
Abstract
Background: IgG4-related disease is an autoimmune process that presents with tumefactive lesions characterized by storiform fibrosis, a dense lymphoplasmacytic infiltrate rich in IgG4+ plasma cells, obliterative phlebitis, and often elevated serum IgG4 levels. Central nervous system IgG4-related disease is very rare and usually occurs in the form of hypertrophic pachymeningitis or hypophysitis. Presentation as a large solitary meningioma-like mass with overlying hyperostosis in a young adult has not been reported before. Case summary: A 16-year-old male presented with focal seizures for 5 months. Imaging showed a large, extra-axial, and contrast-enhancing mass lesion in the left frontoparietal region with focal calvarial thickening. Histopathology revealed a fibrosclerotic lesion involving dura with a polymorphic infiltrate of plasma cells, mature lymphocytes, histiocytes, and occasional eosinophils. Immunohistochemical workup excluded the possibilities of meningioma, lymphoproliferative neoplasms, and histiocytic lesions. Majority of plasma cells were IgG4+ rendering a diagnosis of IgG4-related disease. Further serological and imaging workup did not reveal any evidence of systemic involvement. His serum IgG4 levels were normal. Considering a gross total resection of the lesion, no further treatment was given and the patient has been asymptomatic since. Conclusion: IgG4-related lesions of the CNS are under-recognized and accurate diagnosis, especially in those with isolated CNS disease and normal serum IgG4 levels, necessitates robust histopathological and laboratory workup to exclude mimics. They may occur as large dural masses with hyperostosis and differentiation from lymphoplasmacyte-rich meningiomas, in particular, can be challenging. While steroids are the mainstay of treatment in IgG4-related disease, surgical resection may be curative in solitary lesions presenting with compressive symptoms.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
First described in the pancreas, IgG4-related autoimmune disease (IgG4-RD) is a multi-system disorder of unknown etiology that can involve a variety of extra-pancreatic sites. Central nervous system (CNS) involvement is documented in < 2% of patients with IgG4-RD and commonly takes the form of hypertrophic pachymeningitis (HP) or hypophysitis. IgG4-related HP lesions (IgG4-HP) present as diffuse or nodular thickenings of the cranial and/or spinal dura [1]. In rare instances, they may form tumefactive pseudotumoral masses resembling meningiomas [1, 2], and especially when occurring in the absence of extra-CNS organ involvement, such lesions present a formidable diagnostic challenge.
Clinical summary
A 16-year-old male presented with complaints of focal seizures involving the right side of the body for 5 months. Imaging revealed a large extra-axial contrast-enhancing lesion in the left frontoparietal region with calvarial bone remodeling (Fig. 1). Pre-operative diagnosis was atypical meningioma. The patient underwent left fronto-temporo-parietal craniotomy and tumor excision. Intra-operatively, the tumoral mass was grayish white, firm, and moderately vascular, with poor plane of separation from brain. A gross total excision was achieved. Tumor tissue was processed entirely for histopathology.

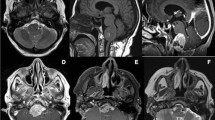
Radiological features of the tumor: magnetic resonance imaging shows a large extra-axial mass in left frontoparietal region with mass effect and midline shift (a axial T1W, b axial T2W, c axial diffusion-weighted image, d axial gradient ECHO). The lesion shows uniform contrast enhancement (e sagittal and f coronal post-gadolinium T1W). Hyperostosis is seen in the overlying calvarium on computed tomography images (g), with bony remodeling better appreciated on the bone window (h)
Microscopy revealed thickened and sclerotic dura infiltrated by plasma cells, mature lymphocytes, and occasional eosinophils. In some areas, storiform fibrosis was evident; however, there was no phlebitis. The inflammatory infiltrate was denser along the dura–brain interface and predominated in histiocytes. Occasional entrapped meningothelial nests were seen adjacent to the brain interface. Lymphoid follicles, emperipolesis, Touton-type giant cells, nuclear grooves in histiocytes, granulomas, giant cells, atypical spindle cells, mitotic activity, or necrosis was not seen. Plasma cells did not show light chain restriction on kappa and lambda immunohistochemistry. The histiocytes were CD68 positive but immunonegative for CD1a, langerin, and S100. Immunostaining for smooth muscle actin and anaplastic lymphoma kinase protein was negative. There was diffuse increase in IgG4-positive plasma cells (30–35/high-power field) with IgG4/IgG ratio of 80% clinching the final diagnosis of IgG4-RD (Fig. 2).

