
Abstract
Background and objectives
The prevalence of chronic obstructive pulmonary disease (COPD) and lung emphysema increases with age and both lung diseases are again risk factors for lung cancer. Since a reduced capacity of fibroblasts for proliferation is a good indicator of tissue aging, we studied the cell proliferation of lung fibroblasts from normal and tumor tissue of lung cancer patients depending on lung comorbidities.
Material and methods
Fibroblasts were isolated from tumor and normal lung tissue of 40 lung cancer patients. Cumulative population doubling (CPD) was determined to assess the proliferation capacity, and the PCR technique was used to measure telomere lengths. Since many patients had previously been exposed to severe air pollution, we also studied the effect of air pollution particles on the fibroblast CPD in vitro.
Results
Fibroblasts from tumor and normal lung tissue had comparable CPDs; however, the CPD of fibroblasts from both tumor and normal lung tissue was significantly reduced in patients also suffering from COPD. This CPD reduction was highest in COPD patients who had already developed emphysema or were smokers. A significant correlation between CPD and telomere length was identified only for fibroblasts of non-COPD patients. Further studies also showed an adverse effect of air pollution particles on the CPD of lung fibroblasts.
Conclusion
Lung cells of COPD patients are characterized by accelerated senescence which must have been initiated prior to lung tumorigenesis and cannot depend on telomere shortening only. In addition to smoking as a known risk factor for COPD and lung cancer, air pollution particles could be another reason for the accelerated senescence of lung cells.
Zusammenfassung
Hintergrund und Zielsetzung
Die Prävalenz der chronisch-obstruktiven Lungenerkrankung (COPD) und des Lungenemphysems nimmt mit dem Alter zu, wobei beide Erkrankungen wiederum das Risiko an Lungenkrebs zu erkranken, steigern. Da eine reduzierte Proliferationskapazität von Fibroblasten einen guten Indikator für die Gewebealterung darstellt, haben wir die Zellproliferation von Lungenfibroblasten aus dem Normal- und Tumorgewebe von Patienten mit Lungenkrebs in Abhängigkeit von pulmonalen Begleiterkrankungen untersucht.
Material und Methoden
Fibroblasten wurden aus Tumor- und Normallungengewebe von 40 Patienten mit Lungenkrebs isoliert. Die kumulative Populationsverdopplung (CPD) wurde als Maß für die Proliferationskapazität ermittelt. Die Telomerlängen wurden per Polymerase-Kettenreaktion (PCR) bestimmt. Da viele Patienten hoher Luftverschmutzung ausgesetzt waren, untersuchten wir zudem den Einfluss von Partikeln aus Industrieanlagen auf die Fibroblasten-CPD in vitro.
Ergebnisse
Die CPD-Werte von Fibroblasten aus Tumor- und Normallungengewebe unterschieden sich nicht. Dagegen waren die CPD von Fibroblasten aus Tumor- sowie Normalgewebe von Patienten mit COPD stark verringert. Diese Verringerung war am stärksten bei den COPD-Patienten, die bereits ein Emphysem entwickelt hatten oder Raucher waren. Die CPD und Telomerlängen korrelierten jedoch nur bei den Fibroblasten von Patienten ohne COPD signifikant. Weitere Untersuchungen zeigten einen nachteiligen Effekt von Luftverschmutzungspartikeln auf die CPD von Lungenfibroblasten.
Schlussfolgerung
Die COPD ist eng mit der beschleunigten Alterung von Lungenzellen verbunden, wobei diese bereits vor der Tumorbildung eingesetzt haben muss und nicht ausschließlich von der Telomerverkürzung abhängig ist. Neben dem Rauchen als bekannter Risikofaktor für COPD und Lungenkrebs könnte auch die Luftverschmutzung Grund für die beschleunigte Zellalterung sein.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Despite apoptosis and senescence of cells being known as anti-cancer mechanisms, their occurrence in age-related diseases is not sufficient to prevent the development of cancer. This problem becomes obvious when looking at the example of chronic obstructive pulmonary disease (COPD), a typical age-related comorbidity in lung cancer patients.
Background and objectives
More than 50 years ago, Hayflick and Moorhead revealed that human diploid cells are able to undergo cell division for a maximum of 60 times until cell division stops [11]. This number of maximum possible cell divisions is also known as Hayflick limit [22]. Main reason for this limit is the shortening of the telomere region at the end of a DNA strand with each cell division. Since telomeres are not only damaged by the cell division-related shortening but also by reactive oxygen species, oxidative stress is another crucial factor reducing the maximum number of possible cell divisions [23]. In addition to telomere damage, DNA damage by double strand breaks cause an irreversible stop in cell division with subsequent senescence or apoptotic death of cells [3].
