
Abstract
The African clawed frog, Xenopus laevis, is able to withstand extremely arid conditions by estivating, in conjunction with dehydration tolerance and urea accumulation. Estivating X. laevis reduce their metabolic rate and recruit anaerobic glycolysis, driven by lactate dehydrogenase (LDH; E.C. 1.1.1.27) enzymes that reversibly convert pyruvate and NADH to lactate and NAD+, to meet newly established ATP demands. The present study investigated purified LDH from the liver of dehydrated and control X. laevis. LDH from dehydrated liver showed a significantly higher K m for l-lactate (1.74 fold), NAD+ (2.41 fold), and pyruvate (1.78 fold) in comparison to LDH from the liver of control frogs. In the presence of physiological levels of urea found in dehydrated animals, the K m values obtained for dehydrated LDH all returned to control LDH K m values. Dot blot analysis showed post-translational modifications may be responsible for the kinetic modification as the dehydrated form of LDH showed more phosphorylated serine residues (1.54 fold), less methylated lysine residues (0.43 fold), and a higher level of ubiquitination (1.90 fold) in comparison to control LDH. The physiological consequence of dehydration-induced LDH modification appears to adjust LDH function in conjunction with urea levels in dehydrated frogs. When urea levels are high during dehydration, LDH retains its normal function. Yet, as urea levels drop during rehydration, LDH function is reduced, possibly shunting pyruvate to the TCA cycle.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The African clawed frog, Xenopus laevis, is a primarily aquatic species of frogs native to South Africa that can survive extreme arid conditions. During seasonally arid periods, the bodies of water in which X. laevis reside can dry up, prompting the frogs to migrate to a new body of water or burrow into the mud and enter a dormant state, termed estivation (Tinsley and Kobel 1996). Estivation is commonly associated with extreme metabolic rate depression (Hillman et al. 2009). During migration and estivation, X. laevis experience dehydration and can tolerate up to 35 % of total body water loss (Romspert 1976). As a consequence of dehydration, anurans often experience hyper-salinity that can, in turn, help hinder further water loss (Malik and Storey 2009; Hillman et al. 2009). One such common osmolyte that increases in X. laevis is urea; plasma urea concentrations increase by 15–30-fold while liver urea concentrations increase by ~20-fold (Balinsky et al. 1967; Malik and Storey 2009). In addition, dehydrated X. laevis experience an increase in blood viscosity and a decrease in blood volume, both of which can impair oxygen transport and create hypoxia stress (Hillman 1978a; Hillman et al. 2009). As such, estivating X. laevis may shift from aerobic to anaerobic metabolism to meet newly established energy demands.
Under hypoxic conditions, metabolic ATP production shifts from primarily oxidative phosphorylation to glycolysis. Pyruvate, generated during glycolysis, is converted to lactate under anaerobic conditions to regenerate the NAD+ needed to sustain glycolysis. Lactate dehydrogenase (LDH; E.C. 1.1.1.27) is responsible for catalyzing the reversible reaction of pyruvate and NADH to lactate and NAD+. X. laevis can estivate for months at a time waiting for favorable environmental conditions to return and would likely need to regulate the build-up of acidic glycolytic end-products such as lactate (Storey and Storey 2004b). Post-translational modifications, such as reversible protein phosphorylation, have been shown to play important roles in regulating key proteins in metabolic pathways in organisms that experience periods of dormancy (Storey and Storey 2004a; Malik and Storey 2009). In fact, post-translational modification of LDH has been reported in previous studies examining dormancy and metabolic rate depression in animals such as the freeze-tolerant North American wood frog (Rana sylvatica) and the anoxia tolerant turtle (Trachemys scripta elegans) (Dawson et al. 2013; Abboud and Storey 2013).
The purpose of this study is to understand the regulation of a key anaerobic metabolic enzyme, lactate dehydrogenase, under dehydration stress in a model organism, X. laevis. Here we report on the purification of LDH from the liver, a central organ involved in metabolism, of control and dehydrated X. laevis and investigate the differences in kinetics, post-translational modifications, and stabilities of LDH from the two conditions. In addition, we examine the effect of urea and protein crowding on the activity/function of LDH from control and dehydrated animals.
Materials and methods
Chemicals and animals
All biochemicals were from BioShop (Burlington, ON, Canada) with a few exceptions; sodium pyruvate was from Sigma (St. Louis, MO), l(+) lactic acid was from Polysciences Inc. (Warrington, PA, USA), potassium phosphate, monobasic was from J.T. Baker Chemical Company (London, UK) and 40× SYPRO Orange dye was from Invitrogen (Carlsbad, CA, USA).
Adult male X. laevis were obtained from the University of Toronto Xenopus colony (Toronto, ON, Canada) and weighed 38.3 ± 1.5 g. Frogs were housed in buckets containing dechlorinated water at room temperature (~20 °C). Animals were acclimated for at least a week prior to the initiation of dehydration experiments. Control animals were sampled from this group. For dehydration exposure, frogs were placed in closed plastic buckets at room temperature; animals were weighed at 12 h intervals until ~30 % of their total body water had been lost. The total body water lost was calculated from the change in mass of the frogs over time:
where, M i is the initial mass of the frog, M d is the mass at any given weighing during the experimental dehydration, and BWCi is the initial body water content of the frog before dehydration, which was experimentally determined to be 0.741 ± 0.019 g H2O per gram body mass (Malik and Storey 2009). The mean percent body water lost from the dehydrated frogs was 34.9 ± 1.6 % and occurred over a 3–4 day time period. It was assumed that no significant weight loss was due to tissue loss over this time period. Frogs from the control and experimental conditions were killed by pithing, and tissues rapidly excised and flash frozen in liquid nitrogen. All tissue samples were stored at −80 °C until use. The Carleton University Animal Care Committee, in accordance with the Canadian Council on Animal Care guidelines, approved all animal handling protocols and experiments performed in this study.
