
Abstract
We found that a female infant presenting with left bundle branch block and left ventricular noncompaction carries uninvestigated gene mutations HCN4(G811E), SCN5A(L1988R), DMD(S2384Y), and EMD(R203H). Here, we explored the possible pathogenicity of HCN4(G811E), which results in a G811E substitution in hyperpolarization-activated cyclic nucleotide-gated channel 4, the main subunit of the cardiac pacemaker channel. Voltage-clamp measurements in a heterologous expression system of HEK293T cells showed that HCN4(G811E) slightly reduced whole-cell HCN4 channel conductance, whereas it did not affect the gating kinetics, unitary conductance, or cAMP-dependent modulation of voltage-dependence. Immunocytochemistry and immunoblot analysis showed that the G811E mutation did not impair the membrane trafficking of the channel subunit in the heterologous expression system. These findings indicate that HCN4(G811E) may not be a monogenic factor to cause the cardiac disorders.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hyperpolarization-activated cyclic nucleotide-gated channel 4 (HCN4) is the dominant type of the alpha subunit of the mixed cation channel expressed in the sinoatrial node (SAN) of the mature human heart [1]. This channel produces the cardiac pacemaker current (If) and is responsible for the regeneration of cardiac action potentials [2, 3]. If channel malfunction due to HCN4 mutations may result in cardiac function disorders including bradycardia, atrial fibrillation, chronotropic incompetence, and exercise-induced premature beats [3,4,5,6,7]. Recently, the spectrum of disorders due to HCN4 mutations has been expanded to the myocardial structure disorders such as left ventricular noncompaction (LVNC), a cardiomyopathy characterized by a noncompacted left ventricular myocardial layer with hypertrabeculations [8, 9]. It is unclear how HCN4 mutations lead to the myocardial structural disorders. If channel malfunction could directly influence primordial cardiomyocytes throughout the immature heart because the expression of HCN4 is not restricted to the SAN at the early developmental stage [10].
In this study, we identified HCN4(G811E), SCN5A(L1988R), DMD(S2384Y), and EMD(R203H), uninvestigated gene mutations from a female infant presenting with left bundle branch block (LBBB) and LVNC. She inherited HCN4(G811E) from her father presenting with mild bradycardia. To investigate whether the G811E mutation produces If channel malfunction leading to cardiac disorders, we compared whole-cell and single-channel ion currents mediated by the channel consisting of the wild-type (WT) and/or mutant HCN4 in heterologous expression cells. Furthermore, we investigated whether the G811E mutation affects the membrane trafficking of HCN4 channel subunit using immunocytochemistry and immunoblot analysis.
Materials and methods
Diagnosis
This study was approved by the Research Ethics Committee of University of Toyama (#22-7). An infant was diagnosed with LVNC by the criteria of Ichida et al.: (1) left ventricular hypertrophy with deep endomyocardial trabeculations predominantly in the distal portion (apex) of the left ventricle, and distributed over one wall segment of the left ventricle, (2) a two-layered endocardium with a noncompacted to compacted ratio of ≥ 2.0, and (3) deep recesses filled with blood from the ventricular cavity visualized by color Doppler imaging [11].
Direct sequencing
After informed consent was obtained from the parents according to the institutional guidelines, DNA was isolated from the infant’s peripheral blood sample using a QIAamp DNA Mini Kit (Qiagen, Hilden, Germany). We performed direct sequencing using an ABI PRISM 3130 genetic analyzer (Life Technologies, Carlsbad, CA, USA).
Next-generation sequencing and in silico analysis
We performed next-generation sequencing (NGS) and in silico analysis on the genomic DNA of the subject infant according to our previous study [12]. We designed a custom AmpliSeq panel using Ion AmpliSeq Designer software (http://www.ampliseq.com) to target all the exons of the previously reported 73 genes associated with cardiomyopathies and cardiac channelopathies (Table 1). NGS was run on an Ion PGM system. Torrent Suite and Ion Reporter Software 5.0 (Life Technologies) were used to perform primary, secondary, and tertiary analyses, including optimized signal processing, base calling, sequence alignment, and variant analysis. In silico analysis using databases and in silico prediction algorithms (Table 2) were performed as follows (Supplemental Figure 1). First, the minor allele frequency (MAF) of all detected variations was consulted across the East Asian (EAS) population of 4327 individuals in the Exome Aggregation Consortium (ExAC) database and the Human Genetic Variation Database (HGVD) containing 1208 Japanese individuals (Table 3). All variations with an MAF of ≥ 0.5% among the EAS population of the ExAC database and HGVD were filtered out by the MAF of the global populations according to ExAC and 1000 Genomes Project (Phase 3) (Table 4). For the resultant mutations, the MAFs of the global population are summarized in Table 4. Second, we excluded the variations that were judged to be benign based on the functional and/or segregation analysis data on the previously reported variations in both the Human Genome Mutation Database (HGMD) and the ClinVar disease mutation database (Table 5). Finally, we evaluated the pathogenic potential of the remaining variations using 4 in silico prediction algorithms, SIFT, MutationTaster2, PolyPhen-2, and CADD with the criteria listed in Table 2. The variations predicted to be pathogenic by 3 or more of these algorithms were regarded as possibly pathogenic mutations (Table 5).
Plasmid construction
WT human HCN4 cDNA (GenBank: NM_005477) was subcloned into pReceiver-M12 vector containing the N-terminal 3× FLAG epitope (GeneCopoeia, Rockville, MD, USA) [FLAG-HCN4(WT)]. FLAG-HCN4(G811E) was generated based on FLAG-HCN4(WT) using a site-directed mutagenesis kit (SMK-101, Toyobo, Osaka, Japan). FLAG-HAins-HCN4(WT) and FLAG-HAins-HCN4(G811E) were generated by inserting the HA (GGGSYPYDVPDYAGGG) sequence into the extracellular segment 3–segment 4 (S3–S4) loop (between S365 and E366, after nucleotide position 1095) of HCN4(WT) and HCN4(G811E), respectively.
Cell culture and transfection
HEK293T cells were cultured in Dulbecco’s modified Eagle’s medium (11995-040, Life Technologies) supplemented with 10% fetal bovine serum (FBS) at 37 °C in 5% CO2. For electrophysiological analyses, cells were transiently transfected with a 2:3 mixture of pCAGGS-EGFP and either FLAG-HCN4(WT), FLAG-HCN4(G811E), or a 1:1 mixture of FLAG-HCN4(WT) and FLAG-HCN4(G811E) (total 250 ng per dish) using TransIT-293 (Mirus Bio, Madison, WI, USA). For immunocytochemistry and immunoblot analysis, cells were transfected with 250 ng/dish of FLAG-HAins-HCN4(WT) or FLAG-HAins-HCN4(G811E).
