
Abstract
Combustion reactions provide the principal energy source of the world yet they are creating a burden through pollution. A detailed understanding of the complicated chemistry of flames is essential for optimizing energy output while minimizing detrimental side effects. We present a measurement technique based on time domain, resonance-enhanced multiphoton ionization via Rydberg states followed by laser-induced plasma backscattering of microwaves and demonstrate this non-intrusive, in-situ technique by selectively identifying 2,3-dimethylbut-2-ene in a complex 2,3-dimethylbut-2-ane flame environment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Although low-carbon energy sources such as solar, wind, geothermal and nuclear energy are playing increasingly important roles, combustion maintains its status as the world’s principal energy source. Yet while producing massive amounts of energy, the combustion of fossil fuels generates large quantities of greenhouse gasses and pollutants that alter the atmosphere and contribute to climate change [1]. That, as well as the ubiquitous demand for increased efficiency, motivates research to better understand the chemical mechanisms of combustion [2].
The complex reaction mechanisms of combustion reactions call for advanced techniques to identify the chemical species that only exist transiently and at high temperatures. Researchers often apply intrusive extraction methods coupled with techniques such as Electron Spin Resonance (ESR) spectroscopy and Mass Spectrometry (MS) to probe the products of flame reactions [3,4,5]. The well-established MS approach utilizes photoionization and time-of-flight detection for molecular identification. Using the Advanced Light Source, researchers at Lawrence Berkeley National Laboratory have optimized the resolution and extended the range of applications by combining MS with tunable and bright vacuum-ultraviolet synchrotron light beams [6,7,8,9]. While the combination of MS with tunable ionization has enabled some isomeric discrimination and multiplexed species detection, the broad Franck–Condon envelopes associated with molecules at high temperatures limit the range of applicability. Additionally, the extraction of species from the flame region results in probe-induced perturbations.
Recent years have witnessed the development of non-intrusive laser spectroscopy techniques as diagnostic tools to characterize chemicals without the need for intrusive extraction that might interfere with the reaction intermediates or chemical products. Examples include laser spectroscopy techniques such as laser-induced incandescence, cavity ring down spectroscopy and Raman scattering [3, 10, 11]. While these methods are not intrusive, their applicability to flames remains limited because vibrational spectra are difficult to resolve for polyatomic molecules at high temperatures.
Photoionization via Rydberg states yields molecular structure sensitive fingerprint spectra even in large molecules, giving rise to a spectral identification by Rydberg fingerprint spectroscopy (RFS) [12,13,14,15,16]. The measurement gives the binding energy, which is the energy difference between an electron placed at a Rydberg state and being infinitely far away from the ion core. Figure 1 illustrates the Coulomb potential and a schematic wave function of an electron in a molecular Rydberg state. The molecular potential shares a similarity with a hydrogen atom at large distance yet is significantly different close to the ion core. This change in the potential energy landscape results in a phase shift of the molecular wave function compared with that of hydrogen atoms, which in turn results in a difference in the binding energy of the Rydberg electron. The phase shift depends on the number, the type and the geometrical arrangement of atoms in the ion core, as well as the distribution of charges. As a result, the Rydberg binding energy is sensitive to the molecule structure. A spectral measurement of the binding energy of a single Rydberg state, or the difference between binding energies of two different Rydberg states, therefore offers a spectroscopic way to identify molecular substances.

Rydberg state scheme. The binding energy (BE) of a Rydberg state depends sensitively on the shape and charge distribution of the ion core because the wavefunction probes the entire potential energy surface of the system. In comparison to hydrogen atoms (blue), the potential energy surface of molecular ion cores depends on the geometrical arrangement of atoms and electrons, leading to binding energies that are typically smaller than in hydrogen. Because the BE depends sensitively on the molecular structure, their measurement can identify molecular species
Electrons in Rydberg states, unlike valence electrons, do not significantly influence the chemical bonds in a molecule. Consequently, the Frank–Condon envelopes for transitions from the Rydberg state to the associated ion states fall within a narrow band, giving rise to well-structured spectra even when the molecules are hot. Additionally, the complexity of Rydberg spectroscopy does not increase with the molecular size, which is an advantage for large molecules. Recent decades witnessed the rapid development of RFS, including its capability to differentiate isomeric compounds [17, 18]. Additional studies have extended RFS to distinguish conformericforms of various molecules [19,20,21,22,23,24,25]. The sensitivity to core geometric structures and the applicability to large, hot molecules combine to make Rydberg spectroscopy an attractive tool to investigate flame chemistry.
