
Abstract
An optical sensor was developed for simultaneous detection of sulfur dioxide (SO2) and sulfur trioxide (SO3) at elevated temperatures using a quantum cascade laser (QCL) spectrometer. The SO2/SO3 concentrations and the SO2 conversions were measured during the heterogeneous catalytic reaction with S-101 intermediate-temperature vanadium catalysts at pressures of 3–20 kPa and temperatures of 550–1000 K. The SO2 spectra in the ν3 band and the SO3 spectra in the ν3 + ν4 band near 7.16 µm at elevated temperatures were well resolved in the strongly overlapping spectra by measuring the SO2 and SO3 high-temperature individual absorption spectra that were extended from room-temperature spectra in the HITRAN database. A SO2/SO3 separation unit was designed to extract and measure the remaining SO2 after conversion at room temperature, with the results comparing well with the optically measured concentrations in the high-temperature catalytic reactions. The system was then used to investigate the pressure and temperature dependencies of the heterogeneous conversions of SO2. SO3 was first detected at temperatures higher than 590 K with the SO2 conversion increasing to 92% at 797 K and then decreasing to less than 3% at 1014 K. The measured conversions show a logarithmic growth with increasing pressures at each temperature. The difference between the measured heterogeneous conversion and the theoretical conversion calculated using a thermodynamic model was also analyzed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Sulfur dioxide (SO2) and sulfur trioxide (SO3), collectively known as SOx, are both major pollutants associated with emissions from combustion of sulfur-containing fuels which require significant air pollution treatment [1]. SO2 is produced in various processes, such as the burning of fossil fuels (coal and oil) [2,3,4], thermal decomposition of sulfates [5] and mineral and soil analyses [6]. SO3, also known as H2SO4 or sulfuric acid mist, is virtually nonexistent in nature and is always formed together with SO2 via homogeneous gas-phase reactions or heterogeneous reactions in the presence of catalysts at elevated temperatures [7,8,9]. SO2 and SO3 pose health hazards to humans and animals, particularly to respiratory systems and eyes due to their corrosivity and acidity. SO3 can also lead to low-temperature corrosion by increasing the flue gas dew point, which can damage equipment in coal-fired power plants. In addition, SO3 is the main component of acid rain, which can cause deforestation, damage aquatic life and corrode building materials and paints [10, 11]. Thus, simultaneous measurements of SO2 and SO3 in gas mixtures containing these sulfur oxides are often required for pollution control.
Many techniques have been developed to measure SO2 concentrations for industrial emission controls and occupational health. Electrochemical SO2 gas sensors [12] have been commonly used in industry due to their low cost but they have the disadvantages of slow response times, poor sensitivity, complex operation and continuous maintenance. In contrast to many conventional techniques, optical techniques offer the advantages of fast response times, high sensitivities and continuous monitoring. Optical techniques include tunable laser absorption spectroscopy (TLAS) [13,14,15,16], differential optical absorption spectroscopy (DOAS) [17,18,19,20,21], Laser-induced fluorescence (LIF) [22] and correlation spectroscopy (COSPEC) [23].
SO3 measurements are more challenging than SO2 measurements since the gaseous SO3 reacts with water vapor (H2O) to form gaseous sulfuric acid when the temperature drops below 500 ℃. In addition, SO3 is a liquid at room temperature and a solid at standard conditions. Thus, SO3 gas is difficult to measure directly and SO3 measurements are often based on measurements of vapor or liquid H2SO4 [24]. Current techniques for monitoring SO3 depend mainly on chemical sensors due to its high reactive nature, such as the controlled condensation method [25], the isopropanol absorption method [26, 27], the acid dew point meter [28] and the salt method [29]. However, real-time monitoring is difficult with chemical methods and the losses of SO3/H2SO4 during sampling often lead to errors between the measured concentrations and those in the target areas. In addition, the interference from SO2 at high concentrations is a key problem for chemical sensors, especially for all the isopropanol methods [26]. Optical sensors have also been developed to measure SO3 based on the absorption spectroscopy. Fourier transform infrared (FTIR) spectroscopy [30, 31] and ultraviolet absorption spectroscopy [32, 33] have both been used to measure SO3 concentrations. However, the absorption characteristics of SO3 are needed for the reference spectrum which is difficult due to the challenging problem of producing SO3 gas with known concentrations [26], so these methods may not provide reliable quantitative measurements. The spectroscopic structure of SO3 with a large number of transitions has a nearly continuous spectrum even at low pressures which overlaps significantly with the SO2 spectrum, so the individual SO3 spectral lines in gaseous mixtures cannot easily be identified with limited resolution. A high resolution FTIR apparatus is then not practical due to its large expense, large size and its susceptibility to vibrations. In addition, the SO2 and SO3 transitions are strongly overlapped for a long path-length and a wide wavelength scan range, which makes a FTIR with a limited resolution difficult to resolve the spectra of the two molecules and measure the gas properties accurately.