Electrophysiological analyses
Forty to 48 h after transfection, ruptured-patch whole-cell voltage-clamp recordings were made from EGFP-positive cells at 24–26 °C as described elsewhere [13]. The recording chamber was perfused with saline containing (in mM) 140 NaCl, 5.4 KCl, 1.8 CaCl2, 1 MgCl2, 5 HEPES, and 5.5 d-glucose (pH, adjusted to 7.4 with NaOH). A glass recording pipette was filled with (in mM) 134 potassium d-gluconic acid, 7.6 KCl, 9 KOH, 10 NaCl, 10 HEPES, 0.5 ethylene glycol-bis(2-aminoethyl ether)-N,N,N’,N’-tetraacetic acid (EGTA), 1.2 MgCl2, 4 ATP magnesium salt, and 0.2 GTP disodium salt (pH, adjusted to 7.4 with KOH) and had a tip resistance of 3–5 MΩ. Current signals were acquired with an EPC-9/2 amplifier (HEKA, Lambrecht, Germany) controlled by PatchMaster software (version, 2.69 or 2.71; HEKA). Series resistance was compensated electronically by 60%. The command voltages were corrected for a liquid junction potential between the saline and pipette solution. Current signals were low-pass filtered at a corner frequency of 10 kHz and sampled at a rate of 20 or 100 kHz. To assess the cAMP-dependent modulation of HCN4 channel, 8-(4-chlorophenylthio)adenosine 3′,5′-cyclic monophosphate (8-CPT-cAMP; 1 µM; ab120424, Abcam, Cambridge, UK), an cAMP analog was added to the pipette solution and measured I–V relations 5 min after membrane rupture.
Forty-eight hour after transfection, single-channel voltage-clamp recordings were made from EGFP-positive cells in a cell-attached mode at 24–26 °C. To set the membrane potential to ~ 0 mV, the cells were bathed in a K+ (150–153 mM)-rich solution consisting of (in mM) 130 or 140 KCl, 10 NaCl, 10 HEPES, 10 EGTA (pH, adjusted to 7.4 with KOH). A glass recording pipette was coated with silicone elastomer (Sylgard 184, Dow Corning, MI, USA) at its taper and filled with the saline that was used as the bath solution in the whole-cell recordings. The tip resistance ranged 5–10 MΩ. The osmolalities of the bath and pipette solution were equalized (296–300 mOsmol/kg) by adding sucrose to either or both of these solutions. Current signals were low-pass filtered at a corner frequency of 10 kHz and sampled at a rate of 100 kHz.
Immunocytochemistry
To label the extracellular side of HCN4, the cells were incubated with an anti-HA antibody (sc-805, Santa Cruz Biotechnologies, Inc.) (1:1000) in DMEM containing 10% FBS (37 °C, 5% CO2, 1 h). Then, the following steps were performed at 4 °C to minimize the endocytosis of the channel. The cells were fixed with 4% paraformaldehyde for 30 min, followed by blocking with 10% donkey serum for 1 h. To detect the HCN4 proteins on the cell surface, the cells were incubated with an Alexa Fluor 488-conjugated donkey anti-rabbit antibody (1:500) for 1 h. Immunofluorescence was captured using a TCS-SP5 confocal microscope (Leica, Wetzlar, Germany).
Immunoblot analysis
Forty-eight hour after transfection, cells were incubated with an anti-HA antibody (1:500, Y-11; Santa Cruz Biotechnology, Inc.) in DMEM containing 10% FBS (37 °C, 5% CO2, 1 h). To prepare the whole-cell lysates, the cells were lysed with 200 µl of a lysis buffer (Pierce IP Lysis buffer, Thermo Fisher Scientific) and a protease inhibitor on ice for 30 min. After centrifugation (16,000×g, 15 min), the whole-cell lysates were treated with 10 µl of Anti-HA Tag Magnetic Beads (MBL) in a rotator at 4 °C for 1 h. The antibody–protein complex bound to the beads was extracted by incubation with a 2× SDS sample buffer consisting of 0.5% SDS, 100 mM Tris (pH 6.8), 20% glycerol, and 50 mM DTT at room temperature for 30 min. Electrophoresis was performed at a constant voltage of 140 V for 1 h. The protein-separated gel was transferred to a reactivated PVDF membrane (Hybond-P, GE Healthcare). N-terminal FLAG-tagged proteins were detected using Anti-DDDDK-tag mAb-HRP-DirecT (MBL) according to the manufacturer’s instruction.
Statistical analyses
For each cell examined in the whole-cell mode, the amplitude of the linear leak current evoked by a test potential (Etest) step was estimated by accumulation of 10 sets of current responses to a 1/10-scaled voltage step recorded immediately after each trial. The current amplitudes used in the following analyses were corrected for the linear leak components offline. The quasi-steady-state (q-s-s) amplitude of a whole-cell HCN4 channel current was measured as a difference from the mean current level during a 0.1-s pre-stimulus period to the mean current level during the last 0.1-s period of the test hyperpolarizing step and then normalized to the whole-cell membrane capacitance (Cm) estimated from the capacitive current as described elsewhere [13] (q-s-s current density). Whole-cell conductance mediated by fully activated HCN4 channels per unit membrane capacitance (gWC/Cm) was estimated from linear regression to the linear region (− 60 to − 10 mV) of the q-s-s current density–Etest plot. Whole-cell chord conductance mediated by HCN4 channel at each Etest was estimated, employing the mean value in Table 7 as the reversal potential (Erev) of HCN4 channel current. Voltage for half-maximal activation (Vhalf) and slope factor (k) were estimated from the Boltzmann function (gWC = a/[1 + exp(Vhalf − Etest)/k] + b, where a, Etest, and b are scale factor, test potential, and offset) fitted to the chord conductance–Etest plot. Time constant of activation (τact) was estimated from the single-exponential function fitted to the rising time-course of an HCN4 channel current. The peak amplitude of a whole-cell HCN4 channel tail current was measured as a difference from the mean current level during a 0.1-s pre-stimulus period to the peak level of the tail current. The Erev of HCN4 channel current was estimated by linear regression to the peak tail current amplitude–Etest plot for an Etest range from − 60 to − 10 mV. Linear regression and exponential curve fitting were done using Excel (for Mac 2008 or 2011; Microsoft, Redmond, WA, USA) and Igor Pro (versions, 6.22 and 6.32A; WaveMetrics, Lake Oswego, OR, USA).