Traditional approaches [26,27,28,29,30,31] of acquiring Rydberg spectra to study reactions are usually coupled with photoelectron spectroscopy or mass spectrometry. Yet for flame applications both those methods are problematic as they require the extraction of reaction intermediates from the flame. To address this issue, Rudakov et al. [32,33,34] utilized microwave backscattering from photoionization plasma to obtain the Rydberg spectra, following an approach for reading out resonance-enhanced multiphoton ionization (REMPI) that initially was proposed by Miles [35,36,37]. Electrons generated through REMPI via a Rydberg state create a weakly ionized and small-volume plasma. The unbounded electrons inside the plasma oscillate in phase and backscatter the microwave radiation [36, 37]. The intensity of the backscattered microwave radiation reveals the unbounded electrons that are generated in the multi-photon ionization via the Rydberg states and thus reflects the Rydberg-Rydberg transitions spectrum [Fig. 2].

Photoionization schemes. a The synchronous scheme has three beams overlapped in space and in time. The 234.5 nm pulse prepares the molecules in the 3s Rydberg state. Tunable IR promotes molecules from the 3s to higher Rydberg states, and 532 nm ionizes the molecules. b Time domain scheme: Introducing a ~ 10 ps time delay between the excitation pulse and the ionization pulse allows molecules to decay from the higher state back to the 3s Rydberg state, from where they are ionized by a 355 nm ionization pulse
To demonstrate the method and evaluate the benefit of the time-domain approach, we studied the combustion of 2,3-dimethylbut-2-ane, or short diisopropyl (DIP), a member of the C-6 isoalkanes that are of great significance to automotive gasoline combustion [38]. The previously described synchronous scheme, Fig. 2a employs multiple pulsed laser beams with different wavelengths that overlap in space and time. The target molecules are first excited by a 234.5 nm pulse to the 3s Rydberg state and then ionized by IR and 532 nm photons. Since the IR wavelength is tuned through the resonances associated with 3s → 3p Rydberg transitions, the ionization efficiency is strongly amplified when the resonance condition is met. This double resonance ensures a selective detection of a product in a flame because only those molecules that have both resonances contribute to the signal and because the probability that another molecule has exactly the same two resonances is small. To further increase the specificity, one could add more resonance steps, as there are many additional Rydberg states that could be employed for a multi-photon ionization scheme. This synchronous scheme has achieved great success in selectively identifying methyl radicals with sharp resonances in methane, propane and hexane fuel rich flames (with different wavelengths than those shown in Fig. 2), but failed to provide clear spectra of more complex reaction intermediates. The background coming from ionization of other flame components represents an important challenge. Propene, 2-methylbut-2-ene, CH4, HCHO and 2,3-dimethylbut-l-ene are the major components of DIP flames, with smaller amounts of 2,3-dimethylbut-2-ene, 3-methylbut-1-ene, but-2-ene and, most likely, a large number of other molecules [39]. Higher laser intensities are required in the synchronous scheme because the high-lying Rydberg states are short-lived. High intensities result in low specificity since many molecules may be off-resonantly ionized along with the target substance. Therefore, the transient species generated in the DIP flame cannot be accurately diagnosed by the synchronous method. Nevertheless, we apply this approach to measure spectra of pure 2,3-dimethylbut-2-ene (also known as tetramethylethylene, TME) as a reference. The time-domain scheme, Fig. 2b, introduces a time delay between the pump pulse and the ionization pulse. This scheme has previously been applied in the different context of remote sensing of N,N-dimethylisopropyl-amine (DMIPA) by Rudakov et al. [34]. It utilizes the fact that the lifetime of the 3s Rydberg state is dependent on the amount of internal vibrational energy deposited to the molecule. In our experiment, the target molecules are again excited to the 3s Rydberg state using a 234.5 nm pulse. A tunable IR pulse further promotes the molecule from the 3s state to higher lying Rydberg states. Internal conversion brings the molecules rapidly back to the 3s state by converting electronic energy into vibrational energy. This large amount of vibrational energy shortens the 3s lifetime [34, 40, 41] so that, when a 355 nm pulse ionizes the molecules after a 10-picosecond delay, only those molecules that were not lifted to the higher Rydberg states remain. This leads to a characteristic decrease in the signal when the IR laser is on resonance with a higher Rydberg state. The time-domain scheme requires lower laser pulse intensities since only long-lived electronic states are probed. The double resonance time-domain scheme is also specific to the molecular species as two resonance conditions must be met. Since the time-domain scheme requires a target molecule featuring a long-lived 3s state as well as a short-lived, higher excited state, this technique allows us to discriminate against those molecules with short excited state lifetimes.