The use of laser absorption spectroscopy (LAS) for quantitative SO3 measurements has also been both theoretically [34, 35] and experimentally [36,37,38,39,40] investigated. A QCLs based spectrometer operated at 7–8 μm has been developed to measure SO2 and SO3 concentrations in the lab [36,37,38] and combustor exhaust streams [39]. However, drift in the zero-absorbance baseline resulting from equipment vibrations and pollutants limits the accuracy of SO3 absorbance measurements. Most importantly, the lack of the SO3 high-temperature spectroscopic parameters in spectral databases (e.g. HITRAN) prevented the authors from simultaneously obtaining the corrected baseline and the SO3 absorbance in the significantly overlapping spectra of the gaseous mixtures. The use of the 4 μm band SO3 absorption lines for in situ SO3 measurements has been investigated using a mid-infrared laser source based on difference frequency generation [40]. Despite less interference from H2O, CO2 and other combustion flue gases, the absorption in the ν1 + ν3 band at 4 μm is much weaker than the ν3 + ν4 band at 7 μm with a lower detection limit.
This paper describes simultaneous detection of SO2 and SO3 using a QCL sensor operated at 7.16 μm using absorption spectroscopy. SO3 was simultaneously produced and measured using heterogeneous catalytic oxidation of SO2 at various temperatures and pressures. High-temperature SO2 and SO3 absorbance were measured and a semi-empirical model was developed based on the HITRAN database and the high-T absorbance so that the SO2 and SO3 absorption spectra could be accurately resolved in the overlapping spectra of the gaseous mixtures in the measurements. In addition, a SO2/SO3 gas separation unit was designed to extract and measure the remaining SO2 at room temperature after the catalytic reaction to further validate the measurement accuracy from the heterogeneous conversions at elevated temperatures. The dependencies of the conversions on temperature and pressure were then investigated and compared with the theoretical results from a thermodynamic model.
2 Spectral window selection
Figure 1 shows the room-temperature (296 K) line strengths of SO2, SO3 and H2O, obtained from the HITRAN database [41] for wavelengths of 1–23 μm. The absorption line strengths of SO2 in the ν3 band and SO3 in the ν3 + ν4 band are both strongest in the 7.0–7.7 μm region compared with other combination-overtone bands in the 3.0–4.0 μm region. Although SO3 also has strong fundamental bands (ν2 and ν5) in the far-infrared region, access to these bands is difficult due to the limited availability of lasers and detectors operating at such long wavelengths. Thus, a QCL operating at 7.16 μm with a scan wavelength range from 1393 to 1399 cm−1 was selected to simultaneously measure SO2 and SO3.The H2O interference from both the ambient air (room-temperature) and the target gaseous mixtures (high-temperature) are the main problems in the SO2 and SO3 measurements. Figure 2a shows the simulated spectra of the three molecules at 600 K and 1 atm from 1300 to 1400 cm−1 with the laser tuning range indicated in the yellow square region. Although the SO3 has stronger absorption around 1398 cm−1 as shown in Fig. 2, the H2O interference is significant, especially with high H2O concentrations and a long path length exposed to the atmosphere. Thus, the spectral window between 1396.5 and 1397.4 cm−1 with no H2O interference was selected for simultaneous measurements of SO2 and SO3.