Senescent cells influence the tissue microenvironment by releasing soluble factors that in most cases are pro-inflammatory. The term “senescence-associated secretory phenotype” has therefore been introduced to emphasize the adverse extracellular effect of senescent cells [6]. Moreover, senescent cells are highly susceptible to undergo cell death by apoptosis. For how long senescent cells can still maintain their viability depends on the function of the cell cycle inhibitory protein p21Cip1 [25]. Since there is no single cellular hallmark for specific indications of senescence, several senescence-related parameters including acid beta-galactosidase need to be verified to demonstrate cellular senescence [3].
The high susceptibility of senescent cells to apoptosis makes an in vitro cultivation after isolation from tissue specimens almost impossible; however, it is possible to isolate and culture pre-senescent cells in order to identify their remaining proliferation capacity. This is a frequently used method to indirectly indicate the presence of senescent cells in situ and is often combined with other methods, such as the detection of acid beta-galactosidase [3]. Moreover, proliferation studies of primary cells isolated from tissue specimens indicate the cellular regeneration capacity of this tissue.
A suitable cell type for this type of study is the fibroblast, which was already used by Hayflick and Moorhead in their previous experiments [11]. Fibroblasts are a cellular part of the connective tissue responsible for generation and modulation of the extracellular matrix. In this function, fibroblasts contribute to the normal tissue architecture, but, if dysregulated, also to an abnormal architecture leading to various pathologies, such as several structural lung diseases. Typical lung diseases associated with dysregulated fibroblasts are chronic obstructive pulmonary disease (COPD), emphysema as a result of long-term COPD or other causative pathologies, idiopathic lung fibrosis, and lung carcinoma [14, 16].
In the field of cancer research, fibroblasts originating from tumor tissue are specifically named “tumor-associated fibroblasts” because of their activated cell state in tumor tissue and their crucial role in tumor progression [9]. Since COPD and emphysema are known risk factors for lung cancer, with high risk to be developed by smokers [17], and their prevalence is age-dependent, we studied the proliferation capacity of isolated lung fibroblasts from both normal and tumor tissue of lung cancer patients partially also suffering from COPD, emphysema and/or other lung pathologies.
Material and methods
Isolation and culture of lung fibroblasts
Fibroblasts were isolated from tumor tissue and paired normal lung tissue of 40 lung cancer patients who had undergone pulmonary resection surgery. All patients had developed a non-small cell lung carcinoma at different tumor stages (TNM I–III). In addition to lung cancer, patients often suffered from other lung diseases and were smokers in most cases (Table 1). In order to isolate fibroblasts, tissue specimens were minced, transferred into phosphate-buffered saline supplemented with collagenase P (Roche Diagnostics, Mannheim, Germany), EDTA-free trypsin solution and antibiotics/antimycotics, and then incubated for 30 min at 37 °C. Thereafter, the cell suspension was filtered, centrifuged, resuspended in Dulbecco’s modified Eagle’s medium (DMEM), supplemented with 10% foetal calf serum (FCS) and antibiotics/antimycotics, and seeded in a culture dish. After 24 h incubation at 37 °C in a 10% CO2 atmosphere, non-adherent cells were washed off and adherent cells were cultured in DMEM with 10% FCS and antibiotics. After reaching cell confluency, which took at least 10 days, the first cell passage was performed by use of trypsin-EDTA solution. When cells were passaged the first times, they were reseeded in new cell culture dishes for cell proliferation studies as well as cryoconservation.
For determination of the cell proliferation, fibroblasts were seeded at a density of 1 × 104 cells/cm2 and then cultured for 4 days. The number of living/dead cells was counted by use of an automated cell counter. Cell population doubling (PD) for each cell passage was calculated according to the formula: PD = (ln cell number at day 4 − ln cell number day 1)/ln2. The cumulative population doubling (CPD) is the sum of all PDs at a given time. If not indicated otherwise, cell culture reagents were purchased from Thermo Fisher Scientific (Waltham, MA, USA) and plastic consumables from Corning Life Sciences (Corning, NY, USA). In order to keep external factors on the cell proliferation to a minimum, enzymes, FCS and cell culture plates were used from the same lot number. The use of patient material was approved by the local ethics committee, and informed consent of all the patients was obtained. Additionally, we studied the cell proliferation of human fibroblasts derived from a fetal lung (WI-38; ATCC cell bank). Air pollution fly ash taken from an incineration plant for household rubbish (TFA98 pre-treated in DMEM with ultrasound, particle size of 200–300 nm [7]) was a generous gift of the Karlsruhe Institute of Technology (Dr. Silvia Diabatè).