Preparation of tissue extracts
Frozen liver tissue samples were homogenized 1:10 in ice cold homogenization buffer [20 mM potassium phosphate (pH 7.2), 15 mM β-glycerophosphate, 1 mM EGTA, 1 mM EDTA, 15 mM β-mercaptoethanol] using a Polytron homogenizer (Brinkmann Instruments, Rexdale, ON, Canada). Immediately following the addition of ice-cold homogenization buffer to the tissue sample, a few crystals of phenylmethylsulfonyl fluoride (PMSF) were added, usually to 1–2 mL of homogenization solution, to prevent protease activity. Tissue homogenates were centrifuged at 10,000×g at 4 °C for 30 min, the supernatant collected and held at 4 °C until use.
Lactate dehydrogenase purification
A 1.6 mL sample of liver extract (1:10 in homogenization buffer) containing ~29 mg of total protein was applied to a DEAE+ column previously equilibrated with homogenization buffer. Following application of the crude extract, the column was washed with 30 mL of homogenization buffer and 3.31 mL fractions collected. From each fraction, a 1:20 dilution was prepared in homogenization buffer and 5 μL from each diluted fraction was assayed to detect LDH activity. The active fractions were pooled and applied to a Cibacron Blue 3GA column equilibrated with homogenization buffer. The Cibacron Blue 3GA column was washed with 30 mL of homogenization buffer to remove unbound protein and a linear gradient of 0–6 mM sodium pyruvate and NADH applied to elute bound protein. Fractions of 1.24 mL were collected and 5 μL from each fraction was assayed to detect LDH activity. The fractions containing peak activity were pooled, diluted 1:10, and applied to a second Cibacron Blue 3GA column in a similar fashion, although in this case, protein was eluted with a linear gradient of 0–2 M KCl in homogenization buffer. The fractions were assayed for LDH activity, and the active fractions pooled. Protein concentrations of all samples and fractions were measured using the Bio-Rad protein assay dye reagent (Bio-Rad, Hercules, CA, USA) with serial dilutions of bovine serum albumin as the standard according to the manufacturer’s instructions. The purity of the LDH sample was checked by denaturing and reducing gel electrophoresis followed by Coomassie blue staining as described below. LDH from the liver tissue of control and dehydrated frogs were purified in the same manner.
Lactate dehydrogenase enzyme assay
The activity of LDH was measured as the rate of production or consumption of NADH using a plate based assay by measuring absorbance at 340 nm using a Thermo Labsystems Multiskan spectrophotometer (Thermo Scientific, Waltham, MA, USA). Optimal assay conditions for LDH in the lactate-oxidizing direction were 100 mM potassium phosphate buffer pH 7.2, 60 mM l-lactate and 2 mM NAD+ in a total volume of 200 μL, with 5 μL of 1:1 (dehydrated) or 1:5 (control) diluted purified LDH per assay. The optimal conditions for LDH in the pyruvate-reducing direction were 100 mM potassium phosphate pH 7.2, 1 mM pyruvate, and 0.2 mM NADH in a total volume of 200 μL containing 5 μL of 1:1 (dehydrated) or 1:5 (control) diluted purified LDH. The K m and IC50 values for lactate, NAD+ or pyruvate were determined by holding the co-substrate constant at 2 mM NAD+, 60 mM lactate, or 0.2 mM NADH. In a second set of experiments, these assays were preformed in the same manner but in the presence of 150 mM urea, 1 % polyethylene glycol (PEG-8000), or a combination of 150 mM urea and 1 % PEG-8000. 1 % PEG-8000 was chosen for studies in molecular crowding as a low concentration of a high molecular weight PEG has been demonstrated to mimic molecular crowding in vitro. A higher percentage of PEG was avoided in our experiments as increasing percentages of PEG have been shown to decrease protein solubility in aqueous solutions (Atha and Ingham 1981). One unit of LDH activity in the pyruvate-reducing direction is defined as the amount of LDH that consumed 1 μmol of NADH per minute at 25 °C. Data were analyzed using the Kinetics v.3.5.1 program [developed by (Brooks 1992)].
Gel electrophoresis and Coomassie blue staining
Purified LDH fractions were separated on 10 % SDS-PAGE gels under reducing conditions by electrophoresis for 40 min at 185 V. Following gel electrophoresis, gels were immersed in Coomassie blue stain (0.025 % Coomassie blue powder, 33.3 % distilled water, 50 % methanol, 16.7 % glacial acetic acid) overnight at room temperature with gentle rocking. Coomassie blue stain was removed and the gel was de-stained with multiple exchanges of de-stain solution (33.3 % distilled water, 50 % methanol, 16.7 % glacial acetic acid) at room temperature with gentle rocking. Gels were visualized under light and images captured using a ChemiGenius Bio-Imaging system with GeneSnap software (Syngene, Frederick, MD, USA). The apparent molecular weight of X. laevis LDH was calculated by plotting Rf versus log MW of the standards in the protein ladder.