For analyzing the data of the single-channel recordings, the trace was low-pass-filtered at a corner frequency of 500 Hz offline. The amplitude of a single-channel current was measured as a difference from the mean current level during a 0.1-s period prior to the single-channel event to the mean steady-state current level during the event. Single-channel conductance mediated by HCN4 channel was estimated by linear regression of the single-channel current amplitude–Etest plot. The open probability of HCN4 channel was measured as the fractional ratio of the total duration of single-channel events to the total recording time (22.5 s). The duration was estimated from the number of the data points where the current level exceeded the midpoint between the basal current level and the steady-state current level of single-channel events.
Each numerical data group is expressed as mean ± SEM throughout the text, table, and figures unless otherwise stated. When the majority of the data groups appeared to have normal distributions (P > 0.05, Shapiro–Wilk test), an unpaired t test was used to examine the differences; otherwise, Wilcoxon rank sum test was used. A difference with a P below 0.05 is regarded significant. Statistical examination was performed with JMP Pro (version, 11; SAS Institute, Cary, NC, USA).
Results
Clinical background
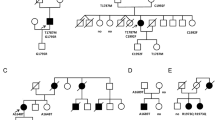
A 3-day-old female infant was diagnosed to have a muscular ventricular septal defect (VSD) from heart murmur. At 4 months of age, complete LBBB was found (Fig. 1a). At 6 months of age, the VSD was closed spontaneously while LVNC (Fig. 1b) and heart failure were observed. Heart failure could not be improved with diuretics and an angiotensin converting enzyme inhibitor. Left ventricular dilation was also observed with cardiac catheterization. The cardiac function of the infant measured at 8 months of age is summarized in Table 6. At the age of 5 years, intermittent LBBB with an occasionally normal or narrow QRS pattern was observed. Her father (~ 50 bpm) and paternal grandmother showed mild bradycardia while the father has never experienced any cardiac events. In addition, the corrected QT intervals of the infant (425 ms) and her father (419 ms) and mother (407 ms) were within the normal ranges (Fig. 1c).

Clinical observations of an infant with LVNC and her father. Twelve-lead electrocardiograms (4 months of age) (a) and echocardiograms of the left ventricle (6 months of age) (b) of the infant. Arrow in b, trabeculation. c Pedigree of the infant’s family. Arrow, the proband. Gray symbols, individuals with bradycardia. Square and circle symbols, male and female, respectively. QTc corrected QT interval calculated by Fredericia’s formula (when the heart rate is higher than 80) or Bazett’s formula (when the heart rate is lower than 80). d Schematic topology of HCN4(G811E); S segment. e Partial amino acid sequences of various mammals’ homologs aligned at the position corresponding to G811 of human HCN4
Genetic analysis
In search of genetic mutations underlying the development of LVNC in the infant, we performed NGS targeted for the 73 major genes related to cardiomyopathies and cardiac channelopathies (Table 1). The NGS analysis revealed that the infant possesses 26 variants in 19 of the 73 genes (Table 3). To pick up the variants that could be relatively important for the development of LVNC, we filtered the variants based on the MAF (Tables 3, 4) and the pathogenicity predicted by multiple in silico algorithms (Supplemental Figure 1, Tables 2, 5). As a result, SCN5A(L1988R) (NM_198056.2:c.5963T>G), HCN4(G811E) (NM_005477.2:c.2432G>A), DMD(S2384Y) (NM_004006.2:c.7151C>A), and EMD(R203H) (NM_000117.2:c.608G>A,) were screened. Sanger sequencing of the infant and her family’s genomic DNA showed that these mutations did not occur de novo and that the infant inherited the SCN5A, HCN4, and EMD mutations from her father and the DMD mutation from her mother (Fig. 1c, Supplemental Figure 2). SCN5A encodes the main subunit of NaV1.5 channel responsible for the cardiac fast Na+ current. SCN5A(L1988R) is reported predominantly in the East Asian populations. SCN5A(L1988R) is not likely to be an arrhythmogenic factor because this variant is found in the healthy controls of Japanese [14] and Korean [15] cohorts. Consistently, this mutation was predicted to be non-pathogenic by three of the four in silico algorithms (Table 5). Furthermore, our voltage-clamp measurement in HEK293T cells show that the L1988R mutation did not change the density or time- or voltage-dependence of the Na+ current mediated by SCN5A channel (Supplemental Figure 3). On the other hand, in silico analyses suggested HCN4(G811E), DMD(S2384Y), and EMD(R203H) as possibly pathogenic mutations (Table 5). In addition, the infant had no missense mutation in the previously reported LVNC-related genes (Table 1) [16,17,18].
HCN4, DMD, and EMD encode the main subunit of If channel, dystrophin, and emerin, respectively. In the following analyses, we focused on whether and how HCN4(G811E) affects If channel function (for the possible contributions of DMD(S2384Y) and EMD(R203H) to the infant’s cardiac symptoms, see “Discussion”). This mutation causes a G811E amino acid substitution in the C-terminus of HCN4 channel subunit (Fig. 1d). G811 is conserved in the corresponding genes of many mammals (Fig. 1e).
Electrophysiological properties of HCN4(G811E)-containing channels
We analyzed whole-cell hyperpolarization-activated HCN4 channel currents in cells transfected with HCN4(WT), HCN4(G811E), or a 1:1 mixture of these genes (WT, GE, and GE/WT cells, respectively) in a ruptured-patch whole-cell mode. The GE and GE/WT cells mimicked the cellular environments of the homo- and heterozygous HCN4(G811E) carriers, respectively.
To analyze the activation gating of the channels, the Em was jumped from a holding potential of − 20 mV to a more negative Etest (Fig. 2a). HCN4 channels were activated with Etest’s more negative than − 70 mV in all the cells (Fig. 2b). The absolute whole-cell HCN4 channel conductance per unit membrane capacitance when the channels were fully activated (gWC/Cm) was estimated from the slope of the linear region of the current density–Etest plot. This parameter was significantly smaller in the GE and GE/WT cells than in the WT cells (Fig. 2b inset, Table 7). There was no significant difference in the gWC/Cm between the GE and GE/WT cells (Fig. 2b inset, Table 7). This result suggests that homozygous or heterozygous inheritance of HCN4(G811E) causes the loss of function of HCN4 channel.