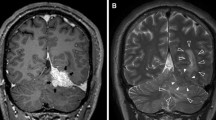
Histopathological features of the tumor: microscopy shows thickened, sclerotic dura (a hematoxylin and eosin (H&E), × 40), and leptomeninges and superficial brain cortex (b H&E, × 40) with a mixed inflammatory infiltrate. Focal storiform fibrosis is seen (c H&E, × 100). The infiltrate is composed of plasma cells, lymphocytes, and occasional eosinophils (d H&E, × 400), with prominence of foamy histiocytes along the brain–dura interface (e H&E, × 200). Very rare meningothelial cells are seen (arrow, f H&E, × 400). There is diffuse increase in IgG4+ plasma cells in both dural (g IHC, × 400) and leptomeningeal (h IHC, × 400) components. The reactive meningothelial nests do not label for MIB-1 (arrow, i, IHC, × 400)
On further workup, hemogram, renal function tests, liver function tests, erythrocyte sedimentation rate, autoimmune antibody profile (anti-double stranded DNA, anti SS-A and SS-B, ANA, ANCA, rheumatoid factor), viral markers (HBV, HCV, HIV), Mantoux test, and post-operative serum IgG4 levels (1.29 g/L; biological reference range, 0.049–1.985) were found to be within normal limits. Whole-body imaging did not reveal any abnormality. Contrast MRI performed 4 months after surgery showed no residual lesions. Patient is under close follow-up since then for the last 16 months and is asymptomatic.
Discussion
IgG4-RD presenting with tumefactive masses is well-known, and such lesions are most commonly described in the orbit, salivary glands, lungs, kidneys, lymph nodes, and retroperitoneum [1]. Intracranial IgG4-related pseudotumors are exceptional and may occur as intracranial/spinal dural-based, sellar, intraventricular, and even intra-parenchymal masses [1]. Meningioma-like masses, including the one in our patient, likely fall within the spectrum of IgG4-HP. While IgG4-HP usually present as diffuse dural thickening raising suspicion for vasculitic disorders, rheumatoid arthritis, sarcoidosis, lymphomatous neoplasms, and tuberculosis [3], presentation as single or multiple nodular meningioma-like masses are also reported [1, 2].
IgG4-HP is being increasingly recognized [3, 4] and emerged as the second most common etiology of HP following ANCA-related vasculitic disorders in a recent Japanese study [1]. IgG4-HP predominates in males (male/female ratio, 10:1) and occurs at a mean age of 56 years [1], ranging from as young as 19 years [5] to as old as 78 years [6]. Our patient is the youngest patient diagnosed with CNS IgG4-RD. Up to 30% of patients lack evidence of systemic involvement at presentation, and up to 45% patients do not show elevations in serum IgG4 levels [1]. Although recent observations have found that intrathecal IgG4 level estimation may be a more sensitive diagnostic marker for IgG4-HP [7], most patients invariably undergo biopsy or surgical excision for a definitive diagnosis.
The classic histopathological triad of IgG4-RD includes lymphoplasmacytic infiltration, storiform fibrosis, and obliterative phlebitis [4]. Among these, phlebitis has been observed less commonly in IgG4-HP and was absent in our case [1]. IgG4+ plasma cells are essential for diagnosis and according to the latest consensus, presence of > 10 IgG4+ plasma cells/high-power field along with one or more of the triad of classical histological features can provide a probable diagnosis of IgG4-related HP [4]. However, one must bear in mind that IgG4+ plasma cells are not specific for IgG4-RD and have been noted in other inflammatory and infectious conditions, including ANCA-related vasculitis, tuberculosis, and Rosai–Dorfman disease (RDD) [1, 8]. Thus, additional clinical, serological, and radiological evidences in the form of other organ involvement and an elevated serum IgG4 level are required for confirmation of IgG4-HP. Consequently, in patients with normal serum IgG4 and without systemic involvement, a robust histopathological and laboratory workup would be necessary.
The histopathological differential diagnosis of dural-based masses with rich inflammatory infiltrates includes lymphoplasmacyte-rich/chordoid meningiomas, RDD, and inflammatory myofibroblastic tumor. As detailed in Table 1, histopathological and immunohistochemical assessment in conjunction with clinical and radiological features help in differentiation. Notably, the young age of our patient, and the presence of bony remodeling and hyperostosis, hitherto described only once before with IgG4-HP [2], made differentiation from a lymphoplasmacyte-rich meningioma particularly difficult. However, the absence of a neoplastic meningothelial component and diffuse increase in IgG4+ plasma cells helped in ruling it out [10].
There are no consensus guidelines on the treatment of CNS IgG4-RD [1]. Glucocorticoids are the mainstay of treatment, with some patients receiving second-line steroid-sparing drugs or B cell-depleting agent rituximab in the face of steroid resistance/toxicity [1]. Surgical decompression may be required only in the presence of compressive symptoms, particularly common in spinal lesions. In instances such as ours with complete excision of lesion and absence of symptomatic systemic lesions, additional steroid therapy is not necessary [2].