Absorption line strengths (HITRAN 2020) of the infrared SO2, SO3 and H2O spectrum at 296 K

taken from the HITRAN/HITEMP database
Simulated infrared spectra of SO2, SO3 and H2O calculated at T = 600 K and P = 1 atm with the select spectral window shown in the lower panel where the interference from the H2O in air also presented. The targeted spectral region is outlined in yellow. The spectral line parameters were
3 Theory of absorption spectroscopy
The transmission, τ(v), of monochromatic light at frequency, v, through a uniform absorbing gas medium is expressed by the Beer-Lambert law [42]:
where It and I0 are the incident and transmitted light intensities and α(ν) is the spectral absorbance which, in the vicinity of a single transition, is related to the thermophysical gas properties through:
where S(T) [cm−2 atm−1] is the transition linestrength. P [atm] is the gas pressure, χ is the mole fraction of the absorbing species, L [cm] is the light path length and φ(v) [cm−1] is the lineshape function.
The individual spectral lineshapes of SO2 and SO3, φ(v), are modeled here by the Voigt profile, a convolution of Lorentzian and Gaussian profiles accounting for collisional and Doppler broadening that is given by [43]:
where \(A = \sqrt {\ln 2/\pi } /\Delta v_{D}\), \(x = \sqrt {\ln 2} (v - v_{0} )/\Delta v_{D}\), \(y = \sqrt {\ln 2} \Delta v_{C} /\Delta v_{D}\), ν0 [cm−1] is the transition linecenter, and ∆νD [cm−1] is the Doppler half width at half maximum (HWHM), given by:
where M [g·mol−1] is the molecular weight of the absorbing species. The collisional width scales with the collision frequency of the absorbing molecule, A, and is modeled as the product of the pressure and the sum of the mole fraction weighted collisional broadening coefficients of each perturbing species, B [43]:
where γA−B (T) [cm−1 atm−1] is the transition-dependent collisional broadening coefficient at temperature T.
4 Experimental details
4.1 Experimental apparatus
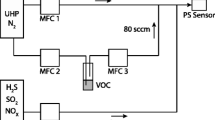
The schematic in Fig. 3 shows the experimental apparatus for the SOx measurements which consists of two gas cells, an inlet gas manifold for mixing the gases and an outlet line to the vacuum pump to facilitate the gas flow. A high temperature optical gas cell was used to simultaneously produce SO3 and measure the SO2/SO3 at temperatures from 550 to 1000 K and pressures from 3 to 20 kPa. The cell body was housed in a tube furnace (Kejia Furnace, KJ-200827 T) with two 20 cm BaF2 rods as optical windows that could withstand the thermal gradient in the furnace and allow optical access to the 11.43 cm long test section. The water cooling channels at both ends maintained the caps at low temperatures so that they could be sealed by fluoro-rubber O rings [44, 45]. The gas pressure within the cell was monitored by a pressure transducer (Inficon, CDG025D) for measurements below 1000 Torr with an accuracy of 0.25%. The cell temperature was monitored by multiple K-type thermocouples (Omega, TJ80-CAXL-116U) attached to the wall of the test region with an accuracy of 0.75%. The gas temperature was determined by the averaging the three values. At the highest test temperature of 1014 K, the average temperature uniformity is ± 5 K with an uncertainty of 0.62% and the inherent uncertainty in each thermocouple (0.75%) exceeds the non-uniformities at all the experimental temperatures. Thus, the measured temperature uncertainty in the experiments can be always regarded as 0.75%.