The G811E mutation reduced whole-cell HCN4 channel conductance. a Representative whole-cell current responses of the labeled cells to voltage stimuli schematically shown above the traces. The corresponding stimulus and response are drawn in the same color. To minimize cell damage, the durations of strongly hyperpolarizing voltage steps were shortened. b Mean quasi-steady-state (q-s-s) HCN4 channel current density plotted against Etest. The data were obtained from 10 WT, 11 GE, and 10 GE/WT cells in this and following panels. Asterisk P < 0.05 compared with the WT cells (unpaired t test). Inset, mean whole-cell HCN4 channel conductances at full activation. Asterisk and NS, P < 0.05 and P > 0.05, respectively (unpaired t test). c Mean chord whole-cell HCN4 channel conductance plotted against Etest. The data are normalized to the maximum for each cell. Sigmoid curves, Boltzmann functions with Vhalf’s and k’s whose values are set to the means of the corresponding cells (Table 7)
The Vhalf and k estimated from the Boltzmann function fitted to the chord conductance–Etest plot (Fig. 2c) were not significantly different between the WT, GE, and GE/WT cells except for a slight difference in k between the WT and GE cells (Table 7). This result suggests that the G811E mutation does not much affect the voltage-dependence.
The τact estimated from the single-exponential function fitted to the rising phase of HCN4 channel current was not significantly different between the WT, GE, and GE/WT cells for all the Etest’s (− 140 to − 80 mV) (not illustrated; as an example, the mean τact at a Etest of − 140 mV is shown in Table 7). This result suggests that the G811E mutation does not affect the time-dependence of activation.
To examine whether the G811E mutation affects the ion-selectivity of HCN4 channel, we measured the reversal potential (Erev) of HCN4 channel current. To this end, we activated HCN4 channel with a conditioning hyperpolarizing step of a fixed potential and then measured the peak amplitude of an HCN4 channel tail current, varying Etest (Fig. 3a). The Erev estimated from the linear function fitted to the peak HCN4 channel tail current amplitude–Etest relation was not significantly different between the WT, GE, and GE/WT cells (Table 7). This result suggests that the G811E mutation does not alter the ion-selectivity.

The G811E mutation did not alter the ion-selectivity or time-dependence of deactivation of HCN4 channel. a To estimate the Erev of HCN4 channel current (Table 7), we measured its tail currents at various Etest’s using the voltage protocol schematically shown above the traces. Each set of traces indicates the representative whole-cell current responses of the labeled cells. b, c Evaluation of the time-dependence of deactivation of HCN4 channels. b We measured hyperpolarization-activated currents before and after a − 40-mV voltage step to deactivate HCN4 channel for 1–6 s. Schematic, voltage protocol. A set of traces, representative whole-cell current responses of the labeled cells. c Mean fraction of the HCN4 channel that remained active at the end of the − 40-mV step (Fact) plotted against the duration of − 40-mV step. Fact was given by an equation Fact = (Iinst,2 − Iinst,1)/(Iq-s-s − Iinst,1), where Iinst,1, Iinst,2, and Iq-s-s are the levels of the instantaneous currents evoked by the first and second hyperpolarizing steps and the mean level over the last 0.1-s of the first hyperpolarizing step, respectively. Exponential curves, single-exponential functions with τdeact’s whose value are set to the means of the corresponding cells (Table 7)
To examine whether the G811E mutation affects the time-dependence of deactivation of HCN4 channel, we measured the fractional population of the HCN4 channels that remained active after a – 40 mV voltage step lasting for 1-6 s (Fact, see Fig. 3 legend) (Fig. 3b). The time constant of deactivation (τdeact) estimated from the single-exponential function fitted to the plot of Fact against − 40-mV step duration was not different between the WT and GE cells (Fig. 3c, Table 7). This result suggests that the G811E mutation does not affect the time-dependence of deactivation.
It is known that cAMP binding to the cNBD of HCN4 subunit facilitates the activation of If channel [19]. This cAMP-dependent modulation is thought to be important for the sympathetic regulation of heart beat [20]. A previous study showed that the C-terminus region distal to the cNBD (amino acids 719–1012) may influence the cAMP-dependent modulation [21]. Because the three-dimensional model of the whole C-terminus of HCN4 is not available, it is unclear whether the site of G811E mutation is located close enough to interact with the cNBD and to affect the cAMP-dependent modulation in the assembled tetramer. Thus, here we directly examined the effect of the G811E mutation on the cAMP-dependent modulation through an electrophysiological approach. In both the WT and GE cells, a 5-min intracellular perfusion with a 8-(4-chlorophenylthio)adenosine 3′,5′-cyclic monophosphate (8-CPT-cAMP, 1 µM)-containing pipette solution positively shifted the Vhalf but not the k (Fig. 4a, b, Table 7). A similar positive shift is reported for If in an SAN-derived macropatch exposed to cAMP [22, 23]. The extent of this positive shift was similar between the WT and GE cells because there was no significant difference in Vhalf between the 8-CPT-cAMP-treated WT and GE cells (Table 7). The intracellular perfusion with the 8-CPT-cAMP-containing solution also accelerated activation, changing the \({\tau}_{\rm {act}}\) in both the WT and GE cells (Table 7). A similar acceleration is reported for If in an SAN-derived macropatch exposed to cAMP [23]. The extent of this acceleration was similar between the WT and GE cells because there was no significant difference in the act between the 8-CPT-cAMP-treated WT and GE cells (Table 7). These results suggest that the G811E mutation does not affect the cAMP-dependent modulation of activation gating.

The G811E mutation did not affect the cAMP-dependent modulation of activation gating. a, b Mean normalized whole-cell HCN4 channel conductance of the WT (a) and GE (b) cells intracellularly perfused with the pipette solution with or without 8-CPT-cAMP (1 µM) plotted against Etest. HCN4 channel was activated with the same voltage stimuli as in Fig. 2a. The data in the presence of 8-CPT-cAMP were obtained from 11 WT and 9 GE cells. The data in the absence of 8-CPT-cAMP were reproduced from Fig. 2. Sigmoid curves, Boltzmann functions with Vhalf’s and k’s whose values are set to the means of the corresponding cells (Table 7)
To explore the possible cause of the reduction in gWC/Cm due to the G811E mutation, we made single-channel recordings from the WT and GE cells in a cell-attached mode. HCN4 single-channel currents could be discerned as events occurring more frequently with more negative Etest’s (Fig. 5a). The plot of the mean single-channel current amplitude against Etest of the GE cells was similar to that of the WT cells (Fig. 5b). Single-channel conductance mediated by HCN4 channels was not significantly different between the WT and GE cells (32.7 ± 2.0 pS, n = 3 and 34.4 ± 1.2 pS, n = 3, respectively) (Fig. 5c). As well, the open probability of the channels was not significantly different between the WT and GE cells at a Etest of − 80 mV (1.32 ± 1.09%, n = 3 and 1.45 ± 0.97%, n = 3, respectively) or − 140 mV (3.70 ± 0.85%, n = 3 and 4.09 ± 1.83%, n = 3, respectively) (Fig. 5d). These results suggest that the reduction in gWC/Cm due to the G811E mutation cannot be ascribed to a difference in unitary activity between the WT and HCN4(G811E)-containing channels.

