fig. 3a). ELISA analysis showed that PCSK9 also regulated secretion of IL-1β and IL-18 in HL-1 cardiomyocytes (Fig. 3b, c). To investigate the role of the NLRP3 inflammasome in the interaction between PCSK9 and pyroptosis, we used siRNA to knockdown NLRP3 inflammasome signaling. The transfection efficiency of different siRNAs was measured by western blot. As shown in Fig. 3d, siRNA transfection markedly inhibited the expression of the target proteins. Compared with hrPCSK9 treatment, knockdown of NLRP3, ASC, caspase-1, IL-1β, and IL-18 significantly inhibited GSDMD-NT level and LDH release (Fig. 3e, f) indicating that the NLRP3 inflammasome is involved in the interaction between PCSK9 and pyroptosis.

PCSK9 regulates NLRP3 inflammasome signaling in HL-1 cardiomyocytes. a hrPCSK9 and PCSK9CRISPRa induce, while PCSK9−/− inhibits expression of NLRP3 inflammasome signaling and secretion of IL-1β (b) and IL-18 (c). Cells were exposed to normoxia or hypoxia for 48 h. d Confirmation of siRNA transfection efficiency by western blot. siRNA-C: siRNA control. e and f NLRP3 inflammasome signaling inhibition by siRNA transfection blocks hrPCSK9 induced GSDMD-NT expression and LDH release. HL-1 cardiomyocytes were transfected for 48 h with siRNAs targeting NLRP3 inflammasome signaling factors. Indicated groups of cells were treated with hrPCSK9 (4 µg ml−1) in normoxia or hypoxia for another 12 h. Data is representative of five biological replicates (n = 5) from two independent experiments shown as mean ± SD. The differences between two groups was tested by unpaired t test. Multiple comparisons were analyzed by one-way ANOVA followed by Tukey post hoc comparisons test. *p < 0.05, **p < 0.01, ***p < 0.001 vs. control
PCSK9−/− inhibits pyroptosis in the border zone of infarct area
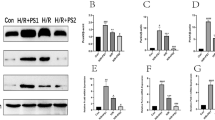
To further investigate the effects of PCSK9 on NLRP3 inflammasome signaling in vivo, we established chronic myocardial ischemia for 1 week in both WT and PCSK9−/− mice. H&E staining showed that 1-week LCA ligation markedly induced infarct size compared with sham control (Fig. 4a). The border zone (peri-infarct) region was defined as the 2-mm area encircling the area of pathologic infarction (Fig. 4a). We studied the expression of NLRP3 inflammasome signaling in serum and heart tissue from the border zone between the infarct area and the unaffected area. As expected, expression of NLRP3 inflammasome signaling molecules (NLRP3, ASC, Caspase-1 p20, IL-1β, and IL-18) were highly expressed in the border zone compared with the remote region (Fig. 4b). The in-vivo studies (consistent with our in-vitro studies) showed that, compared with WT mice, PCSK9−/− mice had markedly lower expression of NLRP3 inflammasome signaling molecules in heart tissue and had lower serum levels of IL-1β and IL-18 (Fig. 4c–f).

PCSK9−/− inhibits NLRP3 inflammasome signaling in the zone bordering infarct area or in serum at 1 week of ischemia (bar = 25 µm). a H&E staining for the remote and border zone regions in the heart. b Expression of NLRP3 inflammasome signaling in the remote and border zone regions. c PCSK9−/− inhibits expression of NLRP3 inflammasome signaling in the border zone. d Quantitative analysis by Image J for NLRP3 inflammasome signaling expression from (c). e and f PCSK9−/− inhibits the secretion of IL-1β and IL-18. Data is representative of seven biological replicates (n = 7) from two independent experiments shown as mean ± SD. The differences between groups was tested by unpaired t test. Multiple comparisons were analyzed by one-way ANOVA followed by Tukey post hoc comparisons test. *p < 0.05, **p < 0.01, or ***p < 0.001 vs. control or as indicated
Recent studies showed that activation of the NLRP3 inflammasome and related pyroptosis were strongly associated with the pathological process of myocardial infarction [27]. As a result of our study confirming the role of PCSK9 in regulating NLRP3 inflammasome signaling (Fig. 3), we further investigated the role of PCSK9 in regulating pyroptosis during chronic myocardial ischemia in mice. Our preliminary data showed that GSDMD-NT levels in the border zone of the ischemic heart and GSDMD-NT and LDH levels in the serum were significantly lower in PCSK9−/− mice than in WT mice (Fig. 5a–c). Similar to PCSK9−/−, MitoT pretreatment markedly reduced expression of GSDMD-NT in the border zone of the ischemic heart (Fig. 5d) and levels of GSDMD-NT and LDH in the serum (Fig. 5e, f).

