
Abstract.
A rare case of congenital duodenal atresia (DA) associated with a choledochal cyst (CC) is reported. At 38 weeks of gestation, a 1,610-g girl was born by cesarean section with a prenatal diagnosis of congenital DA. After the disease was confirmed by radiographs, she underwent a duodenoduodenostomy for complete separation of the duodenum with an annular pancreas. Thirty-two months after the initial operation, she developed upper abdominal pain and acholic stools. Abdominal ultrasonography demonstrated a CC and dilated intrahepatic bile ducts. Magnetic resonane cholangiopancreatography showed an anomalous arrangement of the choledochus and the main pancreatic duct. A diffusely dilated extrahepatic bile duct was resected, and a hepaticoduodenostomy was performed after a cholecystectomy. The patient was discharged without complications. We could not find a similar case report in the English literature. Although it is not reported that there is a close relation of DA and CC in embryologic development, the presence of this combination should be considered.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Anomalous bile-duct anatomy associated with duodenal atresia (DA) has been reported [1], but congenital DA and choledochal cyst (CC) are a rare combination. We assume these anomalies are rarely associated because of different embryologic origins. We report a case of combined DA and CC.
Case report
A female was born by cesarean section at 38 weeks of gestation with polyhydramnios and was small for gestational age. Her birth weight was 1,610 g and Apgar scores of 5 and 8 were recorded at 1 and 5 min, respectively. Abdominal X-ray films were confirmatory of DA. On the same day she underwent an emergency operation for complete separation of the duodenum with an annular pancreas. A transverse incision in the lowest portion of the proximal duodenum and a vertical incision in the uppermost portion of the distal segment were made. Vater’s ampulla was in the distal duodenum and the common bile duct (CBD) was not dilated. A duodenoduodenostomy (diamond-shaped anastomosis) was performed. The postoperative course was uneventful.
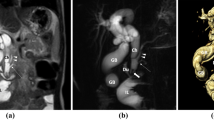
Thirty-two months after the initial operation, she was readmitted with a 3-day history of abdominal pain. Jaundice was noted with a total serum bilirubin level of 5.8 mg/dl and serum alkaline phosphatase 576 IU/1. Abdominal ultrasonography and computed tomography demonstrated a CC with dilatation of the intrahepatic duct, and magnetic resonance cholangiopancreatography showed an anomalous arrangement of the CBD and main pancreatic duct (Fig. 1). At laparotomy, the CBD showed diffuse fusiform dilatation. An operative cholangiogram demonstrated the CBD connected to the high junctional point of the main pancreatic duct, which constituted a long common channel (Fig. 2). The extrahepatic bile duct was resected from the porta hepatis to the level of confluence of the main pancreatic duct and closed with ligatures, and a hepaticoduodenostomy was performed. Histologic studies of the wall of the CC demonstrated chronic inflammation with fibrosis. The patient recovered uneventfully and is doing well.

MR-cholangiopancreatography showing choledochal cyst and anomalous arrangement of choledochus and main pancreatic duct. Arrow at common channel

Operative cholangiogram demonstrates common bile duct entering high junctional point of pancreatic duct
Discussion
The incidence of congenital DA is reported to be 1 in 5,000 to 10,000 births, and congenital CC is seen in roughly 1 in 15,000 births. Consequently, the combination of these anomalies is quite rare: we could not find such a case report in the English literature. We assume that the low incidence is probablu due to the difference of embryologic stages.
During the 2nd month of gestation, there is exuberant growth of the intestinal epithelial lining, obliterating the lumen until the size of the gut has increased sufficiently to accommodate it. As the gut grows, vacuolization of the solid core of cells begins and the lumen is recanalized by the end of the 8th to 10th week. Failure of coalescent vacuolization is thought to produce obstruction of the duodenum [2].
Various theories have been offered to explain the occurrence of CC. Babbitt suggested that there was an etiologic relationship between CC and anomalies of the pancreaticobiliary ductal (PBD) system [3]. Todani et al., from an analysis of endoscopic retrograde cholangiopancreatography and other cholangiographic imaging studies, suggested that almost all patients with CC had an anomalous arrangement of the PBD system [4]. In this condition, the pancreatic duct communicates with the CBD in a proximal direction, well outside the circular muscle of the ampulla of Vater. This would permit reflux of pancreatic enzymes upward into the CBD during fetal development, resulting in damage to the ductal wall, which causes dilatation of the bile duct.
In human embryos at 30 days, the pancreas is formed by two buds originating from the endodermal lining of the duodenum: a dorsal bud in the mesoduodenum and a ventral bud below the primordial liver. The ventral bud, also called the pancreatic anlage, is composed of two lobes (right and left). The two parts of the ventral bud are intimately related to the termination of the common hepatic duct, and each undergoes very different development. The left ventral bud and its duct usually regress completely. The right ventral bud rotates clockwise along with the CBD, and finally fuses with the dorsal bud to form the pancreas in the 7-week-old embryo. The right part of the ventral bud develops into the lower part of the head of the pancreas, which remains in close relation to the terminal of the common duct. Adda et al. reported that the main pancreatic duct is formed from the duct of the right ventral bud and the distal part of the dorsal duct [5]. In normal embryology, by the time the ventral bud fuses with the dorsal bud, the ventral hepatic duct and CBD have already formed their normal relationship within Vater’s ampulla. Therefore, an anomalous arrangement of the PBD system is formed by the 7th week of gestation.
There thus does not appear to be a close etiologic relationship between DA and CC because of the differences in timing of the embryologic development. At the first operation, we did not detect the pancreaticobiliary abnormality because of the lack of dilatation of the CBD. An anomalous arrangement of the PBD system has been reported to be associated with CC, but Ando et al. reported that ductal malunion that resulted in a common channel arrangement of the pancreatic and biliary ducts had occasionally been noted in infants and children without CC [6], which suggests that the CBD became gradually dilated after the first operation because of the congenital pancreaticobiliary anomaly. We would emphasize that although a close relationship in embryologic development of DA and CC has not been reported, physicians should be aware of the possible combination.
References
Knechtle SJ, Filston HC (1990) Anomalous biliary ducts associated with duodenal atresia. J Pediatr Surg 25: 1266–1269
Stauffer UG, Schwoebel M (1998) Duodenal atresia and stenosis – annular pancreas. In: O’Neill JA Jr, Rowe MI (eds) Pediatric surgery, 5th edn. Mosby Year Book, St Louis, pp 1133–1143
Babbitt DP (1969) Congenital choledochal cysts. New etiological concept based on anomalous relationship of the common bile duct and pancreatic bulb. Ann Radiol 12: 231–240
Todani T, Watanabe Y, Fujii T, Uemura S (1984) Anomalous arrangement of the pancreaticobiliary ductal system in patients with a choledochal cyst. Am J Surg 147: 672–676
Adda G, Hannoun L, Loygue J (1984) Development of the human pancreas: variations and pathology. A tentative classification. Anat Clin 5: 275–283
Ando H, Ito T, Nagaya M, Watanabe Y, Seo T, Kaneko K (1995) Pancreaticobiliary maljunction without choledochal cysts in infants and children: clinical features and surgical therapy. J Pediatr Surg 30: 1658–1662
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Sugimoto, T., Yamagiwa, I., Obata, K. et al. Choledochal cyst and duodenal atresia: a rare combination of malformations. Ped Surgery Int 20, 724–726 (2004). https://doi.org/10.1007/s00383-002-0783-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00383-002-0783-6