Single-channel activity was similar between HCN4(WT) and HCN4(G811E) channels. a Representative current responses of the labeled cells recorded at the labeled Etest’s in a cell-attached mode. A downward deflection indicates an inward single-channel current. b Mean single-channel current amplitude plotted against Etest. Each point indicates the mean of 10–12 events of 3 WT cells or 10–13 events of 3 GE cells. Lines, linear functions fitted to the collection of all the data obtained from theses WT and GE cells. c Mean single-channel conductances. P > 0.05 between the cells (Wilcoxon rank sum test). d Mean open probabilities at the labeled Etest’s. P > 0.05 between the WT and GE cells (unpaired t test)
Cell surface expression of HCN4(WT) and HCN4(G811E)
To examine the cell surface expression levels of the HCN4 subunits, we constructed the expression plasmids with an HA-tag sequence at their extracellular S3–S4 loops to allow efficient staining of the extracellular side of the subunits. We performed immunofluorescent labeling without permeabilization to minimize staining of the intracellularly residing subunits. In both the HCN4(WT)- and HCN4(G811E)-expressing cells, the fluorescent signal was detected on the plasma membrane and its intensity was not discerned between these cells (Fig. 6a).

Cell surface expression level was similar between HCN4(WT) and HCN4(G811E) subummits. a Confocal (upper) and bright-filed (lower) microscopic images of HEK293T cells expressing HA-tagged HCN4(WT) and HA-tagged HCN4(G811E). Scale bars 50 µm. b, c Immunoblot analysis of the surface expression levels of HCN4 subunits using anti-FLAG antibody. b Immunoblots of the whole-cell extracts (input) and bead-bound fractions of the cells transfected with FLAG-HAins-HCN4(WT) or FLAG-HAins-HCN4(G811E) (WT or GE, respectively.) c Comparison of the density ratio of the bead-bound fraction to the whole-cell extracts. The values are expressed, taking the density ratio of the HA-tagged HCN4(WT)-expressing cells as 1. Column and error bar, mean and + SD, respectively; n, 3
To quantify the cell surface expression level, we performed densitometry on the immunoblots of the whole-cell extracts (Fig. 6b, input) and the surface-expressed proteins that were isolated as the anti-HA antibody-bound fractions (Fig. 6b, beads). We calculated the density ratio of the bead-bound fraction to the whole-cell extracts for each of the cell groups [1 group of HCN4(WT)-expressing cells and 3 groups of HCN(G811E)-expressing cells]. The relative density ratio of the HCN4(G811E)-expressing cells was 0.91 ± 0.30 (mean ± SD) when the density ratio of the HCN4(WT)-expressing cells was taken as 1. These results suggest that HCN4(G811E) subunit can be trafficked to the cell surface as efficiently as HCN4(WT) subunit.
Discussion
The G811E mutation may slightly reduce HCN4 channel current density although the underlying mechanism remains unclear
In this study, we identified HCN4(G811E), SNC5A(L1988R), DMD(S2384Y), and EMD(R203H) in a female infant presenting with LBBB and LVNC (Fig. 1a–c). HCN4(G811E) was inherited from her father (Supplemental Figure 2) and possibly her paternal grandmother who showed mild bradycardia, a symptom often found in mutant HCN4 carriers [5, 8, 9, 24,25,26]. We performed functional analyses to examine whether the G811E mutation causes a change in the channel function that may lead to the cardiac disorders.
The whole-cell recordings showed that the G811E mutation slightly reduced whole-cell HCN4 channel conductance (Fig. 2b, Table 7) while the G811E mutation did not affect the voltage- or time-dependence of activation, ion-selectivity, time-dependence of deactivation, or cAMP-dependent modulation of activation gating (Figs. 2, 3, 4). The whole-cell HCN4 channel conductance at full activation was similar between the GE and GE/WT cells (Fig. 2b, Table 7). This result suggests that the G811E mutation might exert a similar action in homozygous and heterozygous HCN4(G811E) carriers. The single-channel recordings showed that a functional HCN4 channel consisting of 4 WT or mutant subunits mediates the same amount of ion current (Fig. 5). Immunocytochemistry and immunoblot analysis showed that the G811E mutation did not impair the membrane trafficking of HCN4 subunit (Fig. 6).
Although there was a difference in the whole-cell HCN4 channel conductance, the different was small at membrane potentials near the activation threshold (Fig. 5b). Because If exerts its action on cardiac action potentials at these levels of the membrane potential (see below), the effect of the G811E mutation on the action potentials might be negligible. Therefore, G811E mutation might not be a monogenic factor to cause the cardiac disorders of the infant. It should be noted that the effect of G811E mutation on HCN4 channel function described above was assessed in the heterologous expression system; the mutation could exert different effects on HCN4 channel function in the cardiomytocytes in vivo.
In this study, we could not clarify how the G811E mutation produces the difference in the whole-cell HCN4 channel conductance. This issue awaits future investigations.
It is noteworthy that the threshold voltage for activation measured in this study was relatively negative in all the cell groups (~ − 70 mV, Fig. 2b). This was presumably because our recordings were made at room temperature. As shown in previous studies in heterologous expression systems, the I–V plot of HCN4 current measured at room temperature is typically shifted by − 25 mV from that measured at a temperature of 37 °C [8, 9]. Thus, the threshold voltage of HCN4(G811E)-containing channel and the WT channel is thought to be around − 45 mV at a body temperature. HCN4(G811E)-containing channel as well as the WT channel may operate in vivo because HCN4 channel is activated at voltages observed during the diastolic depolarization (− 40 to − 60 mV) in human SAN pacemaker cells under a physiological condition (37 °C) [27].
SCN5A(L1988R) may not be an arrhythmogenic factor
Our whole-cell recordings showed that the L1988R mutation did not change the density or kinetics of SCN5A channel current. The previous studies report that SCN5A(L1988R) is a common minor variant found in the healthy populations in East Asia (see “Results”). These facts suggest that SCN5A(L1988R) may not be an arrhythmogenic factor.