Dot blotting
Equal amounts of purified LDH from liver tissues of control and dehydrated animals were applied to nitrocellulose membranes (Bio-Rad, Hercules, CA, USA) using a Bio-Dot micro-filtration apparatus (Bio-Rad, Hercules, CA, USA) using a previously developed protocol (Dawson et al. 2013). Briefly, nitrocellulose membranes placed between the plates of the Bio-Dot microfiltration apparatus were pre-wet with tris-buffered saline (TBS; 100 mM tris, 1.4 M NaCl, pH 7.6). Sample was applied to the wells of the Bio-Dot microfiltration apparatus and allowed to flow through by gravity for 1 h. Membranes were washed twice with 200 μL of TBS using vacuum suction. The membrane was removed from the apparatus, washed three times for 5 min each in TBS containing 0.05 % Triton-X (TBST) before being blocked with 10 mL of warm 1 mg mL−1 PVA in TBST for 15 s. Membranes were washed three times with TBST for 5 min each before the primary antibody was applied: (1) rabbit anti-phosphoserine (Cat # 618100, Invitrogen, Carlsbad, CA, USA); (2) rabbit anti-phosphothreonine (Cat. # 718200, Invitrogen, Carlsbad, CA, USA); (3) rabbit anti-phosphotyrosine (Cat # 615800, Invitrogen, Carlsbad, CA, USA); (4) rabbit anti-pan-acetyl (CR)-R (Cat. # sc-8663-R, Santa Cruz Biotechnology, Santa Cruz, CA, USA); (5) rabbit anti-ubiquitin (ab19247, Abcam, Cambridge, UK); or (6) rabbit anti-methylated lysine (SPC-158F, StressMarq, Biosciences Inc., Victoria, BC, Canada). Primary antibodies were applied to membranes and allowed to incubate overnight at 4 °C with gentle rocking. Membranes were washed with TBST (3 × 5 min), and incubated with a 1:5,000 dilution of goat anti-rabbit IgG-peroxidase secondary antibody for 15 min at room temperature. Blots were washed with TBST (3 × 5 min) prior to chemiluminescence visualization on the ChemiGenius Bioimaging System (Syngene, Frederick, MD, USA). The dot intensities were quantified using the GeneTools software (Syngene, Frederick, MD, USA). To confirm equal protein loading of samples, Coomassie blue staining was performed on the membranes and used to standardize immunoblotting dot intensities. Protein-normalized dot intensities were then normalized to that of the signal intensities from the control LDH samples.
Differential scanning fluorimetry
Purified control and dehydrated LDH were concentrated 16× using 10 K Amicon Ultra-4 centrifugal filters (Merck Millipore Ltd., Tullagreen, Carrigtwohill, Co., Cork, IRL) by centrifugation for 10 min at 7,500×g. Differential scanning fluorimetry was used to assess protein unfolding as previously described (Niesen et al. 2007; Biggar et al. 2012). Briefly, 15 μL of concentrated sample, 2.5 μL of DSF buffer (100 mM potassium phosphate, pH 7, 150 mM NaCl, without or with 1.2 M urea) and 2.5 μL of 40× SYPRO Orange dye (Invitrogen, Carlsbad, CA, USA) were added to wells of a 96 well plate before being sealed with optical adhesive film (BioRad, Hercules, CA, USA). The thermocycling program run on the MyIQ2 qRT-PCR thermocycler (BioRad, Hercules, CA, USA) consisted of 1 °C increases in temperature every 30 s starting from an initial temperature of 15 °C, ending with 95 °C. Fluorescence of SYPRO Orange was measured every 5 s (excitation filter: 485 ± 30 nm, emission filter: 625 ± 30 nm) using the MyIQ2 software (version 3.0.6070, BioRad, Hercules, CA, USA). Data were analyzed using OriginPro 8.5 software (OriginLab Corporation, North Hampton, MA, USA) employing the Boltzmann distribution curve to yield the temperature at which half of the protein was unfolded (T m).
Statistics
Data for all kinetic parameters and dot blots were analyzed using the Student’s t test (two-tailed). Data for kinetic parameters in response to urea and PEG were analyzed using one-way analysis of variances (ANOVAs) followed by a Tukey post hoc test. DSF data were analyzed using a two-way ANOVA with a Tukey post hoc test. A probability of <0.05 was considered significant.
Results
Purification of LDH from the liver of control and dehydrated Xenopus laevis
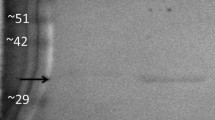
Purification of LDH from the liver of control and dehydrated X. laevis was accomplished by chromatography with a DEAE+ column (unbound fraction), a Cibacron blue affinity column (0–6 mM NADH and sodium pyruvate gradient), and finally a second Cibacron blue affinity column (0–2 M KCl gradient) (Table 1). Collection of the flow through fractions from the DEAE+ column resulted in 1.08-fold purification of recovered activity for the LDH from the liver of control (control LDH) and dehydrated (dehydrated LDH) frogs. Elution from the Cibacron blue column using a metabolite gradient gave 31.9-fold purification, while elution from the second Cibacron blue column using a KCl gradient resulted in a 26.4-fold purification (Table 1). The second Cibacron blue column using a KCl gradient was required to remove a final contaminating protein, resulting in a 10,820 mU mg−1 specific activity of LDH. As a result of this three-step purification scheme, LDH was purified to >95 % homogeneity, as determined by separation by gel electrophoresis and staining with Coomassie blue (Fig. 1). The purified LDH had an apparent molecular weight of 37.3 kDa (Fig. 1), consistent with the predicted molecular weight of 36.5 kDa for X. laevis (Genbank accession number NP_001080702).