Schematic of the experimental apparatus for simultaneous measurements of SO2/SO3
A catalytic cavity full of vanadium catalysts (Sanfeng Catalyst, S101 intermediate-temperature catalyst) and an empty cavity were mounted symmetrically alongside the test section with both connected to the heated cell inside the furnace. The S-101 catalysts have an average bulk density of 0.635 kg/L and the length and the diameter of each catalyst particle are both 5 mm. The total mass and free surface area of the catalysts in the catalytic cavity were 41 g and 100 cm2. The catalytic cavity was used as a heterogeneous reactor and the empty cavity can be used to measure the absorption signals before the catalytic reactions at each experimental temperature and pressure. The catalytic reactor and the heated optical gas cell were designed as an integrated unit placed in one furnace. Compared with the separate apparatus in previous studies [32, 49], the SO3 loss in the gas transmission from the catalytic reactor to the measurement system can be avoided in our design. The empty cavity could also play a role as a homogeneous reactor to be compared with the heterogeneous reaction although the homogeneous conversions did not occur in our experimental temperature ranges (600–1000 K), which will be further discussed in Sect. 5.3. Two needle valves were used to switch between the catalytic and empty cavities that were connected to a local gas delivery manifold leading to source gas bottles, mass flow controllers (MFC, Sevenstar, CS200D) and a mixing tank with Teflon beads used to prepare the gas mixtures of SO2 (99.9%) /N2 (99.9%) /O2 (99.9%) (Jinghui Gas) for the experimental measurements.
The gas mixtures with a total flow of 400 mL/min first flowed through the catalytic cavity where the SO2 catalytic oxidation occurs with SO3 produced at high temperatures. Then, the gases flowed into the heated optical gas cell for the measurements. The gas mixtures were subsequently cooled by a condenser with the liquefied SO3 absorbed in a quartz cell full of silica wool. The remaining SO2 flowed into the room-T gas cell where the concentration was measured. The temperature of the remaining SO2 was measured by another thermocouple placed on the room-T gas cell with temperatures displayed around 18–20 ℃ (room-T: 19 ℃) in the experiments. There were no high-temperature lines observed in the SO2 spectra measured in the room-T gas cell, which also validated the accuracy of the measured temperature of the gas mixtures after being cooled. The residual SO2 was finally reacted with a NaOH solution and exhausted by a vacuum pump. The residence time (3.9 s) during the flow through the catalytic cavity can be calculated by using the total flow (400 mL/min) divided by volume (26 mL) of the cavity. The residence time from 0.23 to 3.7 s for the SO2 catalytic oxidation reaction has been investigated in the references [46, 47] where a residence time of 3.7 s was demonstrated long enough for this reaction. Thus, a residence time of 3.9 s in our work is long enough compared to the chemical reaction time. In addition, the gas flow maintains constant in our experiments and the conversion is independent on the flow rate.
A tunable quantum cascade laser (HAMAMATSU, LE0981QCL) operating near 7.16 µm was used for the high spectral resolution investigation of SO2 and SO3. The QCL was tunable from 1393 to 1399 cm−1 and had a nominal output power of 50 mW at 1397 cm−1 working at a current of 0.84 A and temperature of 24.5 ℃ driven by a laser current controller (QCL1500 LAB, Wavelength) and a temperature controller (TC10 LAB, Wavelength). A 1 kHz sawtooth waveform injection current generated by the signal generator (Keysight, 33510B) was used to tune the QCL across a 1 cm−1 range to resolve the SO2 and SO3 spectra. The laser beam was split using a ZnSe beam splitter with the two beams then propagated through the heated gas cell and the room-temperature gas cell. The laser beams then passed through an optical iris and a bandpass spectral filter (Spectrogon, NB-7230–180 nm) to minimize thermal emissions before being focused onto thermo-electrically cooled photovoltaic (PV) detectors (VIGO System PVI-4TE-10.6). The relative frequency of the laser light was measured by placing a germanium etalon with a free spectral range of 0.0164 cm−1 in the path of the beam using an optical flip-mount. The visible light coinciding with the QCL light was used to help adjust the light path. The regions of the optical path outside the furnace were purged by pure N2 to prevent the detectable absorbance in the ambient.