Possible contribution of DMD(S2384Y) and EMD(R203H) to the development of the cardiac disorders
At the present, it is difficult to experimentally examine the pathogenicities of DMD(S2384Y) and EMD(R203H) because in vitro assays to examine the cellular function of the product proteins have not been established. Instead, here we summarize the current knowledge of DMD and EMD mutations obtained in clinical and molecular studies. DMD encodes dystrophin, a very large protein that connects the cytoskeleton of a muscle fiber to the extracellular matrix. In general, DMD mutations causing defects in dystrophin may lead to dystrophinopathies including Duchenne muscular dystrophy and Becker muscular dystrophy [28]. Such a mutation could also cause LVNC because some dystrophinopathy patients manifest LVNC [29,30,31]. As to the particular case of DMD(S2384Y), this mutation causes an amino acid substitution at the central rod domain of dystrophin. This domain is thought to be dispensable for the protein’s function because deletions in this domain cause only very mild phenotypes [28]. Consistently, the infant’s mother carrying DMD(S2384Y) but not HCN4(G811E) or EMD(R203H) did not show any muscular and myocardial abnormalities. EMD encodes emerin, a nuclear protein that mediates membrane anchorage to the cytoskeleton. Although the precise function of emerin is unclear [32], it is known that emerin binds various partner proteins [33] and that some EMD mutations cause Emery–Dreifuss muscular dystrophy, which is characterized by muscular atrophy and cardiomyopathy with conduction disturbance [34]. However, it is reported that phenotypes due to missense mutations in EMD are usually mild as compared with severe phenotypes due to mutations causing protein truncation [32]. Consistently, the infant’s father did not show LVNC presentation despite EMD is an X-chromosomal gene and he possesses only the mutational variant in his genome. These facts suggest that DMD(S2384Y) or EMD(R203H) by itself might not be enough to develop LVNC.
Polygenic mechanisms may underlie the cardiac disorders
Our findings suggest that HCN4(G811E) might not be a monogenic factor to cause the cardiac disorders while could produce cardiac disorders with the aid of DMD(S2384Y), EMD(R203H), and/or other gene mutations that were not included in the panel of our exome analysis.
References
Shi W, Wymore R, Yu H, Wu J, Wymore RT, Pan Z, Robinson RB, Dixon JE, McKinnon D, Cohen IS (1999) Distribution and prevalence of hyperpolarization-activated cation channel (HCN) mRNA expression in cardiac tissues. Circ Res 85:e1–e6
Verkerk AO, Wilders R (2014) Pacemaker activity of the human sinoatrial node: effects of HCN4 mutations on the hyperpolarization-activated current. Europace 16:384–395
DiFrancesco D (2015) HCN4, sinus bradycardia and atrial fibrillation. Arrhythm Electrophysiol Rev 4:9–13
Duhme N, Schweizer PA, Thomas D, Becker R, Schroter J, Barends TR, Schlichting I, Draguhn A, Bruehl C, Katus HA, Koenen M (2013) Altered HCN4 channel C-linker interaction is associated with familial tachycardia-bradycardia syndrome and atrial fibrillation. Eur Heart J 34:2768–2775
Schweizer PA, Duhme N, Thomas D, Becker R, Zehelein J, Draguhn A, Bruehl C, Katus HA, Koenen M (2010) cAMP sensitivity of HCN pacemaker channels determines basal heart rate but is not critical for autonomic rate control. Circ Arrhythm Electrophysiol 3:542–552
Schulze-Bahr E, Neu A, Friederich P, Kaupp UB, Breithardt G, Pongs O, Isbrandt D (2003) Pacemaker channel dysfunction in a patient with sinus node disease. J Clin Investig 111:1537–1545
Hategan L, Csanyi B, Ordog B, Kakonyi K, Tringer A, Kiss O, Orosz A, Saghy L, Nagy I, Hegedus Z, Rudas L, Szell M, Varro A, Forster T, Sepp R (2017) A novel ‘splice site’ HCN4 Gene mutation, c.1737 + 1 G>T, causes familial bradycardia, reduced heart rate response, impaired chronotropic competence and increased short-term heart rate variability. Int J Cardiol 241:364–372
Milano A, Vermeer AM, Lodder EM, Barc J, Verkerk AO, Postma AV, van der Bilt IA, Baars MJ, van Haelst PL, Caliskan K, Hoedemaekers YM, Le Scouarnec S, Redon R, Pinto YM, Christiaans I, Wilde AA, Bezzina CR (2014) HCN4 mutations in multiple families with bradycardia and left ventricular noncompaction cardiomyopathy. J Am Coll Cardiol 64:745–756
Schweizer PA, Schröter J, Greiner S, Haas J, Yampolsky P, Mereles D, Buss SJ, Seyler C, Bruehl C, Draguhn A, Koenen M, Meder B, Katus HA, Thomas D (2014) The symptom complex of familial sinus node dysfunction and myocardial noncompaction is associated with mutations in the HCN4 channel. J Am Coll Cardiol 64:757–767
Liang X, Evans SM, Sun Y (2015) Insights into cardiac conduction system formation provided by HCN4 expression. Trends Cardiovasc Med 25:1–9
Ichida F, Tsubata S, Bowles KR, Haneda N, Uese K, Miyawaki T, Dreyer WJ, Messina J, Li H, Bowles NE, Towbin JA (2001) Novel gene mutations in patients with left ventricular noncompaction or Barth syndrome. Circulation 103:1256–1263
Hata Y, Kinoshita K, Mizumaki K, Yamaguchi Y, Hirono K, Ichida F, Takasaki A, Mori H, Nishida N (2016) Postmortem genetic analysis of sudden unexplained death syndrome under 50 years of age: a next-generation sequencing study. Heart Rhythm 13:1544–1551
Yamaguchi Y, Nishide K, Kato M, Hata Y, Mizumaki K, Kinoshita K, Nonobe Y, Tabata T, Sakamoto T, Kataoka N, Nakatani Y, Ichida F, Mori H, Fukurotani K, Inoue H, Nishida N (2014) Glycine/serine polymorphism at position 38 influences KCNE1 subunit’s modulatory actions on rapid and slow delayed rectifier K+ currents. Circ J 78:610–618