PCSK9−/− inhibits pyroptosis in the zone bordering the infarct area or in serum at 1 week of ischemia. a–c PCSK9−/− inhibits GSDMD-NT expression, secretion of GSDMD, and LDH in serum. d MitoTEMPOL (MitoT) administration inhibits GSDMD-NT expression in the border zone of the ischemic heart. e and f MitoTEMPOL administration inhibits serum levels of GSDMD and LDH. g MitoTEMPOL pretreatment and PCSK9−/− inhibit GSDMD-NT expression in the border zone of the ischemic heart (bar = 30 µm). MitoTEMPOL at 10 mg kg−1 in 0.3 ml of saline was injected intraperitoneally every 2 days. Data is representative of 5–7 biological replicates (n = 5–7) from two independent experiments shown as mean ± SD. The differences between groups was tested by unpaired t test. Multiple comparisons were analyzed by one-way ANOVA followed by Tukey post hoc comparisons test. **p < 0.01 or ***p < 0.001 vs. as indicated
Previously, we showed that PCSK9 is highly expressed in the border zone between the infarct area and the unaffected area of the heart [7]. Consistent with this, our data showed that GSDMD-NT, a marker of pyroptosis, was present at high levels in the border zone after 1 week of LCA ligation. Importantly, GSDMD-NT levels in the border zone were markedly lower among mice pretreated with mitochondria-targeted antioxidant MitoTEMPOL and mice with PCSK9−/− (Fig. 5g). We formerly showed that PCSK9−/− reduced infarct size and improved cardiac function and this data further suggest that pyroptosis mediated by PCSK9-induced mtDNA damage plays a key role during myocardial ischemia.
PCSK9 and pyroptosis are highly expressed in patients with chronic myocardial ischemia
The demographic details of the study population are shown in Table 1. As expected, patients with chronic myocardial ischemia had higher systolic blood pressure and higher diastolic blood pressure than control subjects. Prevalence of hypertension, dyslipidemia, and diabetes was greater in patients than in control subjects. Aspirin or statins, renin-angiotensin inhibitors, calcium channel blockers, and beta-blockers were frequently used in those with chronic myocardial ischemia. To investigate whether PCSK9 and pyroptosis are present in the serum of human patients with chronic myocardial ischemia, we examined serum levels of PCSK9 and proteins related to pyroptosis (i.e., caspase-1, IL-1β, IL-18, GSDMD-NT, and LDH) among healthy control subjects (n = 20) and patients with chronic myocardial ischemia (n = 21). The preliminary clinical data showed that serum levels of these proteins were higher among the patients than the controls (Fig. 6a–f). To determine whether PCSK9 is present in ischemic heart tissue of humans, we immunohistochemically stained multiple heart sections from patients who had died of acute myocardial infarction (1–7-day post-myocardial infarction). Anti-PCSK9, Caspase-1, IL-1β, and GSDMD staining showed that all these proteins were present at high levels in the border zone between the ischemic areas and remote unaffected areas in each heart (Fig. 6g). These observations are consistent with our findings in mice (Fig. 5g).