Purified liver LDH from control Xenopus laevis. Coomassie-stained 10 % SDS-PAGE gel (reducing conditions) showing molecular weight ladder (left) and the purified liver control LDH enzyme (right)
Kinetic activity of purified LDH from control and dehydrated liver
Analysis of the kinetic activity of LDH from the liver of control and dehydrated animals showed distinct differences. For the forward reaction, the K m for pyruvate of LDH nearly doubled from the control to the dehydrated condition from 0.074 ± 0.004 to 0.132 ± 0.005 mM (Table 2, P < 0.001, Fig. 2c). Similarly, the K m for lactate and K m for NAD+ of dehydrated LDH enzyme were nearly double that of the K m values for the control LDH (Table 2, P = 0.01 and P = 0.001, and Fig. 2a, b, respectively).

Michaelis-Menten curves for forward and reverse reactions catalyzed by purified control and dehydrated LDH. Lactate (a), NAD+ (b), and pyruvate (c). Michaelis-Menten curves for control LDH (open circles) and dehydrated LDH (black circles). Plots are fitted with a three-parameter Hill coefficient curve using SigmaPlot 11. Data are mean ± SEM, n = 8 individual determinations on purified enzyme samples
Post-translational modifications of LDH from control and dehydrated frogs
Immunoblotting using a dot blot apparatus was used to assess the possible differences in the post-translational modifications of liver LDH from control and dehydrated conditions. Phosphorylation via serine residues of dehydrated LDH was 1.54 ± 0.04 fold higher compared to control LDH (1 ± 0.08, P < 0.001, Fig. 3a). The phosphorylation levels of threonine and tyrosine residues between control LDH and dehydrated LDH did not differ (P > 0.05, Fig. 3a). In addition to phosphorylation, additional post-translational modifications of control LDH and dehydrated LDH were examined and included: acetylation, methylation of lysine residues, and ubiquitination. Levels of methylated lysine for dehydrated LDH were 0.57 ± 0.09 fold that of control LDH levels (1 ± 0.09, P = 0.007, Fig. 3b). Conversely, ubiquitination of dehydrated LDH was 1.91 ± 0.17 fold higher in comparison to the level of ubiquitination of control LDH (1 ± 0.19, P = 0.007, Fig. 3b). The acetylation state of control LDH and dehydrated LDH did not differ (P > 0.05, Fig. 3b).

Quantification of post-translational modifications of purified control and dehydrated LDH. Fold change in phosphorylation (a) and other post-translational modification levels (b) of purified control LDH and dehydrated LDH are shown. Chemiluminescence signal intensities were normalized to protein amount, and the control LDH reference group set to an arbitrary value of 1. Data are mean ± SEM, n = 6 individual determinations on purified enzyme samples. Asterisks indicate significant differences from the corresponding control LDH levels, Student’s t test, two-tailed, P < 0.05
Influence of urea and polyethylene glycol on K m values for control and dehydrated LDH forward and reverse reactions
To assess whether urea and protein crowding would have an effect on the kinetic characteristics of control and dehydrated LDH, the K m values for lactate, NAD+ and pyruvate of control and dehydrated LDH were measured in the presence of (1) 150 mM urea to simulate the physiological concentration of built-up urea in a dehydrated frog, (2) 1 % PEG to simulate the crowding of enzymes in a dehydrated cell due to smaller cell volumes, or (3) a combination of 150 mM urea and 1 % PEG. While the presence of 150 mM urea did not have an effect on the K m values for lactate (Fig. 4a), NAD+ (Fig. 4c) or pyruvate (Fig. 4e) of control LDH (P > 0.05), the presence of 150 mM urea significantly reduced the K m for lactate (Fig. 4b), NAD+ (Fig. 4d) and pyruvate (Fig. 4f) of dehydrated LDH to 8.02 ± 0.51 mM (P = 0.006), 0.38 ± 0.02 mM (P < 0.001), and 0.061 ± 0.005 mM (P > 0.001), respectively. These values, when compared to that of control LDH K m values (7.72 ± 0.77, 0.37 ± 0.03, and 0.074 ± 0.005 mM, respectively), were not significantly different (P > 0.05).

Influence of urea and polyethylene glycol on the K m values of the forward and reverse reactions for control and dehydrated LDH. The K m of lactate for control LDH (a), K m of lactate for dehydrated LDH (b), K m of NAD+ for control LDH (c), K m of NAD+ for dehydrated LDH (d), K m of pyruvate for control LDH (e), and K m of pyruvate for dehydrated LDH (f) were determined for enzyme alone (white bars), enzyme with 150 mM urea (gray bars), enzyme with 1 % PEG (black bars), and enzyme with 150 mM urea and 1 % PEG (black and white striped bars). Data are mean ± SEM, n = 8 individual determinations on purified enzyme samples. Letters indicate statistical difference; bars with the same lettering are not statistically different from each other while those bars with different letters are statistically different from each other, one-way ANOVA with Tukey’s post hoc test, P < 0.05
The effect of 1 % PEG on the K m values varied between control and dehydrated LDH and depended on the kinetics being examined. For example, the K m for pyruvate for control LDH in the presence of PEG increased to 0.11 ± 0.02 mM compared to that of 0.074 ± 0.005 mM for control LDH alone (P = 0.013, Fig. 4e), whereas there were no differences between the K m for lactate (Fig. 4a) and K m for NAD+ (Fig. 4c) values for control LDH in the absence or presence of 1 % PEG (P > 0.05). For dehydrated LDH, the presence of 1 % PEG lowered the K m for lactate to 9.34 ± 0.80 mM (P = 0.049, Fig. 4b) and K m for pyruvate to 0.081 ± 0.004 mM (Fig. 4f) from 13.44 ± 1.70 to 0.133 ± 0.005 mM, respectively. The presence of 1 % PEG did not affect the K m for NAD+ (Fig. 4f, P > 0.05).