4.2 Experimental procedure
The experiments were performed at various temperatures from 550 to 1000 K using an experimental procedure having four steps at each temperature: (i) The valve to the catalytic cavity was closed and that to the empty cavity was opened. The SO2 spectrum for the initial concentration was measured in both the heated gas cell and the room-T gas cell before the catalytic oxidation at various pressures from 3 to 20 kPa. The pressure was adjusted by changing the needle valve in front of the vacuum pump to adjust the pump capacity so that the pressure could be adjusted to the expected value and maintained stable during the data acquisition. (ii) The valve to the empty cavity was closed and that to the catalytic cavity was opened. SO3 was produced via the heterogeneous catalytic reaction of SO2 with the combined SO2/SO3 spectrum then measured in the heated gas cell. The remaining SO2 was extracted and measured in the room-T gas cell at the same pressures as in step (i). (iii) The valves to the two cavities were both closed and the gas source was shut off. The SO2 was exhausted from the heated gas cell but SO3 could still be observed for some time due to its continued evolution from the catalyst surface. The signals were recorded as only the SO3 absorption with no SO2 interference after the SO2 was no longer observed in the room-T gas cell. However, the SO3 absorption was difficult to measure at temperatures higher than 850 K using this method due to the lower conversions with larger uncertainties. (iv) The experimental apparatus was treated with a flow of dry air to remove any traces of absorbed H2O, SO2 and SO3 for 3–5 h in the entire system, especially the gas cells and the catalytic cavity. The background signals were measured until no SOx absorption was observed.
Figure 4 shows the data processing flow chart. The SO2 spectra for the initial gas before the catalytic reactions were measured in step (i) to give the relationship between the SO2 absorbance and the gas properties (i.e. temperature, pressure and concentration) for the subsequent measurements. The SO2/SO3 spectra and the corresponding concentrations were simultaneously measured in the heated gas cell by the operations in step (ii) to determine the temperature and pressure dependencies of the SO2 conversions. The conversions were also determined by measuring the SO2 concentrations before and after the catalytic reaction in the room-T gas cell, which provided well-defined initial concentrations in the simultaneous fit of the SO2/SO3 absorbance and validated the measurement accuracy at high temperatures. The SO2 concentrations measured in the room-T gas cell were corrected since the gas compositions changed with the loss of all the SO3 and part of the O2 from the gas compositions in the heated gas cell.

Flow chart for measuring the SO2 conversion by simultaneous measurements of the SO2/SO3 spectra
5 Results and discussion
5.1 SOx absorption spectra
In the experiments using direct absorption spectroscopy, the transmitted light intensity, It, is directly measurable, but the incident light intensity baseline, I0, must be accurately inferred to determine the measured absorbance and the gas properties of interest. However, the baseline is difficult to accurately estimate in these experiments due to the time and wavelength dependent intensity distortions, thermal emissions and, most importantly, the baseline distortion resulting from pollution and refractive index variations of the optical windows during the chemical reactions. The offset and distortion of I0 can be seen in Fig. 5. In addition, the target absorption features between 1396.5 and 1397.4 cm−1 are an unresolved grouping of strong lines sitting atop a quasi-continuum of weaker lines with no zero-absorbance regions observed at higher pressures. Moreover, there is no spectroscopic parameters for the high-T SO3 spectral lines in either the spectroscopic databases or previous studies and some SO2 spectral lines at high temperatures are also not included in the HITRAN databases. The measured overlapped spectra are difficult to be resolved without knowledge of the individual absorbance of each gas species, especially at higher pressures with no non-absorption zones to determine the baseline accurately. Thus, accurate SO2 and SO3 individual absorbance must be both measured at high temperatures. With knowledge of the spectral models of the two molecules, the absorbance and the corrected baseline were obtained simultaneously from the measured transmitted intensity, It, the measured incident intensity, I0, and the SO2/SO3 high-T spectral model. The baseline, I0, was measured in the step (iv) and a 3rd order polynomial was used to correct the baseline in the fitting procedure considering the baseline drift. However, the sensor itself is still subject to zero-baseline shift if applied to in-situ measurement applications. The effects of different types of baseline shifts on the measured absorbance and gas properties were analyzed in our previous work [48]. The direct absorption spectroscopy (DAS) is most sensitive to the horizontal shifting of the baseline (time synchronization) so that the measurement accuracy will be reduced when the sensor running out of synchronization in a long-time running. However, the DAS is insensitive to a vertical shifting (offsets) and the slope shifting of the baseline commonly associated with dynamic background radiation in the in-situ measurements and these shifts can be easily corrected by using a polynomial.