To conclude, CNS IgG4-RD is extremely rare and most commonly manifest as HP. The latter may present as an isolated tumor-like mass with normal serum IgG4. Elevated IgG4+ plasma cells are necessary but not adequate for diagnosis, and a wide variety of inflammatory, infectious, and neoplastic conditions need exclusion. Accurate diagnosis, documentation, and long-term follow-up of CNS IgG4-RD is necessary for improved understanding of its biology.
References
Baptista B, Casian A, Gunawardena H, D’Cruz D, Rice CM (2017) Neurological manifestations of IgG4-related disease. Curr Treat Options Neurol 19:14
Lin CK, Lai DM (2013) IgG4-related intracranial hypertrophic pachymeningitis with skull hyperostosis: a case report. BMC Surg 13:37
Wallace ZS, Carruthers MN, Khosroshahi A, Carruthers R, Shinagare S, Stemmer-Rachamimov A, Deshpande V, Stone JH (2013) IgG4-related disease and hypertrophic pachymeningitis. Medicine (Baltimore) 92:206–216
Deshpande V, Zen Y, Chan JK et al (2012) Consensus statement on the pathology of IgG4-related disease. Mod Pathol 25:1181–1192
Radotra BD, Aggarwal A, Kapoor A, Singla N, Chatterjee D (2016) An orphan disease: IgG4-related spinal pachymeningitis: report of 2 cases. J Neurosurg Spine 25:790–794
Hwang G, Jin SY, Kim HS (2016) IgG4-related disease presenting as hypertrophic pachymeningitis and compressive optic neuropathy. Joint Bone Spine 83:601–602
Della-Torre E, Galli L, Franciotta D, Bozzolo EP, Briani C, Furlan R, Roveri L, Sessa M, Passerini G, Sabbadini MG (2014) Diagnostic value of IgG4 indices in IgG4-related hypertrophic pachymeningitis. J Neuroimmunol 266:82–86
Menon MP, Evbuomwan MO, Rosai J, Jaffe ES, Pittaluga S (2014) A subset of Rosai-Dorfman disease cases show increased IgG4-positive plasma cells: another red herring or a true association with IgG4-related disease? Histopathology 64:455–459
Zhu HD, Xie Q, Gong Y, Mao Y, Zhong P, Hang FP, Chen H, Zheng MZ, Tang HL, Wang DJ, Chen XC, Zhou LF (2013) Lymphoplasmacyte-rich meningioma: our experience with 19 cases and a systematic literature review. Int J Clin Exp Med 6:504–515
Lal A, Dahiya S, Gonzales M, Hiniker A, Prayson R, Kleinschmidt-DeMasters BK, Perry A (2014) IgG4 overexpression is rare in meningiomas with a prominent inflammatory component: a review of 16 cases. Brain Pathol 24:352–359
Sandoval-Sus JD, Sandoval-Leon AC, Chapman JR, Velazquez-Vega J, Borja MJ, Rosenberg S, Lossos A, Lossos IS (2014) Rosai-Dorfman disease of the central nervous system: report of 6 cases and review of the literature. Medicine (Baltimore) 93:165–175
Denis DJ, Elayoubi K, Weil AG, Berthelet F, Bojanowski MW (2013) Inflammatory myofibroblastic tumors of the central nervous system that express anaplastic lymphoma kinase have a high recurrence rate. Surg Neurol Int 4:70
Yamamoto H, Yamaguchi H, Aishima S, Oda Y, Kohashi K, Oshiro Y, Tsuneyoshi M (2009) Inflammatory myofibroblastic tumor versus IgG4-related sclerosing disease and inflammatory pseudotumor: a comparative clinicopathologic study. Am J Surg Pathol 33:1330–1340
Prayer D, Grois N, Prosch H, Gadner H, Barkovich AJ (2004) MR imaging presentation of intracranial disease associated with Langerhans cell histiocytosis. AJNR Am J Neuroradiol 25:880–891
Zhu M, Yu BB, Zhai JL, Sun G (2016) Case of Langerhans cell histiocytosis that mimics meningioma in CT and MRI. J Korean Neurosurg Soc 59:165–167
Lachenal F, Cotton F, Desmurs-Clavel H, Haroche J, Taillia H, Magy N, Hamidou M, Salvatierra J, Piette JC, Vital-Durand D, Rousset H (2006) Neurological manifestations and neuroradiological presentation of Erdheim-Chester disease: report of 6 cases and systematic review of the literature. J Neurol 253:1267–1277
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Authors declare no conflict of interests for this article.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Nambirajan, A., Sharma, M.C., Garg, K. et al. Large dural-based mass with bony hyperostosis in a 16-year-old male: IgG4-related disease mimicking lymphoplasmacyte-rich meningioma. Childs Nerv Syst 35, 1423–1427 (2019). https://doi.org/10.1007/s00381-019-04187-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00381-019-04187-z