Maekawa K, Saito Y, Ozawa S, Adachi-Akahane S, Kawamoto M, Komamura K, Shimizu W, Ueno K, Kamakura S, Kamatani N, Kitakaze M, Sawada J (2005) Genetic polymorphisms and haplotypes of the human cardiac sodium channel alpha subunit gene (SCN5A) in Japanese and their association with arrhythmia. Ann Hum Genet 69:413–428
Lee YS, Olaopa MA, Jung BC, Lee SH, Shin DG, Park HS, Cho Y, Han SM, Lee MH, Kim YN (2016) Genetic variation of SCN5A in Korean patients with sick sinus syndrome. Korean Circ J 46:63–71
Hussein A, Karimianpour A, Collier P, Krasuski RA (2015) Isolated noncompaction of the left ventricle in adults. J Am Coll Cardiol 66:578–585
Ohno S, Omura M, Kawamura M, Kimura H, Itoh H, Makiyama T, Ushinohama H, Makita N, Horie M (2014) Exon 3 deletion of RYR2 encoding cardiac ryanodine receptor is associated with left ventricular non-compaction. Europace 16:1646–1654
Towbin JA, Lorts A, Jefferies JL (2015) Left ventricular non-compaction cardiomyopathy. Lancet 386(9995):813–825
Lolicato M, Bucchi A, Arrigoni C, Zucca S, Nardini M, Schroeder I, Simmons K, Aquila M, DiFrancesco D, Bolognesi M, Schwede F, Kashin D, Fishwick CW, Johnson AP, Thiel G, Moroni A (2014) Cyclic dinucleotides bind the C-linker of HCN4 to control channel cAMP responsiveness. Nat Chem Biol 10:457–462
DiFrancesco D (2010) The role of the funny current in pacemaker activity. Circ Res 106:434–446
Liao Z, Lockhead D, St Clair JR, Larson ED, Wilson CE, Proenza C (2012) Cellular context and multiple channel domains determine cAMP sensitivity of HCN4 channels: ligand-independent relief of autoinhibition in HCN4. J Gen Physiol 140:557–566
DiFrancesco D, Tortora P (1991) Direct activation of cardiac pacemaker channels by intracellular cyclic AMP. Nature 351:145–147
DiFrancesco D (1999) Dual allosteric modulation of pacemaker (f) channels by cAMP and voltage in rabbit SA node. J Physiol 515:367–376
Laish-Farkash A, Glikson M, Brass D, Marek-Yagel D, Pras E, Dascal N, Antzelevitch C, Nof E, Reznik H, Eldar M, Luria D (2010) A novel mutation in the HCN4 gene causes symptomatic sinus bradycardia in Moroccan Jews. J Cardiovasc Electrophysiol 21:1365–1372
Macri V, Mahida SN, Zhang ML, Sinner MF, Dolmatova EV, Tucker NR, McLellan M, Shea MA, Milan DJ, Lunetta KL, Benjamin EJ, Ellinor PT (2014) A novel trafficking-defective HCN4 mutation is associated with early-onset atrial fibrillation. Heart Rhythm 11:1055–1062
Nof E, Luria D, Brass D, Marek D, Lahat H, Reznik-Wolf H, Pras E, Dascal N, Eldar M, Glikson M (2007) Point mutation in the HCN4 cardiac ion channel pore affecting synthesis, trafficking, and functional expression is associated with familial asymptomatic sinus bradycardia. Circulation 116:463–470
Baruscotti M, Barbuti A, Bucchi A (2010) The cardiac pacemaker current. J Mol Cell Cardiol 48:55–64
Ferlini A, Neri M, Gualandi F (2013) The medical genetics of dystrophinopathies: molecular genetic diagnosis and its impact on clinical practice. Neuromuscul Disord 23:4–14
Arbustini E, Weidemann F, Hall JL (2014) Left ventricular noncompaction: a distinct cardiomyopathy or a trait shared by different cardiac diseases? J Am Coll Cardiol 64:1840–1850
Kimura K, Morita H, Daimon M, Kawata T, Nakao T, Lee SL, Hirokawa M, Ebihara A, Nakajima T, Ozawa T, Yonemochi Y, Aida I, Motoyoshi Y, Mikata T, Uchida I, Komori T, Kitao R, Nagata T, Takeda S, Komaki H, Segawa K, Takenaka K, Komuro I (2015) Prognostic impact of venous thromboembolism in patients with Duchenne muscular dystrophy: prospective multicenter 5-year cohort study. Int J Cardiol 191:178–180
Parent JJ, Moore RA, Taylor MD, Towbin JA, Jefferies JL (2015) Left ventricular noncompaction cardiomyopathy in Duchenne muscular dystrophy carriers. J Cardiol Cases 11:7–9
Meinke P, Nguyen TD, Wehnert MS (2011) The LINC complex and human disease. Biochem Soc Trans 39:1693–1697
Yuan J, Xue B (2015) Role of structural flexibility in the evolution of emerin. J Theor Biol 385:102–111
Parmar MS, Parmar KS (2012) Emery-Dreifuss humeroperoneal muscular dystrophy: cardiac manifestations. Can J Cardiol 28(4):516.e1–516.e3
Monserrat L, Hermida-Prieto M, Fernandez X, Rodríguez I, Dumont C, Cazón L, Cuesta MG, Gonzalez-Juanatey C, Peteiro J, Álvarez N, Penas-Lado M, Castro-Beiras A (2007) Mutation in the alpha-cardiac actin gene associated with apical hypertrophic cardiomyopathy, left ventricular non-compaction, and septal defects. Eur Heart J 28:1953–1961
Girolami F, Iascone M, Tomberli B, Bardi S, Benelli M, Marseglia G, Pescucci C, Pezzoli L, Sana ME, Basso C, Marziliano N, Merlini PA, Fornaro A, Cecchi F, Torricelli F, Olivotto I (2014) Novel alpha-actinin 2 variant associated with familial hypertrophic cardiomyopathy and juvenile atrial arrhythmias: a massively parallel sequencing study. Circ Cardiovasc Genet 7:741–750
Stöllberger C, Finsterer J, Blazek G, Bittner RE (1996) Left ventricular non-compaction in a patient with Becker’s muscular dystrophy. Heart 76:380
Williams T, Machann W, Kuhler L, Hamm H, Muller-Hocker J, Zimmer M, Ertl G, Ritter O, Beer M, Schonberger J (2011) Novel desmoplakin mutation: juvenile biventricular cardiomyopathy with left ventricular non-compaction and acantholytic palmoplantar keratoderma. Clin Res Cardiol 100:1087–1093
Blinder JJ, Martinez HR, Craigen WJ, Belmont J, Pignatelli RH, Jefferies JL (2011) Noncompaction of the left ventricular myocardium in a boy with a novel chromosome 8p23.1 deletion. Am J Med Genet A 155A:2215–2220