Similar to the addition of PEG, the addition of both urea and PEG had varying effects on the K m values depending on the condition the enzyme was purified from and the kinetic activity being measured. The combination of urea and PEG increased the K m for NAD+ of control LDH to 0.60 ± 0.09 mM from that of 0.37 ± 0.03 mM (P = 0.015, Fig. 4c), while the K m for lactate (Fig. 4a) and K m for pyruvate (Fig. 4e) were not significantly different from that of control LDH alone (P > 0.05). For dehydrated LDH, the combination of urea and PEG did not affect the K m for lactate compared to dehydrated LDH alone or dehydrated LDH with PEG, but was significantly higher than dehydrated LDH with urea (P = 0.04, Fig. 4b). The K m for NAD+ for dehydrated LDH in the presence of urea and PEG was significantly lower compared to dehydrated LDH (P = 0.014) and dehydrated LDH with PEG (P = 0.017), but not significantly different from that of dehydrated LDH with urea (P > 0.05) (Fig. 4d). Lastly, the K m for pyruvate of dehydrated LDH with urea and PEG was significantly lower than that of dehydrated LDH (P = 0.003), was significantly higher than that of dehydrated LDH with urea (P = 0.001), but was not significantly different from dehydrated LDH with PEG (P > 0.05, Fig. 4f).
Structural stability of LDH
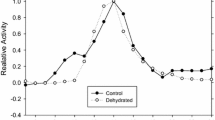
Differential scanning fluorimetry was used to assess the thermal stability of LDH purified from the liver of control and dehydrated frogs by measuring the T m values. The T m for control LDH was 68.8 ± 0.03 °C and was not significantly different than the T m of 68.9 ± 0.30 °C for dehydrated LDH (Fig. 5, P < 0.05, n = 4). In addition, since a physiological concentration of urea (150 mM in the cells of dehydrated frogs) had an effect on the K m values of the forward and reverse reaction, the effect of urea on the thermal stability of control and dehydrated LDH enzymes was examined. For control LDH in the presence of urea, the T m of 67.5 ± 0.10 °C was significantly lower than that of the T m for control LDH without urea (Fig. 5, P < 0.001, n = 4). Similarly, the T m of dehydrated LDH was significantly lowered to 66.8 ± 0.08 °C in the presence of urea (Fig. 5, P < 0.001, n = 4). The T m values for control LDH and dehydrated LDH both in the presence of urea, however, were not significantly different (P > 0.05).

Thermal stability of control and dehydrated LDH in the absence and presence of urea as measured by differential scanning fluorimetry. The melting curves and graphical representation of mean T m values (inset) of control LDH (a) and dehydrated LDH (b) examined in the absence and presence of 150 mM urea are shown. Data are mean ± SEM, n = 4 individual determinations on purified enzyme samples. Asterisks indicate statistical significance compared to the corresponding enzyme in the absence of urea, two-way ANOVA with Tukey’s post hoc test, P < 0.05
Discussion
Various species of frogs have well-developed tolerances for dehydration. X. laevis undergo estivation that is frequently characterized by a substantial loss of body water (as much as 35 %) as the soil loses water (Romspert 1976). Studies focused on long-term estivation in X. laevis show a reduction in oxygen consumption and suppression of the activities of various metabolic enzymes (Merkle 1989; Merkle and Hanke 1988; Onishi et al. 2005). Metabolic adaptations supporting such facultative anaerobiosis include: substantial reserves of fermentable substrate (glycogen), particularly in liver, along with substantial changes in the activities of glycolytic enzymes in all organs and metabolic rate depression. Anaerobic glycolysis, the conversion of hexose phosphates to lactate, helps support the ATP requirements of the estivating frog. As a result, there is a requirement for strict regulatory control over glycolytic rate, likely critical for the maintenance of homeostasis during long-term estivation. Interestingly, lactate levels in a dehydrating frog have been shown to increase (fivefold) under dehydration conditions (Churchill and Storey 1994). This increase in lactate has been attributed to hypoxia induced by a decrease in blood-flow as a result of increased blood viscosity in dehydrated animals (Hillman 1978a; Hillman et al. 2009).
Lactate dehydrogenase has been studied in other aquatic animals including T.s. elegans (Xiong and Storey 2012) and R. sylvatica (Abboud and Storey 2013) in which significant changes in kinetic parameters were discovered between the control and stressed state. Most pertinent to this study, R. sylvatica LDH only demonstrated kinetic differences when animals experienced dehydration stress (Abboud and Storey 2013). Similarly, in this study, a nearly twofold increase (Table 2) in the Michaelis constant for all substrates (forward and reverse direction) was observed for liver LDH in X. laevis during dehydration, suggesting a broad reduction in the catalytic efficiency of LDH during dehydration stress.