2 Experimental setup
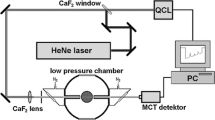
Our experimental setup consists of two OPGs pumped by a 10 Hz, 21 ps, 70 mJ Nd:YAG laser (Ekspla PL2251C) and a microwave homodyne transceiver system, Fig. 3. The harmonics generation produces three beams with wavelengths of 355 nm, 532 nm, and 1064 nm. The 1064 nm output is upconverted to produce 532 nm (synchronous scheme) or 355 nm (time-domain scheme) via LBO crystals. An optical parametric generator (OPG) is pumped by the 355 nm to generate 234.5 nm. The 532 nm-pumped OPG produces tunable radiation ranging from 1000 to 2400 nm. The three outputs are overlapped in space using a set of dichroic mirrors and focused into the target region by a lens (f = 250 mm). The laser power density at the beam waist is estimated as ~ 3·1010 W/cm2 for the OPG outputs and ~ 1·1012 W/cm2 and ~ 1·1011 W/cm2 for the 532 nm and 355 nm wavelengths, respectively. All laser pulse durations are about 10 ~ 15 ps.

Schematic diagram of the experimental setup. The Nd:YAG laser is upconverted to three wavelengths at 532 nm, 1064 nm, 355 nm. The 532 nm-pumped OPG1 produces tunable radiation ranging from 1000 ~ 2400 nm. The 1064 nm is upconverted using LBO crystals to 532 nm or 355 nm for the synchronous and time-domain schemes, respectively. The 355 nm-pumped OPG2 produces the 234.5 nm beam
The distance detection of the photoionization is obtained by Rayleigh microwave backscattering from the laser-induced plasma. The intensity of the microwave scattering signal is measured as the IR wavelength is tuned in the range from 1070 to 2400 nm with a step size of 10 nm. The microwave homodyne detection system has been discussed in a prior publication [33]. The output of an 11 GHz 20 dBm microwave source is split into two parts. One is used to illuminate the laser-induced plasma through a microwave horn while the other part is used for the homodyne mixer. The microwave horn also collects microwaves that are backscattered from the plasma. The received signals are amplified and mixed with the source, yielding a signal at the difference frequency that is amplified again and recorded by a digitizer.
The samples studied consisted of DIP for the flame and TME for taking a reference spectrum. The DIP combustion experiments were conducted in a flat, fuel-rich flame produced by a McKenna Burner (Holthuis & Associates). DIP was premixed with air to a 2:1 equivalence ratio. Room temperature measurements of TME, as a reference, were performed in a vacuum cell containing a droplet of TME, which has a vapor pressure of 131.56 mmHg at 26.13 °C [42].
3 Results and discussion
The spectrum of room-temperature vapor of TME measured with the synchronous scheme is compared to the time-domain measurement and the DIP flame spectrum in Fig. 4. The synchronous spectrum is inverted to make it comparable to the time-domain measurement, where a characteristic decrease in signal results from the resonance excitation with the IR light. The comparison of the spectra indicates that in each case we are observing the same molecular species.

Rydberg–Rydberg transition spectra probed by microwave backscattering from a laser-induced plasma. The synchronous scheme (Scheme 1) is applied to the pure TME vapor at room temperature, (a), and inverted to match the time-domain spectra (Scheme 2), (b). Panel (b) shows the spectra of cold TME (synchronous scheme), cold TME obtained with the time-domain scheme, and the spectrum of a DIP flame using the time-domain scheme. The similarity of the spectra indicates the presence of TME in the DIP flame
According to a prior study [43, 46, 47] the binding energy of TME in the 3s Rydberg state is 2.99 eV. The energy difference between the ionization potential (8.27 eV) [43] and the 3s Rydberg state excitation energy is therefore 5.28 eV, so that a single 532 nm (2.33 eV) photon cannot ionize the molecule from this state. The binding energy of the 3p Rydberg states can be estimated to be 1.86–2.23 eV [46, 47], so that a single 532 nm photon can ionize the molecule from that state. As the IR wavelength is tuned through the resonances associated with the 3s → 3p Rydberg transitions, the ionization efficiency, as well as the intensity of the backscattered microwaves, strongly increases at the resonances. Therefore, the 3s → 3p Rydberg transitions are observed as peaks in the spectrum with the synchronous method. The inverted spectrum displays the resonances as dips.