PCSK9, NLRP3 inflammasome signaling, and pyroptosis in patients with chronic myocardial ischemia. Compared with healthy control subjects (n = 20), patients with chronic myocardial ischemia (CMI, n = 21) have high levels of a PCSK9, b caspase-1, c IL-1β, d IL-18, e GSDMD, and f LDH. Eligible patients were 56–79 years and had received a diagnosis of chronic myocardial ischemia about 1 month before blood samples were collected. g PCSK9, Caspase-1, IL-1β, and GSDMD are highly expressed in the border zone of the human ischemic heart. Data is shown as mean ± SD from two independent experiments. The differences between groups was tested by unpaired t test. ***p < 0.001, ****p < 0.0001 vs. healthy control
Discussion
Myocardial ischemia is associated with the induction of a sterile inflammatory response that leads to further injury [12]. The NLRP3 inflammasome is a multimeric protein complex that initiates an inflammatory form of cell death (pyroptosis) and triggers the release of proinflammatory cytokines IL-1β and IL-18 [15]. Increasing evidence suggests that PCSK9 is a key signaling molecule in regulating inflammatory response [18, 23]. Similar to NLRP3 inflammasome, PCSK9 regulates infarct size and cardiac function in both myocardial ischemia, reperfusion, and chronic myocardial ischemia [7, 20]. However, it remains largely unknown what the role of PCSK9 is in regulating NLRP3 inflammasome activation and pyroptosis during chronic myocardial ischemia. Herein, we demonstrated for the first time that PCSK9 induces mtDNA damage, activates NLRP3 inflammasome, and further aggravates pyroptosis in cardiomyocytes. We demonstrated that both PCSK9 and pyroptosis related proteins are highest expressed in the zone bordering the infarcted area. These results suggest that PCSK9-induced proinflammatory programmed cell death may be an initial response during chronic myocardial ischemia.
NLRP3 inflammasome activation depends on two functionally distinct steps: “priming” and “activation” [12]. For priming, it involves the direct engagement of Toll-like receptors (TLRs) by pathogen-associated or damage-associated molecular patterns resulting in the rapid activation of nuclear factor kappa B (NF-κB) which stimulates the pro-IL-1β synthesis and increased expression of NLRP3 [12]. For NLRP3 inflammasome activation, increases in mtROS and subsequent mtDNA damage are considered two common inducers [12]. We showed that there is crosstalk between PCSK9 and damaged mtDNA in vascular smooth muscle cells [5] and TLR4 -NF-κB signaling plays an important role in PCSK9 secretion [13]. Therefore, we hypothesized that the axis of PCSK9-mtDNA damage-NLRP3 inflammasome activation-pyroptosis may play a key role in myocardial infarction. For three separate conditions (hrPCSK9, PCSK9−/−, and PCSK9CRISPRa), our data showed that PCSK9 is involved in mtDNA damage in cardiomyocytes. A study with mitochondria-targeted antioxidant MitoTEMPOL pretreatment further confirmed the role of PCSK9 in regulating mtDNA damage.
Cardiomyocyte loss is a major contributor to the decreased cardiac function observed in myocardial infarction. We speculated if PCSK9 might regulate viability in cardiomyocytes. As determined by MTT assay, hrPCSK9 was found to drastically decrease, while PCSK9−/− increased viability in cardiomyocytes during hypoxia. Since PCSK9 is considered as a pro-inflammatory factor, we further investigated the mechanisms of PCSK9-mediated viability in cardiomyocytes with pyroptosis, a caspase-1-dependent programmed cell death. As expected in the condition of hypoxia, PCSK9 itself activates the NLRP3 inflammasome and markedly elevates the release of IL-1β and IL-18. The role of PCSK9 in regulating of pyroptosis was confirmed by GSDMD-NT expression and LDH release. This data is the first to show the role of PCSK9 in regulating viability in cardiomyocytes and indicates that PCSK9 may be a novel therapeutic to slow or treat pyroptosis-mediated myocardial infarction.