Determination of telomere length and others
Genomic DNA was isolated with TRIzol™ Reagent (Thermo Fisher Scientific) and spectrophotometrically quantified. For DNA isolation we used fibroblasts which had been cryoconserved at passage 2 and then recultured for one cell passage. As described by Cawthon [5] telomere lengths were assessed by quantitative PCR in the iCycler iQ™ Real-Time PCR Detection System (Bio-Rad Laboratories, Munich, Germany) using primers for human telomere (sense: ACACTAAGGTTTGGGTTTGGGTTTGGGTTTGGGTTAG TGT, antisense: TGTTAGGTATCCCTATCCCTATCCCTATCCCTATCCCTAACA) or albumin as a single copy gene and GoTaq® qPCR mix (Promega, Madison, WI, USA). The PCR reliability was tested by use of genomic DNA prepared from different passages of lung fibroblasts as well as by use of different DNA dilutions. Telomere lengths were calculated per external standard curve containing different concentrations of genomic DNA with lowest concentration set to 1 relative unit.
Cell staining for acid beta-galactosidase was performed as previously described [8]. Lysates of sub-confluent fibroblasts were subjected to a standard immunoblot procedure using antibodies against p21Cip1 (Merck-Millipore, Burlington, MA, USA) or p16Ink4a (BD Bioscience [Franklin Lakes, NJ, USA], Sigma [St. Louis, MO, USA]). Pro-inflammatory factors released from confluent fibroblasts (3.5 × 105 cells per 10 ml DMEM) in 24 h were detected with BD™ Cytometric Bead Array sets in a BD FACS Array Luminex 100/200 device equipped with FCAP ArrayTM Software 2.0 (BD Bioscience).
Statistics
End CPD values were used for comparisons of two or many groups and then tested for significance (t-test or ANOVA test followed by Holm-Sidak method). The correlation coefficient of linear regression analysis was tested for significance by t-test. P values ≤ 0.05 indicate significant differences. All statistical calculations and data presentations were performed by use of the SigmaStat 3.5 and SigmaPlot 10 software (Systate Software, Erkrath, Germany).
Results
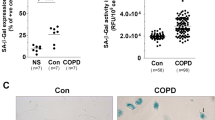
Serial cultivation of human lung fibroblasts causes decreased proliferation rates (PD) and more cell death with advancing time in vitro (Fig. 1a). Over time fibroblasts also undergo cellular senescence as indicated by typical changes in the cell morphology (flattening, increased size) and positive staining for acid beta-galactosidase (Fig. 1b). Human lung fibroblasts highly express the cell cycle inhibitor p21Cip1 (Fig. 1b) but p16Ink4a, another cell cycle inhibitory protein involved in cellular senescence, was hardly detected (Fig. 1b). Among known pro-inflammatory factors we identified the monocyte chemotactic protein-1 (MCP-1) to be increased in senescent lung fibroblasts (Fig. 1b). In order to identify the proliferation capacity of isolated lung fibroblasts we calculated the CPDs at given time points and the end CPDs. Comparative analyses of lung fibroblasts isolated from tumor and paired normal tissue of lung cancer patients indicated that the CPDs of these fibroblasts were independent of the respective tissue origin (Fig. 1c). In most cases, we observed similar end CPDs for fibroblasts of normal and tumor tissue of the same patient (Fig. 1c).

a Serial cultivation of lung fibroblasts derived from normal or tumor tissue of one selected lung cancer patient and its influence on PD and cell death with increasing time in vitro. b Expression level of senescence-associated cell cycle inhibitors (p21Cip1, p16Ink4a) and pro-inflammatory factors (IL-6, IL-8, MCP-1) in human lung fibroblasts at low and high CPD. Microscopic images indicate changes in cell morphology and expression of the acid-β galactosidase (blue stain) with higher CPD . c CPD of lung fibroblasts isolated from normal and tumor tissue of lung cancer patients per time each cultured in vitro. The ratio of the end CPD determined for fibroblasts of normal and tumor tissue of the same person is given separately. Data are given as mean ± standard deviation (for n Table 1). The time of fibroblasts cultured in vitro is always given as mean calculated for all cell preparations because the time points of first passages varied between the cell preparations
Since the tumor stage did not influence the CPDs of tumor-derived fibroblasts, we then analyzed the impact of other lung diseases. These analyses revealed a strong influence of COPD on the proliferation capacity of fibroblasts from both normal lung and paired lung tumors (Fig. 2a). Measurements of telomere length in lung fibroblasts taken at an early cell passage showed a positive correlation between telomere length and end CPD (Fig. 2b); however, this correlation is based more on the data of fibroblasts derived from non-COPD than those from COPD lungs (Fig. 2b).