Example wavelength detector signals in both the inflow gas cell (blue) and the room-T gas cell (red)
The individual SO2 absorbance can be obtained when the gas mixtures flow through the empty cavity where only the SO2 spectrum diluted in N2/O2 is present. The measured SO2 absorbance (blue dashed line) at 1014 K and 3.44 kPa is shown in Fig. 6 in which the green and the black absorbance are calculated from the spectroscopic parameters of the transitions from HITRAN2016 and HITRAN2020. It is observed that some high-temperature spectral lines from the hot band emerge in the measured SO2 absorbance compared with the HITRAN2016 prediction while the calculated absorbance from the recently updated HITRAN2020 is more consistent with the measured values, especially for the high-T transitions although there are still several measured weak lines not covered by the HITRAN2020. The absorption increase of these high-T transitions with increasing temperature was also exactly observed in our experimental temperature range. The Voigt profile described in Sect. 3 was used to calculate all the transitions with the best-fit spectra shown in red in Fig. 6a. At high temperatures and moderate pressures with ∆νC /∆νD around 1 [42], the collisional narrowing effect should be taken into account but the congested spectral structures and limited signal to-noise ratio (SNR) preclude the use of more advanced lineshapes than the Voigt. The lower panel in Fig. 6 shows the best-fit residuals scaled by the peak absorbance with a peak residual less than 7.5% and the standard deviation of 8.02 × 10−3 at the peak absorbance of 0.88.

Measured SO2 absorbance (blue) at 3.38 kPa and 1014 K with the corresponding semi-empirical model fits (red) with high-T “equivalent lines” included alongside HITRAN (black) predictions. The stem plots indicate the transitions included in HITRAN 2016
Since the high-T SO3 spectrum is much more congested than that of SO2, the individual lines can only be partially resolved at much lower pressures with much smaller collisional widths of the spectral lines. Thus, the individual SO3 spectra were derived in step (iii) at a pressure of only 0.75 kPa as shown in Fig. 7 in which only several lines were consistent with those in HITRAN and most of the transitions were observed for the first time at high temperatures. There were no spectra of other interfering species (H2O, H2SO4) in the measured wavelength range [50]. The gas source of SO2 has been shut off and SO2 was exhausted from the heated gas cell as described in step (iii) so that only SO3 was present. These measured new SO3 lines were called “equivalent lines” here and the positions of these “equivalent lines” were approximately determined at such a low pressure. The linestrength and broadening coefficients of the high-T “equivalent lines” for the Voigt profile were derived by fitting the measured SO3 spectra at known temperatures and pressures. The collisional broadening coefficients for all the “equivalent lines” are constrained to the same value to reduce the degrees of freedom in the fitting procedure. In this way, a semi-empirical model based on the HITRAN database combined with these “equivalent lines” was developed to describe the high-T SO3 absorbance. The best-fit SO3 residuals in the lower panel give a similar peak value as that of SO2. The semi-empirical model was also validated by using the low-P absorbance to predict the measured high-P absorbance with the residuals within 10%.

Measured SO3 absorbance (blue) at 0.75 kPa and 742 K with the semi-empirical model fits with high-T “equivalent lines” included (red) alongside the HITRAN prediction (black). The stem plots indicate the transitions included in HITRAN
5.2 Calibration and measurements
In each case, the SO2 absorbance was measured while the SO2 flowed through the empty cavity before the catalytic oxidation of SO2 to determine the temperature and pressure dependencies of the peak absorbance at 1397.16 cm−1, which are shown in Fig. 8a, to calibrate the SO2 absorbance for the subsequent SO2 and SO3 measurements. The SO2 absorbance increases almost linearly with increasing pressure but falls with increasing temperature as shown in Fig. 8b, c which show the absorbance at three pressures and three temperatures to illustrate the absorbance variations.