This study provides an alternative hypothesis that there is a bidirectional interaction between PCSK9 and inflammatory response. On one hand, endotoxin-free recombinant PCSK9 increased the expression of pro-inflammatory molecules (e.g., IL-1β, TNFα, MCP-1, and IL-6) in primary macrophages and THP-1 macrophages [18, 23]. In clinical trials, PCSK9 inhibitors have not shown any alteration in plasma C-reactive protein level [18, 21, 24]; however, there is lessened inflammatory response in the arterial wall that could attenuate atherosclerotic plaque development [18]. The ATHEOROREMO-IVUS study showed a correlation of serum PCSK9 levels with coronary plaque inflammation and the absolute volume of necrotic core tissue [3]. On the other hand, pro-inflammatory stimuli such as TNFα and LPS enhance the expression of PCSK9 in vascular endothelial cells, smooth muscle cells, and macrophages [5, 7]. Treatment of human hepatocytes with resistin, found in inflammatory zone 3, is associated with intense PCSK9 expression [11, 16]. We recently showed that TLR4-MyD88-NF-κB signaling plays an important role in the regulation of PCSK9 expression in the aorta [13]. We provided evidence that NLRP3 inflammasome via IL-1β plays an important role in determining PCSK9 secretion particularly in the presence of a high-fat diet [8]. In this study, our data showed PCSK9 is involved in NLRP3 inflammasome activation in cardiomyocytes and regulates inflammatory response during chronic myocardial ischemia. Almontashiri et al. reported elevated serum PCSK9 levels in patients with myocardial infarction in two independent retrospective angiographic studies [1]. Consistently, our data showed that both PCSK9 and pyroptosis signaling are highly expressed in patients with myocardial infarction. Further studies need to investigate whether aspirin, statins, renin-angiotensin inhibitors, calcium channel blockers, or beta-blockers play a role in regulating circulating PCSK9 and pyroptosis.
In summary, this study provides the first definitive data on the link between PCSK9 and Caspase-1-dependent pyroptosis in cardiomyocytes and ischemic heart. Several recent studies have shown the roles of PCSK9 in the heart [16, 30]. Palee et al. observed that PCSK9 inhibitors, administration before ischemia, exerts cardio-protection demonstrated by the attenuation of infarct size and cardiac arrhythmia during cardiac ischemia or reperfusion injury [20]. Miñana et al. reported that in patients with their first ST-segment elevation and myocardial infarction with reduced ejection fraction at index admission that was treated with primary percutaneous coronary interventions showed circulating PCSK9 was associated with lower left ventricular ejection fraction at 6 months [17]. These findings shed light on the potential contribution of PCSK9 to myocardial infarction.
References
Almontashiri NA, Vilmundarson RO, Ghasemzadeh N, Dandona S, Roberts R, Quyyumi AA, Chen HH, Stewart AF (2014) Plasma PCSK9 levels are elevated with acute myocardial infarction in two independent retrospective angiographic studies. PLoS ONE 9:e106294. https://doi.org/10.1371/journal.pone.0106294
Buja LM, Entman ML (1998) Modes of myocardial cell injury and cell death in ischemic heart disease. Circulation 98:1335–1357. https://doi.org/10.1161/01.cir.98.14.1355
Cheng JM, Oemrawsingh RM, Garcia-Garcia HM, Boersma E, van Geuns RJ, Serruys PW, Kardys I, Akkerhuis KM (2016) PCSK9 in relation to coronary plaque inflammation: results of the ATHEROREMO-IVUS study. Atherosclerosis 248:117–122. https://doi.org/10.1016/j.atherosclerosis.2016.03.010
Cheng SB, Nakashima A, Huber WJ, Davis S, Banerjee S, Huang Z, Saito S, Sadovsky Y, Sharma S (2019) Pyroptosis is a critical inflammatory pathway in the placenta from early onset preeclampsia and in human trophoblasts exposed to hypoxia and endoplasmic reticulum stressors. Cell Death Dis 10:927. https://doi.org/10.1038/s41419-019-2162-4