The time-domain scheme uses the fact that excited electronic state lifetimes are dependent on the internal vibrational energy [34, 40, 41]. Exciting TME using wavelengths between 215.9 nm and 232.4 nm results in 0.47 ~ 0.06 eV of energy to be deposited into vibrational motions upon internal conversion. According to Wu and his colleagues’ measurement [44], this shortens the lifetime of the TME 3s Rydberg state from about 30 ps into a range of 5.6 ps to 22 ps. In the present experiments, TME is excited with 234.5 nm photons which, extrapolating slightly beyond Wu’s data set, should result in a lifetime of about 28 ps. The promotion of an electron to higher Rydberg states with the tunable IR pulse, followed by internal conversion back to the 3s Rydberg state, deposits an additional 0.76–1.13 eV of energy. With this additional vibrational energy, the lifetime of the 3s Rydberg state is shortened to ~ 5 ps [44]. Since the TME binding energy of the 3s Rydberg state is 2.99 eV, the third, time-delayed pulse at 355 nm (3.49 eV) can ionize the molecule out of this state. We note that a single 355 nm photon cannot ionize the molecular species created by the decay [45] because their ionization energies are too high. Therefore, the ionization efficiency decreases when the tunable IR radiation meets the resonance condition, resulting in dips in the time-domain scheme spectrum. Additionally, dissociation of molecules excited to the higher Rydberg states on a picosecond timescale could also give rise to a dip.
As the spectra obtained with either scheme of Fig. 2 are caused by the same resonance transitions, they serve as selective and characteristic fingerprints of the molecular species. From the similarity of the spectra in Fig. 4, we conclude that all peaks originate from TME, and that we selectively observe TME in the DIP flame. The signal level is more than 10 times the detection limit as characterized by the noise of the experiment.
The identification of transient species in flames only requires a molecule-specific Rydberg fingerprint spectrum. An assignment of the spectra to spectroscopic transitions is not necessary. Nevertheless, it is interesting to speculate about the identity of the observed peaks. The observed spectrum as a function of the IR photon energy, Fig. 5, representing transitions from the initially excited 3s level to higher Rydberg states, can be fit to three Gaussians centered at 0.79, 0.90 and 1.10 eV. A photoelectron spectroscopy study by Rijkenberg et al. reports the energy difference between 3s and 3pz Rydberg states and between 3s and 3px Rydberg states of TME to be 0.77 eV and 0.83 eV, respectively [46]. Snyder et al. report, based on a magnetic circular dichroism measurement, the 3s to 3pz transition at 0.75 eV, and the transition to 3py at 1.10 eV [47]. Our measurement of the 0.79 eV transition concords well with the reported values for the 3s to 3pz transition of 0.75 and 0.77 eV, respectively, and the peak at 1.10 eV fits exactly with Snyder’s 3s to 3py transition. Our third peak, at 0.90 eV, fits less well with the literature reports, but might conceivably be the 3s to 3px transition. Given the sizeable internal energy of the TME molecules in the flame, which can lead to slight peak shifts in Rydberg-Rydberg transitions, we consider the agreement of our measurement with the literature value quite satisfactory.

Microwave scattering intensity as a function of IR photon energy. Three Gaussians are used to fit the data
In general, REMPI/microwave scattering experiments can be calibrated to allow for a quantitative measurement [48], once the cross sections are known from either calibrations or calculations. In addition, the time-domain scheme requires a lifetime measurement for quantification because the lifetimes of the Rydberg states depend on the temperature and thus varies with the position within the flame. While it would be attractive to measure the temperature together with the species identification, it would require a time-domain measurement at every spatial point.