a End CDP of fibroblasts from normal or tumor lung tissue and mean CPDa per time cultured in vitro depending on the coexistence of COPD. b Influence of COPD on the correlation between the end CPD determined for fibroblasts from normal or tumor lung and their telomere lengths determined at the beginning of the fibroblast cultivation. Linear regression analysis includes the data of all lung fibroblasts, i. e., fibroblasts from normal and tumor tissue (for n see Table 1). c Mean CDPa of lung fibroblasts determined at the end of each fibroblast culture in vitro is given in dependence on COPD and emphysema as well as COPD and cigarette smoking in general or (d) depending on the pack-years. aMean CPD corresponds to the average calculated from the values of normal and tumor tissue of the same person
Lung emphysema often results from previously developed COPD but can also have other causes. In this context, Fig. 2c indicates that the CPD of lung fibroblasts is impaired by COPD but not by emphysema. Due to the primary impact of COPD on the proliferation capacity of lung fibroblasts and the relatively low number of patients analyzed, the influence of other possible lung diseases as well as smoking is difficult to prove statistically. Nevertheless, our data clearly show that the CPD of lung fibroblasts derived from COPD patients either having already developed lung emphysema or being smokers is worst in comparison to other evaluation groups (Fig. 2c). Moreover, we observed a trend for a negative correlation between time and intensity of smoking (pack-years) and the final CPD of lung fibroblasts (Fig. 2d).
Many patients included in our study suffered from anthracosis (Table 1) which is caused by the accumulation of carbon in the lung parenchyma due to repeated exposure to air pollution or inhalation of smoke or coal dust particles. Because most patients were also smokers and many of them suffered from COPD, the sole effect of air pollution on the proliferation capacity of lung cells could not be analyzed. Therefore, in separate experiments we studied the influence of fly ash containing numerous air pollution particles on the CPD of lung fibroblasts. Here we used fibroblasts from fetal human lung tissue because, in contrast to our patients’ fibroblasts, they were not pre-influenced by environmental impacts. These studies showed a reduced CPD of lung fibroblasts in response to fly ash in vitro (Fig. 3). Since fly ash did not enhance cell death with advancing time in vitro (Fig. 3), it indicates a fly ash-mediated CDP reduction by induction of cellular senescence.

Concentration-dependent influence of air pollution fly ash (TAF98) on CPD and cell death of fibroblasts from fetal human lung tissue (WI-38, CPD of 35 at the beginning of the experiment) per time cultured in vitro. Data are given as mean data ± standard deviation (n = 3)
Discussion
Our study showed that COPD is major reason for a reduced proliferation capacity of lung cells and, therefore, a higher susceptibility of cells to undergo senescence and death. Comparative analyses of lung fibroblasts from tumor and non-tumor tissue indicated that the adverse effect of COPD on the proliferation capacity precedes the lung carcinogenesis. Our study additionally showed that permanent lung exposure to air pollution might impair the proliferation capacity of lung cells.
The reduced proliferation capacity of lung fibroblasts from lung cancer patients with COPD corresponds to findings of other studies investigating in situ lung specimens or in vitro isolated cells of lung specimens from COPD patients without lung cancer. In this respect, signs for cellular senescence in COPD-affected lung tissues have not only been found for fibroblasts [12, 18] but also for alveolar epithelial cells [2, 10, 24], endothelial cells [2, 24] and smooth muscle cells [19]. Ito and Barnes were the first to conclude that “COPD is a disease of accelerated lung aging” [13]. Although the COPD dependency of the proliferation of fibroblasts derived from non-tumor lung tissue was no surprise, we did not expect similar but rather lower proliferation capacities of tumor-derived fibroblasts caused by tumor cell-mediated activation of fibroblasts in vivo [15]. Since these findings could not be verified in vitro, it leads to the assumption that lung tumor cells activate adjacent fibroblasts without effect on their proliferation and, therefore, depletion of the remaining possible number of cell divisions. Furthermore, the isolated impact of COPD on the CPD of tumor-derived fibroblasts indicates that the depletion of the possible number of cell divisions must have happened in COPD lungs prior to lung carcinogenesis. The latter is also plausible because the progression of COPD takes more time than the progression of lung cancer.