Ding Z, Liu S, Wang X, Mathur P, Dai Y, Theus S, Deng X, Fan Y, Mehta JL (2016) Cross-talk between PCSK9 and damaged mtDNA in vascular smooth muscle cells: role in apoptosis. Antioxid Redox Signal 25:997–1008. https://doi.org/10.1089/ars.2016.6631
Ding Z, Liu S, Wang X, Theus S, Deng X, Fan Y, Zhou S, Mehta JL (2018) PCSK9 regulates expression of scavenger receptors and ox-LDL uptake in macrophages. Cardiovasc Res 114:1145–1153. https://doi.org/10.1093/cvr/cvy079
Ding Z, Wang X, Liu S, Shahanawaz J, Theus S, Fan Y, Deng X, Zhou S, Mehta JL (2018) PCSK9 expression in the ischaemic heart and its relationship to infarct size, cardiac function, and development of autophagy. Cardiovasc Res 114:1738–1751. https://doi.org/10.1093/cvr/cvy128
Ding Z, Wang X, Liu S, Zhou S, Kore RA, Mu S, Deng X, Fan Y, Mehta JL (2020) NLRP3 inflammasome via IL-1β regulates PCSK9 secretion. Theranostics 10:7100–7110. https://doi.org/10.7150/thno.45939
Hausenloy DJ, Yellon DM (2013) Myocardial ischemia-reperfusion injury: a neglected therapeutic target. J Clin Invest 123:92–100. https://doi.org/10.1172/JCI62874
Karmakar M, Minns M, Greenberg EN, Diaz-Aponte J, Pestonjamasp K, Johnson JL, Rathkey JK, Abbott DW, Wang K, Shao F, Catz SD, Dubyak GR, Pearlman E (2020) N-GSDMD trafficking to neutrophil organelles facilitates IL-1β release independently of plasma membrane pores and pyroptosis. Nat Commun 11:2212. https://doi.org/10.1038/s41467-020-16043-9
Maxwell KN, Fisher EA, Breslow JL (2005) Overexpression of PCSK9 accelerates the degradation of the LDLR in a post-endoplasmic reticulum compartment. PNAS 102:2069–2074. https://doi.org/10.1073/pnas.0409736102
Liu D, Zeng X, Li X, Mehta JL, Wang X (2017) Role of NLRP3 inflammasome in the pathogenesis of cardiovascular diseases. Basic Res Cardiol 113:5. https://doi.org/10.1007/s00395-017-0663-9
Liu S, Deng X, Zhang P, Wang X, Fan Y, Zhou S, Mu S, Mehta JL, Ding Z (2020) Blood flow patterns regulate PCSK9 secretion via MyD88 mediated proinflammatory cytokines. Cardiovasc Res 116:1721–1732. https://doi.org/10.1093/cvr/cvz262
Lu J, Wang X, Wang W, Muniyappa H, Deshmukh A, Hu C, Das K, Mehta JL (2012) Abrogation of lectin-like oxidized LDL receptor-1 attenuates acute myocardial ischemia-induced renal dysfunction by modulating systemic and local inflammation. Kidney Int 82:436–444. https://doi.org/10.1038/ki.2012.186
Man SM, Karki R, Kanneganti TD (2017) Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol Rev 277:61–75. https://doi.org/10.1111/imr.12534
Melone M, Wilsie L, Palyha O, Strack A, Rashid S (2012) Discovery of a new role of human resistin in hepatocyte low-density lipoprotein receptor suppression mediated in part by proprotein convertase subtilisin/kexin type 9. J Am Coll Cardiol 59:1697–1705. https://doi.org/10.1016/j.jacc.2011.11.064
Miñana G, Núñez J, Bayés-Genís A, Revuelta-López E, Ríos-Navarro C, Núñez E, Chorro FJ, López-Lereu MP, Monmeneu JV, Lupón J, Sanchis J, Bodí V (2020) Role of PCSK9 in the course of ejection fraction change after ST-segment elevation myocardial infarction: a pilot study. ESC Heart Fail 7:117–122. https://doi.org/10.1002/ehf2.12533
Momtazi-Borojeni AA, Sabouri-Rad S, Gotto AM, Pirro M, Banach M, Awan Z, Barreto GE, Sahebkar A (2019) PCSK9 and inflammation: a review of experimental and clinical evidence. Eur Heart J Cardiovasc Pharmacother 5:237–245. https://doi.org/10.1093/ehjcvp/pvz022
Muendlein HI, Jetton D, Connolly WM, Eidell KP, Magri Z, Smirnova I, Poltorak A (2020) cFLIP L protects macrophages from LPS-induced pyroptosis via inhibition of complex II formation. Science 367:1379–1384. https://doi.org/10.1126/science.aay3878