4 Conclusions
Our measurements selectively identify 2,3-dimethylbut-2-ene as a transient reaction component in a 2,3-dimethylbut-2-ane flame. The combination of Rydberg fingerprint spectroscopy and microwave backscattering allows us to selectively detect the combustion transient in spite of the complicated chemical environment and the high temperature. The time-domain technique in particular delivers a high degree of discrimination against background signals, allowing for a selective detection of combustion transients in the complex flame situation. Future work will focus on enhancing the selectivity by introducing additional resonance steps to further reduce the possibility that different molecules accidentally share a resonance wavelength. Utilizing femtosecond laser pulses, one can take further advantage of the excited state lifetimes as a discriminator between excitation pathways.
References
S. Cheskis, A. Goldman, Prog. Energy Combust. Sci. 35(4), 365–382 (2009)
J.A. Miller, M.J. Pilling, J. Troe, Proc. Combust. Inst. 30(1), 43–88 (2005)
J. Kiefer, P. Ewart, Prog. Energy Combust. Sci. 37(5), 525–564 (2011)
T. Bierkandt, P. Hemberger, P. Oßwald, D. Krüger, M. Köhler, T. Kasper, Proc. Combust. Inst. 37(2), 1579–1587 (2019)
F.N. Egolfopoulos, N. Hansen, Y. Ju, K. Kohse-Höinghaus, C.K. Law, F. Qi, Prog. Energy Combust. Sci. 43, 36–67 (2014)
T.A. Cool, K. Nakajima, T.A. Mostefaoui, F. Qi, A. McIlroy, P.R. Westmoreland, M.E. Law, L. Poisson, D.S. Peterka, M. Ahmed, J. Chem. Phys. 119(16), 8356–8365 (2003)
C.A. Taatjes, N. Hansen, A. Mcllroy, J.A. Miller, J.P. Senosiain, S.J. Kilppenstein, F. Qi, L. Sheng, Y. Zhang, T.A. Cool, J. Wang, P.R. Westmoreland, M.E. Law, T. Kasper, K. Kohse-Höinghaus, Science 308, 1887–1889 (2005)
N. Hansen, S.J. Klippenstein, C.A. Taatjes, J.A. Miller, J. Wang, T.A. Cool, B. Yang, R. Yang, L. Wei, C. Huang, J. Wang, F. Qi, M.E. Law, P.R. Westmoreland, J. Phys. Chem. A 110(10), 3670–3678 (2006)
N. Hansen, T.A. Cool, P.R. Westmoreland, K. Kohse-Höinghaus, Prog. Energy Combust. Sci 35(2), 168–191 (2009)
C.S. Moreau, E. Therssen, X. Mercier, J.F. Pauwels, P. Desgroux, Appl. Phys. B 78(3), 485–492 (2004)
J.A. Sutton, J.F. Driscoll, Opt. Lett. 29(22), 2620–2622 (2004)
N. Kuthirummal, P.M. Weber, Chem. Phys. Lett. 378(5–6), 647–653 (2003)
J.L. Gosselin, P.M. Weber, J. Phys. Chem. A 109(22), 4899–4904 (2005)
N. Kuthirummal, P.M. Weber, J. Mol. Struct. 787(1–3), 163–166 (2006)
J.D. Cardoza, P.M. Weber, Resolved: electronic states underneath broad absorptions. J. Chem. Phys. 127(3), 036101 (2007)
H. Gudmundsdóttir, Y. Zhang, P.M. Weber, H. Jónsson, J. Chem. Phys. 139(19), 194102 (2013)
F. Rudakov, P.M. Weber, J. Chem. Phys. 136(13), 134303 (2012)
J.D. Cardoza, F.M. Rudakov, N. Hansen, P.M. Weber, J. Electron Spectrosc. Relat. Phenom. 165, 5–10 (2008)
M.P. Minitti, P.M. Weber, Phys. Rev. Lett. 98(25), 253004 (2007)
J.C. Bush, M.P. Minitti, P.M. Weber, J. Phys. Chem. A 114(42), 11078–11084 (2010)
S. Deb, B.A. Bayes, M.P. Minitti, P.M. Weber, J. Phys. Chem. A 115(10), 1804–1809 (2011)