Accelerated senescence of cells, especially of extracellular matrix-producing fibroblasts, in COPD lungs supposedly is one reason for the development of emphysema in many COPD patients [12, 18, 20, 24]. Supporting this supposition, isolated fibroblasts of COPD patients with additional emphysema showed the least proliferation capacity; however, the accelerated senescence of lung cells cannot be the sole reason for lung emphysema because the proliferation capacity of lung fibroblasts in emphysema patients without COPD was not impaired. It is therefore conceivable that cellular senescence accelerates the progression of emphysema specifically in COPD patients, and by that mainly discriminates it from the lung emphysema caused by other reasons (e. g. pulmonary infection). Since human lung fibroblasts highly express p21Cip1, they seem to use this cell cycle inhibitory protein to reach cellular senescence.
Shortened telomeres could explain lower proliferation capacities of fibroblasts, a coherence which is also supported by the positive correlation identified between telomere length and CPD of each fibroblast culture; however, telomere length and CPD did not correlate in all cases indicating the additional influence of telomere-independent factors, such as double strand DNA breaks induced by cigarette smoke [2]. These seem to be crucial for the cellular senescence in COPD lungs because a CPD dependency on the telomere length could not be identified in those fibroblasts isolated from COPD lungs.
Most COPD patients have a history of smoking abuse. Toxins in cigarette smoke extracts/condensates cause genotoxic damage in respiratory tract cells which in turn induce cellular senescence [3]. Since cellular senescence is characterized by a stop in cell proliferation and the secretion of mostly pro-inflammatory factors such as MCP-1, as shown in this study, tissue repair processes are consequently impaired, while pro-inflammatory processes are promoted. This scenario is well conceivable in COPD lungs and has been supported by current findings [26]; however, most patients included in our study were long-term smokers some of which suffered from COPD but only diagnosed COPD patients showed reduced fibroblast CPDs. This suggests two further assumptions. First, smoking only causes COPD if DNA/senescence-protective mechanisms are exhausted. Second, the tissue microenvironment in COPD lungs enhances cellular senescence. The second possibility is also indirectly supported by our observation indicating that the fibroblast CPDs are independent of the presence of lung emphysema.
In order to investigate the separate effects of COPD and smoking on the proliferation capacity of lung cells, it would be necessary to include more non-smokers as well as healthy subjects into the study. Moreover, our patients came from the industrial region of Halle (Saale) and had possibly been exposed to severe air pollution particles for decades. This might also be a reason for high frequency of diagnosed anthracosis in our study patients. Comparable to compounds of the cigarette smoke, particulate matter air pollutants also causes genotoxic stress [1]. In separate experiments we observed a negative effect of air pollution particles on the proliferation capacity of primary human lung fibroblasts. This observation is in accordance with other studies observing increased cellular senescence after treatment of lung cells with particulate matter air pollutants [4, 21].
Senescence and apoptosis of cells are generally known as important anti-cancer mechanisms preventing the further participation of damaged cells in tissue homeostasis. Despite the fact that COPD-affected lungs are characterized by an increased susceptibility of cells to undergo senescence and apoptosis, COPD is a major risk factor for lung cancer. One critical reason for this might be the tumor-promoting microenvironment generated by senescent fibroblasts [6].