Palee S, McSweeney CM, Maneechote C, Moisescu D, Jaiwongkam T, Kerdphoo S, Chattipakorn SC, Chattipakorn N (2019) PCSK9 inhibitor improves cardiac function and reduces infarct size in rats with ischaemia/reperfusion injury: benefits beyond lipid-lowering effects. J Cell Mol Med 23:7310–7319. https://doi.org/10.1111/jcmm.14586
Raal FJ, Stein EA, Dufour R, Turner T, Civeira F, Burgess L, Langslet G, Scott R, Olsson AG, Sullivan D, Hovingh GK (2015) PCSK9 inhibition with evolocumab (AMG 145) in heterozygous familial hypercholesterolaemia (RUTHERFORD-2): a randomised, double-blind, placebo-controlled trial. The Lancet 385:331–340. https://doi.org/10.1016/S0140-6736(14)61399-4
Rathinam VAK, Zhao Y, Shao F (2019) Innate immunity to intracellular LPS. Nat Immunol 20:527–533. https://doi.org/10.1038/s41590-019-0368-3
Ricci C, Ruscica M, Camera M, Rossetti L, Macchi C, Colciago A, Zanotti I, Lupo MG, Adorni MP, Cicero AFG, Fogacci F, Corsini A, Ferri N (2018) PCSK9 induces a pro-inflammatory response in macrophages. Sci Rep 8:2267. https://doi.org/10.1038/s41598-018-20425-x
Sahebkar A, Di Giosia P, Stamerra CA, Grassi D, Pedone C, Ferretti G, Bacchetti T, Ferri C, Giorgini P (2016) Effect of monoclonal antibodies to PCSK9 on high-sensitivity C-reactive protein levels: a meta-analysis of 16 randomized controlled treatment arms. Br J Clin Pharmacol 81:1175–1190. https://doi.org/10.1111/bcp.12905
Schlüter KD, Wolf A, Weber M, Schreckenberg R, Schulz R (2017) Oxidized low-density lipoprotein (oxLDL) affects load-free cell shortening of cardiomyocytes in a proprotein convertase subtilisin/kexin 9 (PCSK9)-dependent way. Basic Res Cardiol 112:63. https://doi.org/10.1007/s00395-017-0650-1
Sun L, Ma W, Gao W, Xing Y, Chen L, Xia Z, Zhang Z, Dai Z (2019) Propofol directly induces caspase-1-dependent macrophage pyroptosis through the NLRP3-ASC inflammasome. Cell Death Dis 10:542. https://doi.org/10.1038/s41419-019-1761-4
Takahashi M (2019) Cell-specific roles of NLRP3 inflammasome in myocardial infarction. J Cardiovasc Pharmacol 74:188–193. https://doi.org/10.1097/FJC.0000000000000709
Wang X, Guo Z, Ding Z, Mehta JL (2018) Inflammation, autophagy, and apoptosis after myocardial infarction. J Am Heart Assoc 7:e008024. https://doi.org/10.1161/JAHA.117.008024
Wang Y, Shi P, Chen Q, Huang Z, Zou D, Zhang J, Gao X, Lin Z (2019) Mitochondrial ROS promote macrophage pyroptosis by inducing GSDMD oxidation. J Mol Cell Biol 11:1069–1082. https://doi.org/10.1093/jmcb/mjz020
Wree A, Eguchi A, McGeough MD, Pena CA, Johnson CD, Canbay A, Hoffman HM, Feldstein AE (2014) NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology 59:898–910. https://doi.org/10.1002/hep.26592
Zeng C, Duan F, Hu J, Luo B, Huang B, Lou X, Sun X, Li H, Zhang X, Yin S, Tan H (2020) NLRP3 inflammasome-mediated pyroptosis contributes to the pathogenesis of non-ischemic dilated cardiomyopathy. Redox Biol 34:101523. https://doi.org/10.1016/j.redox.2020.101523
Acknowledgements
This study was supported by a seed grant from the Vice-Chancellor of Research & Innovation from the University of Arkansas for Medical Sciences (to Dr. Ding). Additionally, this research was in part supported by the Supporting Plan for Scientific and Technological Innovative Talents in Universities of Henan Province (19HASTIT004 to Dr. Wang) and the National Natural Science Foundation of China (U1804166, 81873459, 81370428 to Dr. Wang).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Rights and permissions
About this article

Cite this article
Wang, X., Li, X., Liu, S. et al. PCSK9 regulates pyroptosis via mtDNA damage in chronic myocardial ischemia. Basic Res Cardiol 115, 66 (2020). https://doi.org/10.1007/s00395-020-00832-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00395-020-00832-w