Vatta M, Mohapatra B, Jimenez S, Sanchez X, Faulkner G, Perles Z, Sinagra G, Lin JH, Vu TM, Zhou Q, Bowles KR, Di Lenarda A, Schimmenti L, Fox M, Chrisco MA, Murphy RT, McKenna W, Elliott P, Bowles NE, Chen J, Valle G, Towbin JA (2003) Mutations in Cypher/ZASP in patients with dilated cardiomyopathy and left ventricular non-compaction. J Am Coll Cardiol 42:2014–2027
Liu Z, Shan H, Huang J, Li N, Hou C, Pu J (2016) A novel lamin A/C gene missense mutation (445 V > E) in immunoglobulin-like fold associated with left ventricular non-compaction. Europace 18:617–822
Wessels MW, Herkert JC, Frohn-Mulder IM, Dalinghaus M, van den Wijngaard A, de Krijger RR, Michels M, de Coo IF, Hoedemaekers YM, Dooijes D (2015) Compound heterozygous or homozygous truncating MYBPC3 mutations cause lethal cardiomyopathy with features of noncompaction and septal defects. Eur J Hum Genet 23:922–928
Hoedemaekers YM, Caliskan K, Majoor-Krakauer D, van de Laar I, Michels M, Witsenburg M, ten Cate FJ, Simoons ML, Dooijes D (2007) Cardiac beta-myosin heavy chain defects in two families with non-compaction cardiomyopathy: linking non-compaction to hypertrophic, restrictive, and dilated cardiomyopathies. Eur Heart J 28:2732–2737
Ouyang P, Saarel E, Bai Y, Luo C, Lv Q, Xu Y, Wang F, Fan C, Younoszai A, Chen Q, Tu X, Wang QK (2011) A de novo mutation in NKX2.5 associated with atrial septal defects, ventricular noncompaction, syncope and sudden death. Clin Chim Acta 412:170–175
Hoedemaekers YM, Caliskan K, Michels M, Frohn-Mulder I, van der Smagt JJ, Phefferkorn JE, Wessels MW, ten Cate FJ, Sijbrands EJ, Dooijes D, Majoor-Krakauer DF (2010) The importance of genetic counseling, DNA diagnostics, and cardiologic family screening in left ventricular noncompaction cardiomyopathy. Circ Cardiovasc Genet 3:232–239
Shan L, Makita N, Xing Y, Watanabe S, Futatani T, Ye F, Saito K, Ibuki K, Watanabe K, Hirono K, Uese K, Ichida F, Miyawaki T, Origasa H, Bowles NE, Towbin JA (2008) SCN5A variants in Japanese patients with left ventricular noncompaction and arrhythmia. Mol Genet Metab 93:468–474
Ronvelia D, Greenwood J, Platt J, Hakim S, Zaragoza MV (2012) Intrafamilial variability for novel TAZ gene mutation: Barth syndrome with dilated cardiomyopathy and heart failure in an infant and left ventricular noncompaction in his great-uncle. Mol Genet Metab 107:428–432
Kapadia R, Choudhary P, Collins N, Celermajer D, Puranik R (2016) Left ventricular non-compaction in Holt–Oram syndrome. Heart Lung Circ 25:626–630
Luedde M, Ehlermann P, Weichenhan D, Will R, Zeller R, Rupp S, Müller A, Steen H, Ivandic BT, Ulmer HE, Kern M, Katus HA, Frey N (2010) Severe familial left ventricular non-compaction cardiomyopathy due to a novel troponin T (TNNT2) mutation. Cardiovasc Res 86:452–460
Chang B, Nishizawa T, Furutani M, Fujiki A, Tani M, Kawaguchi M, Ibuki K, Hirono K, Taneichi H, Uese K, Onuma Y, Bowles NE, Ichida F, Inoue H, Matsuoka R, Miyawaki T, Noncompaction Study C (2011) Identification of a novel TPM1 mutation in a family with left ventricular noncompaction and sudden death. Mol Genet Metab 102:200–206
Graham TP Jr, Jarmakani JM, Canent RV Jr, Morrow MN (1971) Left heart volume estimation in infancy and childhood. Reevaluation of methodology and normal values. Circulation 43:895–904
Acknowledgements
We thank the gene sample provider and her family for their kind cooperation and Kohki Nishide, M.Eng., Hiroyuki Takahashi, M.Eng., and Nozomi Hisajima, M.Eng. for their technical assistance. This study was partly supported by JSPS KAKENHI (Grant numbers JP24590852, JP15K08867 to YH; JP26430012 to TT), AMED (Grant number 15dk020710h0002) to TT, and Presidential Discretionary Funds, University of Toyama 2014 to NN.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None for any authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
380_2018_1116_MOESM1_ESM.eps
Supplementary material 1 Supplemental Fig. 1 Flow of the in silico analysis. The variations identified in NGS were screened by the three-step procedure depicted in the flow chart (EPS 2989 kb)
380_2018_1116_MOESM2_ESM.eps
Supplementary material 2 Supplemental Fig. 2 Direct sequencing confirmed the mutations identified using NGS. Partial DNA sequences of the labeled genes from the proband’s family. The corresponding amino acids are shown above each codon. Integer denotes the position of the first nucleotide or amino acid of each partial sequence (EPS 5142 kb)
380_2018_1116_MOESM3_ESM.eps
Supplementary material 3 Supplemental Fig. 3 The L1988R mutation did not change the density or kinetics of SCN5A channel current. a Representative current responses of cells expressing wild-type SCN5A channel or SCN5A(L1988R) channel (WT and LR, respectively) to the voltage stimuli schematically shown above. Note a similarity in the rising and decaying time-courses between the WT and mutant channel currents. Whole-cell recordings were made as described in Kinoshita et al. (Heart Rhythm 13:1113–20, 2016). b Mean peak current density–voltage plots of the channel currents. n, 20 cells for SCN5A(WT) and 23 cells for SCN5A(L1988R). c Mean whole-cell conductance mediated by the channels estimated from the slope of the line fitted to the current density–voltage relation for each cell. d Relative g–V plots of the channel currents. The data were taken from the same cells as in b (EPS 2594 kb)
Rights and permissions
About this article

Cite this article
Yokoyama, R., Kinoshita, K., Hata, Y. et al. A mutant HCN4 channel in a family with bradycardia, left bundle branch block, and left ventricular noncompaction. Heart Vessels 33, 802–819 (2018). https://doi.org/10.1007/s00380-018-1116-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00380-018-1116-6