The physiological implications of kinetic changes or changes in enzyme function are often hard to establish in an in vitro system in which an enzyme has been purified away from the osmolytes and proteins within a cellular environment. One must take into consideration the changing cellular environment in which an enzyme resides during the prolonged exposure of X. laevis to dehydration. Urea accumulation in the plasma, liver and muscle is one strategy used by estivating amphibians, including X. laevis, to minimize water loss (Janssens 1964; Wray and Wilkie 1995; Romspert 1976; Balinsky et al. 1961; Malik and Storey 2009; Hillman 1978b). To properly elucidate the physiological implications of the present findings, kinetic experiments were performed in the presence of physiological concentrations of urea found in the liver of estivating X. laevis. Dehydrated LDH kinetics reverted back to control values in all cases (Fig. 4) when measured in the presence of 150 mM urea. Urea treatments yielded no significant changes to control LDH, suggesting that this phenomenon is isolated to the dehydrated form of LDH (Fig. 4). There are examples from other animals in which LDH activity is interrelated with urea concentration. For example, elasmobranch fish possess a urea-requiring LDH with markedly increased enzymatic activity compared to non-elasmobranch fish (Yancey and Somero 1978). However, the kinetic activity of elasmobranch fish LDH in the presence of urea decreased to levels typically found in non-elasmobranch fish (Yancey and Somero 1978). Furthermore, there are a number of reports that document low concentrations of urea increasing enzyme activity, such as bovine LDH (Garza-Ramos et al. 1992), human aldose reductase (Burg et al. 1999) and human biliverdin-IXa reductase (Franklin et al. 2013). These studies demonstrate the spectrum of effects that low concentrations of urea have on enzyme activity. To ensure that the effects of urea on LDH from the liver of dehydrated X. laevis were not simply due to a protein-crowding effect of increased osmolytes, a 1 % PEG solution was employed as a control (Fig. 4). While treatment with PEG did result in changes in measured kinetic parameters, these changes varied, suggesting that the effects of urea on dehydrated LDH function are unique and not due to protein crowding.
Previous studies have shown that LDH is subject to reversible phosphorylation (Yasykova et al. 2000; Fan et al. 2011; Xiong and Storey 2012; Abboud and Storey 2013). For example, differential phosphorylation of liver and muscle LDH, along with a subsequent change in kinetic parameters, has been reported in turtles in response to hypoxia (Xiong and Storey 2012; Dawson et al. 2013). The data presented in this study indicate that X. laevis liver LDH is also a phosphoprotein, phosphorylated via serine residues (Fig. 3). Phophorylation of X. laevis liver LDH on serine residues is similar to the phospho-control of turtle LDH in which serine-phosphorylation was suggested to regulate LDH in response to hypoxia (Xiong and Storey 2012; Dawson et al. 2013). The change in the phosphorylation state of LDH during dehydration stress suggests that reversible phosphorylation of LDH may adjust enzyme function in response to changing conditions.
The addition of ubiquitin or methyl groups to frog LDH has unknown effects on enzyme function. Classically, ubiquitination of cellular proteins leads to proteasome-mediated degradation. In addition, previous studies have shown that LDH ubiquitination may be sensitive to oxidative stress, with increased hydrogen peroxide-derived free radicals leading to enhanced ubiquitination and degradation (Onishi et al. 2005). Protein methylation has demonstrated regulatory properties in protein–protein interaction (Bedford and Richard 2005; Martin and Zhang 2005; Huang and Berger 2008), however, there is also speculation as to the role of both protein methylation and ubiquitination in protein stabilization (Bedford and Richard 2005; Varadan et al. 2002; Clarke 1993). In the present study, similar T m values for control and dehydrated LDH were observed despite differences in their methylation and ubiquitination levels, suggesting that these post-translational modifications do not affect liver LDH thermal stability.
The results from this study suggest that the stress form of LDH in X. laevis liver is being modified, relative to control LDH, in a manner that allows for dehydrated LDH to function in the presence of urea at a similar level to that of control LDH. However, in the absence of urea, dehydrated LDH has reduced activity. It is interesting to note that the kinetics of control LDH enzyme were not affected by urea, suggesting the modifications to dehydrated LDH are not a mechanism to maintain LDH activity during dehydration. Rather, our observations suggest that the modifications to LDH during dehydration may, in fact, be a regulatory mechanism in preparation for recovery from stress, marked by the frog re-entering an aquatic habitat, rapidly excreting urea and rehydrating (Balinsky et al. 1961; Jorgensen 1997). Previous studies examining other enzymes, such as antioxidant enzymes, from a number of animal model systems have brought light to an emerging, re-occurring, theme suggestive of protein modification and regulation as a preparatory mechanism for return to a normal post-stress homeostatic state (Hermes-Lima et al. 1998; Storey and Storey 2004c). For example, in R. sylvatica the activity of glutathione peroxidase, an antioxidant enzyme, was increased by ~2 fold in heart, kidney and muscle of frozen frogs, yet returned to basal levels in recovered (thawed) animals (Joanisse and Storey 1996). In these studies, it has been proposed that the increase in anti-oxidant enzyme activity is triggered during the hypoxic stressed state in preparation for an increase in oxygen consumptions and subsequent production of oxidants. In a similar fashion, we hypothesize that the post-translational modification of LDH in dehydrated animals acts as a preparatory mechanism to reduce conversion of pyruvate to lactate in the absence of urea. During exit from estivation and recovery from dehydration, urea is rapidly excreted, resulting in a decrease in intracellular urea concentration. In the absence of urea, LDH from the dehydrated liver of X. laevis was found to be less active, which would suggest the bulk of pyruvate formed via glycolysis may be shunted to the TCA cycle, favouring a return to oxidative phosphorylation. Furthermore, the ubiquitination of dehydrated LDH may enhance proteasome-dependent degradation of the less active form of LDH in recovering animals, as proteasome activity appears to be suppressed in other estivating or hibernating organisms (Ramnanan et al. 2009; Woods and Storey 2005). Thus, ubiquitinated LDH in estivating frogs may be subject to delayed-turnover upon exit from estivation and further facilitate a shift from glycolysis to oxidative phosphorylation. Together, the urea dependence of LDH kinetics and regulation of LDH degradation may be key mechanisms in facilitating aerobic metabolism in rehydrating frogs. The potential facilitation of a return to aerobic metabolism by LDH as outlined in this study is speculative, but could be clarified by future experiments that generate experimental groups of frogs at multiple time points over the course of recovery (rehydration). Our current animal protocols and resources do not allow for us to conduct this additional study, but examining the interactions of enzyme activity, posttranslational modifications and urea effects will be a focus of our lab in the future.