X. Cheng, Y. Zhang, S. Deb, M.P. Minitti, Y. Gao, H. Jónsson, P.M. Weber, Chem. Sci. 5(11), 4394–4403 (2014)
Y. Zhang, S. Deb, H. Jónsson, P.M. Weber, J. Phys. Chem. Lett 8(16), 3740–3744 (2017)
S. Deb, X. Cheng, P.M. Weber, J. Phys. Chem. Lett. 4(16), 2780–2784 (2013)
X. Cheng, Y. Gao, F. Rudakov, P.M. Weber, Chem. Sci. 7(1), 619–627 (2016)
W. Cheng, N. Kuthirummal, J.L. Gosselin, T.I. Sølling, R. Weinkauf, P.W. Weber, J. Phys. Chem. A 109(9), 1920–1925 (2005)
J.D. Cardoza, F.M. Rudakov, P.M. Weber, J. Phys. Chem. A 112(43), 10736–10743 (2008)
F. Rudakov, P.M. Weber, Chem. Phys. Lett. 470(4–6), 187–190 (2009)
C.C. Pemberton, Y. Zhang, K. Saita, A. Kirrander, P.M. Weber, J. Phys. Chem. A 119(33), 8832–8845 (2015)
J.L. Gosselin, M.P. Minitti, F.M. Rudakov, T.I. Sølling, P.M. Weber, J. Phys. Chem. A 110(12), 4251–4255 (2006)
S. Deb, M.P. Minitti, P.M. Weber, J. Chem. Phys. 135(4), 044319 (2011)
F. Rudakov, Z. Zhang, Opt. Lett. 37(2), 145–147 (2012)
F. Rudakov, Y. Gao, X. Cheng, P.M. Weber, Combust. Flame 171, 162–167 (2016)
F. Rudakov, Y. Zhang, X. Cheng, P.M. Weber, Opt. Lett. 38(21), 4445–4448 (2013)
Z. Zhang, M.N. Shneider, R.B. Miles, Plasma Sources Sci. Technol. 30, 103001 (2021)
M.N. Shneider, R.B. Miles, J. Appl. Phys. 98(3), 033301 (2005)
Z. Zhang, M.N. Shneider, R.B. Miles, Phys. Rev. Lett. 98(26), 265005 (2007)
W.J. Pitz, G. Kukkadapu, K. Zhang, M. Mehl, S.W. Wagnon, C.K. Westbrook, Energy Gov. (2018) https://www.energy.gov/sites/default/files/2018/06/f52/acs013_pitz_2018_o.pdf. Accessed 30 July 2021
R.R. Baldwin, G.R. Drewery, R.W. Walker, J. Chem. Soc. Faraday Trans. 80(11), 3195–3207 (1984)
B.A. Jacobson, J.A. Guest, F.A. Novak, S.A. Rice, J. Chem. Phys. 87(1), 269–283 (1987)
S.K. Kulkarni, J.E. Kenny, J. Chem. Phys. 89(7), 4441–4443 (1988)
A. Baghdoyan, J. Malik, V. Fried, J. Chem. Eng. Data 16(1), 96–97 (1971)
P. Masclet, D. Grosjean, G. Mouvier, J. Dubois, J. Electron Spectrosc. Relat. Phenom. 2(3), 225–237 (1973)
G. Wu, A.E. Boguslavskiy, O. Schalk, M.S. Schuurman, A. Stolow, J. Chem. Phys. 135, 164309 (2011)
M. Sato, S. Adachi, T. Suzuki, J. Phys. Chem. A 120(27), 5099–5102 (2016)
R.A. Rijkenberg, W.J. Buma, C.A. van Walree, L.W. Jenneskens, J. Phys. Chem. A 106(21), 5249–5262 (2002)
P.A. Snyder, R.W.C. Hansen, E.M. Rowe, J. Phys. Chem. 100(45), 17756–17761 (1996)
Y. Wu, Z. Zhang, T.M. Ombrello, V.R. Katta, Appl. Phys. B 111(3), 391–397 (2013)
Acknowledgements
This project was funded by the U.S. Department of Energy, Office of Science, Basic Energy Sciences, under Award # DE-SC0017995 and DE-SC0020276 and the Army Research Office, Award # W911NF-19-1-0178.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest that are relevant to the content of this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Xu, X., Rudakov, F. & Weber, P.M. Spectroscopic identification of 2,3-dimethylbut-2-ene transients in 2,3-dimethylbut-2-ane flames. Appl. Phys. B 128, 84 (2022). https://doi.org/10.1007/s00340-022-07803-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00340-022-07803-0