References
Aoki Y (2017) Evaluation of in vivo mutagenesis for assessing the health risk of air pollutants. Genes Environ 39:16
Aoshiba K, Zhou F, Tsuji T et al (2012) DNA damage as a molecular link in the pathogenesis of COPD in smokers. Eur Respir J 39:1368–1376
Bartling B (2013) Cellular senescence in normal and premature lung aging. Z Gerontol Geriatr 46:613–622
Buchner N, Ale-Agha N, Jakob S et al (2013) Unhealthy diet and ultrafine carbon black particles induce senescence and disease associated phenotypic changes. Exp Gerontol 48:8–16
Cawthon RM (2009) Telomere length measurement by a novel monochrome multiplex quantitative PCR method. Nucleic Acids Res 37:e21
Coppe JP, Desprez PY, Krtolica A et al (2010) The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 5:99–118
Diabate S, Günther R, Völkel K et al (2004) Gesundheitseffekte durch inhalierbare Feinststäube aus technischen Verbrennungsanlagen: In vitro Untersuchungen zur Wirkung feiner und ultrafeiner Partikel auf kultivierte Lungenzellen, p 52 (In, http://www.fachdokumente.lubw.baden-wuerttemberg.de/servlet/is/40181/?COMMAND=DisplayBericht&FIS=203&OBJECT=40181&MODE=METADATA)
Dimri GP, Lee X, Basile G et al (1995) A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci Usa 92:9363–9367
Erdogan B, Webb DJ (2017) Cancer-associated fibroblasts modulate growth factor signaling and extracellular matrix remodeling to regulate tumor metastasis. Biochem Soc Trans 45:229–236
Hara H, Araya J, Takasaka N et al (2012) Involvement of creatine kinase B in cigarette smoke-induced bronchial epithelial cell senescence. Am J Respir Cell Mol Biol 46:306–312
Hayflick L, Moorhead PS (1961) The serial cultivation of human diploid cell strains. Exp Cell Res 25:585–621
Holz O, Zuhlke I, Jaksztat E et al (2004) Lung fibroblasts from patients with emphysema show a reduced proliferation rate in culture. Eur Respir J 24:575–579
Ito K, Barnes PJ (2009) COPD as a disease of accelerated lung aging. Chest 135:173–180
Kulkarni T, O’reilly P, Antony VB et al (2016) Matrix remodeling in pulmonary fibrosis and emphysema. Am J Respir Cell Mol Biol 54:751–760
Kuzet SE, Gaggioli C (2016) Fibroblast activation in cancer: when seed fertilizes soil. Cell Tissue Res 365:607–619
Mahale J, Smagurauskaite G, Brown K et al (2016) The role of stromal fibroblasts in lung carcinogenesis: a target for chemoprevention? Int J Cancer 138:30–44
Mouronte-Roibas C, Leiro-Fernandez V, Fernandez-Villar A et al (2016) COPD, emphysema and the onset of lung cancer. A systematic review. Cancer Lett 382:240–244
Müller KC, Welker L, Paasch K et al (2006) Lung fibroblasts from patients with emphysema show markers of senescence in vitro. Respir Res 7:32
Noureddine H, Gary-Bobo G, Alifano M et al (2011) Pulmonary artery smooth muscle cell senescence is a pathogenic mechanism for pulmonary hypertension in chronic lung disease. Circ Res 109:543–553
Nyunoya T, Monick MM, Klingelhutz AL et al (2009) Cigarette smoke induces cellular senescence via Werner’s syndrome protein down-regulation. Am J Respir Crit Care Med 179:279–287
Sanchez-Perez Y, Chirino YI, Osornio-Vargas AR et al (2009) DNA damage response of A549 cells treated with particulate matter (PM10) of urban air pollutants. Cancer Lett 278:192–200
Shay JW, Wright WE (2000) Hayflick, his limit, and cellular ageing. Nat Rev Mol Cell Biol 1:72–76
Toussaint O, Remacle J, Dierick JF et al (2002) From the Hayflick mosaic to the mosaics of ageing. Role of stress-induced premature senescence in human ageing. Int J Biochem Cell Biol 34:1415–1429
Tsuji T, Aoshiba K, Nagai A (2010) Alveolar cell senescence exacerbates pulmonary inflammation in patients with chronic obstructive pulmonary disease. Respiration 80:59–70
Yosef R, Pilpel N, Papismadov N et al (2017) p21 maintains senescent cell viability under persistent DNA damage response by restraining JNK and caspase signaling. Embo J 36:2280–2295
Zhou F, Onizawa S, Nagai A et al (2011) Epithelial cell senescence impairs repair process and exacerbates inflammation after airway injury. Respir Res 12:78
Acknowledgements
The authors appreciate the technical assistance of S. Koitzsch and K. Szymala.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
B. Bartling and H.-S. Hofmann declare that they have no competing interests.
The use of patient material was approved by the local ethics committee, and informed consent of the patients was obtained.
Rights and permissions
About this article

Cite this article
Bartling, B., Hofmann, HS. Reduced proliferation capacity of lung cells in chronic obstructive pulmonary disease. Z Gerontol Geriat 52, 249–255 (2019). https://doi.org/10.1007/s00391-018-1377-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00391-018-1377-9