The kinetic alterations identified in this study suggest that liver LDH from dehydrated and control X. laevis exists in two distinct forms. While the exact natures of the aforementioned posttranslational modifications are not explicitly known, it seems as though post-translational modifications of liver LDH during dehydration results in a global reduction in LDH function. This reduction in function seems to be countered by a natural increase in urea levels during dehydration. Together, this suggests that LDH function may play a role in the maintenance of glycolytic flux during dehydration when urea levels are high, while acting as a metabolic shunt, directing the flow of pyruvate toward oxidative phosphorylation when urea levels are low. This may effect the survival of X. laevis upon rehydration as no net energy would be required to induce a metabolic shift to aerobic metabolism.
Abbreviations
- ANOVA:
-
Analysis of variance
- ATP:
-
Adenosine triphosphate
- BWCi :
-
Initial body water content
- DEAE+ :
-
Diethylaminoethanol
- DSF:
-
Differential scanning fluorimetry
- EDTA:
-
Ethylene diamine tetraacetic acid
- EGTA:
-
Ethylene glycol tetraacetic acid
- IgG:
-
Immunoglobulin G
- K m :
-
Michaelis constant
- LDH:
-
Lactate dehydrogenase
- M d :
-
Measured mass
- M i :
-
Initial mass
- MW:
-
Molecular weight
- NAD+ :
-
Nicotinamide adenine dinucleotide
- NADH:
-
Reduced form of nicotinamide adenine dinucleotide
- PEG:
-
Polyethylene glycol
- PMSF:
-
Phenylmethanesulfonyl fluoride
- PVA:
-
Polyvinyl alcohol
- Rf:
-
Distance migrated over the gel length
- SDS-PAGE:
-
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- SEM:
-
Standard error of the mean
- TBS:
-
Tris-buffered saline
- TBST:
-
Tris-buffered saline with 0.05 % Tween-20
- TCA cycle:
-
Tricarboxylic acid cycle
- T m :
-
Melting point
- V max :
-
Maximum initial velocity of the enzyme catalyzed reaction
References
Abboud J, Storey KB (2013) Novel control of lactate dehydrogenase from the freeze tolerant wood frog: role of posttranslational modifications. PeerJ 1:e12. doi:10.7717/peerj.12
Atha DH, Ingham KC (1981) Mechanism of precipitation of proteins by polyethylene glycols. Analysis in terms of excluded volume. J Biol Chem 256(23):12108–12117
Balinsky JB, Cragg MM, Baldwin E (1961) The adaptation of amphibian waste nitrogen excretion to dehydration. Comp Biochem Phys 3:236–244
Balinsky JB, Choritz EL, Coe CG, van der Schans GS (1967) Amino acid metabolism and urea synthesis in naturally aestivating Xenopus laevis. Comp Biochem Phys 22(1):59–68
Bedford MT, Richard S (2005) Arginine methylation an emerging regulator of protein function. Mol Cell 18(3):263–272
Biggar KK, Dawson NJ, Storey KB (2012) Real-time protein unfolding: a method for determining the kinetics of native protein denaturation using a quantitative real-time thermocycler. Biotechniques 53(4):231–238
Brooks SP (1992) A simple computer program with statistical tests for the analysis of enzyme kinetics. Biotechniques 13(6):906–911
Burg MB, Peters EM, Bohren KM, Gabbay KH (1999) Factors affecting counteraction by methylamines of urea effects on aldose reductase. Proc Natl Acad Sci USA 96(11):6517–6522
Churchill TA, Storey KB (1994) Effects of dehydration on organ metabolism in the frog Pseudacris crucifer––hyperglycemic responses to dehydration mimic freezing-induced cryoprotectant production. J Comp Phys B 164(6):492–498
Clarke S (1993) Protein methylation. Curr Opin Cell Biol 5(6):977–983
Dawson NJ, Bell RA, Storey KB (2013) Purification and properties of white muscle lactate dehydrogenase from the anoxia-tolerant turtle, the red-eared slider, Trachemys scripta elegans. Enzyme Res 2013:784973
Fan J, Hitosugi T, Chung TW, Xie J, Ge Q, Gu TL, Polakiewicz RD, Chen GZ, Boggon TJ, Lonial S, Khuri FR, Kang S, Chen J (2011) Tyrosine phosphorylation of lactate dehydrogenase A is important for NADH/NAD(+) redox homeostasis in cancer cells. Mol Cell Biol 31(24):4938–4950
Franklin E, Mantle T, Dunne A (2013) Activation of human biliverdin-IX alpha reductase by urea: generation of kinetically distinct forms during the unfolding pathway. Biochim Biophys Acta 1834(12):2573–2578
Garza-Ramos G, Fernandez-Velasco DA, Ramirez L, Shoshani L, Darszon A, Tuena de Gomez-Puyou M, Gomez-Puyou A (1992) Enzyme activation by denaturants in organic solvent systems with a low water content. Eur J Biochem 205(2):509–517
Hermes-Lima M, Storey JM, Storey KB (1998) Antioxidant defenses and metabolic depression. The hypothesis of preparation for oxidative stress in land snails. Comp Biochem Physiol B 120(3):437–448
Hillman SS (1978a) Roles of oxygen delivery and electrolyte levels in dehydrational death of Xenopus laevis. J Comp Physiol 128(2):169–175
Hillman SS (1978b) Some effects of dehydration on internal distribution of water and solutes in Xenopus laevis. Comp Biochem Physiol A 61:303–307
Hillman SS, Withers PC, Drewes RC, Hillyard SD (2009) Ecological and environmental physiology of amphibians. Oxford University Press, Oxford
Huang J, Berger SL (2008) The emerging field of dynamic lysine methylation of non-histone proteins. Curr Opin Genet Dev 18(2):152–158
Janssens PA (1964) Urea production + transaminase activity in Xenopus laevis Daudin. Comp Biochem Phys 13(3):217
Joanisse DR, Storey KB (1996) Oxidative damage and antioxidants in Rana sylvatica the freeze-tolerant wood frog. Am J Physiol Reg I 271(3):R545–R553
Jorgensen CB (1997) Urea and amphibian water economy. Comp Biochem Physiol A 117(2):161–170
Malik AI, Storey KB (2009) Activation of extracellular signal-regulated kinases during dehydration in the African clawed frog, Xenopus laevis. J Exp Biol 212(Pt 16):2595–2603
Martin C, Zhang Y (2005) The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol 6(11):838–849
Merkle S (1989) Long-term starvation in Xenopus laevis Daudin-III. Effects on enzymes in several tissues. Comp Biochem Physiol B 94(4):783–788
Merkle S, Hanke W (1988) Long-term starvation in Xenopus laevis Daudin-II. Effects on several organs. Comp Biochem Physiol A 90(3):491–495
Niesen FH, Berglund H, Vedadi M (2007) The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat Protoc 2(9):2212–2221
Onishi Y, Hirasaka K, Ishihara I, Oarada M, Goto J, Ogawa T, Suzue N, Nakano S, Furochi H, Ishidoh K, Kishi K, Nikawa T (2005) Identification of mono-ubiquitinated LDH-A in skeletal muscle cells exposed to oxidative stress. Biochem Biophys Res Commun 336(3):799–806
Ramnanan CJ, Allan ME, Groom AG, Storey KB (2009) Regulation of global protein translation and protein degradation in aerobic dormancy. Mol Cell Biochem 323(1–2):9–20
Romspert AP (1976) Osmoregulation of African clawed frog, Xenopus laevis, in hypersaline media. Comp Biochem Phys A 54(2):207–210
Storey KB, Storey JM (2004a) Metabolic rate depression in animals: transcriptional and translational controls. Biol Rev Cam Philos Soc 79(1):207–233
Storey KB, Storey JM (2004b) Oxygen limitation and metabolic rate depression. In: Functional metabolism: regulation and adaptation. Wiley, New York, pp 415–442
Storey KB, Storey JM (2004c) Physiology, biochemistry and molecular biology of vertebrate freeze tolerance: the wood frog. In: Benson E, Fuller B, Lane N (eds) Life in the frozen state. CRC Press, Boca Raton, pp 243–274
Tinsley RC, Kobel HR (1996) The biology of Xenopus. Oxford University Press, Oxford
Varadan R, Walker O, Pickart C, Fushman D (2002) Structural properties of polyubiquitin chains in solution. J Mol Biol 324(4):637–647
Woods AK, Storey KB (2005) Effects of hibernation on multicatalytic proteinase complex in thirteen-lined ground squirrels, Spermophilus tridecemlineatus. Mol Cell Biochem 271(1–2):205–213
Wray S, Wilkie DR (1995) The relationship between plasma urea levels and some muscle trimethylamine levels in Xenopus laevis––a P-31 and N-14 nuclear-magnetic-resonance study. J Exp Biol 198(2):373–378
Xiong ZJ, Storey KB (2012) Regulation of liver lactate dehydrogenase by reversible phosphorylation in response to anoxia in a freshwater turtle. Comp Biochem Phys B 163(2):221–228
Yancey PH, Somero GN (1978) Urea-requiring lactate-dehydrogenases of marine Elasmobranch fishes. J Comp Physiol 125(2):135–141
Yasykova MY, Petukhov SP, Muronetz VI (2000) Phosphorylation of lactate dehydrogenase by protein kinases. Biochemistry (Mosc) 65(10):1192–1196
Acknowledgments
The authors thank JM Storey for helpful discussions during the preparation of this manuscript. This research was supported by a discovery grant from the Natural Sciences and Engineering Research Council (NSERC) of Canada (6793 to KBS). BAK was supported by a NSERC Post Doctoral Fellowship and NJD was supported by an Ontario Graduate Scholarship.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by I. D. Hume.
B. A. Katzenback and N. J. Dawson contributed equally.
Rights and permissions
About this article
Cite this article
Katzenback, B.A., Dawson, N.J. & Storey, K.B. Purification and characterization of a urea sensitive lactate dehydrogenase from the liver of the African clawed frog, Xenopus laevis . J Comp Physiol B 184, 601–611 (2014). https://doi.org/10.1007/s00360-014-0824-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00360-014-0824-1