a Dependence of the measured SO2 absorbance at 1397.16 cm−1 (Peak) on temperature and pressure in 50% O2 and 47.5% N2. b Pressure dependence of the SO2 peak absorbance at T = 633 K, 852 K and 1014 K taken from (a). c Temperature dependence of the SO2 peak absorbance at P = 4.89 kPa, 12.5 kPa and 20 kPa taken from (a). The dashed lines were polynomial fits of the data shown in (b) and (c)
The catalytic cavity was then opened with SO3 produced in the heterogeneous catalytic reaction and the SO2/SO3 spectra simultaneously measured in the heated gas cell at various temperatures and pressures. No SO3 spectra were observed at temperatures lower than 590 K which indicates that the catalytic oxidation cannot occur at such low temperatures. Figure 9 shows the measured overlapping spectra of SO2 and SO3 at temperatures from 633 to 1014 K and pressures around 3.35 kPa. The individual SO2 and SO3 spectra are well resolved as shown by the orange and green curves in Fig. 9 based on the semi-empirical models described in Sect. 5.1. The SO3 spectra show continuous absorption over the scanned wavelength region from 1396.5 to 1397.4 cm−1 while the SO2 spectra are less congested with much weaker absorption around 1396.52 and 1397.32 cm−1 so these two wavelengths are the best choices for the SO3 measurements with minimum interference by SO2. The SO3 spectrum accounts for the largest proportion in the overlapping spectra at 742 K, which can be seen at the wavenumber of 1397.16 cm−1 and it indicates that the heterogeneous conversion of SO2 is likely largest at this temperature. The proportion of the SO2 spectra in the total SOx spectra decreased from 633 to 742 K then increased from 742 to 1014 K with increasing temperatures and the strongest SO2 absorption and weakest SO3 absorption were obtained at 1014 K. It indicates that the SO2 conversion increases then decreases with increasing temperatures.

Measured overlapping spectra of SO2/SO3 with the corresponding model fits with the uncoupled individual SO2 and SO3 spectra at T = (a) 633 K, (b) 742 K, (c) 908 K, and (d) 1014 K and P ≈ 3.38 kPa
To validate the SO2 measurement accuracy from the overlapping spectra in the high-T gas cell, the remaining SO2 concentrations after the catalytic reaction were also measured in the room-T gas cell by extracting the SO2 from the SOx mixtures to determine the nominal remaining SO2 concentrations. The absorption line near 1397.16 cm−1 was applied to determine the SO2 concentrations in both the high-T and room-T gas cell. A variety of the SO2 concentrations were obtained due to the different conversions at different experimental temperatures and pressures. The concentrations from 0.6 to 2.3% measured by both approaches are plotted in Fig. 10 in which the red circles indicate the measured concentrations in the high-T gas cell and the black line represents the ideal response (y = x) of the measured concentrations in the room-T gas cell with the relative deviations shown in the lower panel. The measured SO2 concentrations at high temperatures compare well with the concentrations measured at room temperature with uncertainties of less than 7% and with most of the concentrations measured at room-T being less than those at high-T. The loss of SO2 due to the slight adsorption on the tube walls and dissolution in a small amount of liquefied H2O when gas mixtures flowing through the cooling system and the silica-wool cell mainly lead to the difference between the concentrations measured in two cells. In addition, the uncertainties (~ 2%) of the optical path-length of the high-T and room-T cell also lead to the difference of the two measured SO2 concentrations, especially for the path-length uncertainty of the high-T cell since there will be changes in the path-length at elevated temperatures due to the thermal expansion of the stainless steel and the BaF2 rods, which leads to larger uncertainties (~ 5%) of the path length and the measured concentrations.

Measured SO2 concentrations in the SO2/SO3 mixtures at high T versus the residual SO2 concentrations measured at room T
The dependencies of the heterogeneous conversions of SO2 on pressure and temperature were both investigated. The conversion calculation was based on the SO2 concentrations measured simultaneously in the high-T cell. The pressure dependence of the conversion is shown in Fig. 11a for pressures from 3 to 20 kPa at 797 K in which the measured SO3 concentrations (green) and SO2 conversions (red) show a logarithmic growth rate with increasing pressure. The SO2 concentrations measured in both the heated gas cell (blue) and the room-T gas cell (orange) are also shown with a high degree of consistency. The temperature dependence of the conversions from 600 to 1000 K at 17.5 kPa are shown in Fig. 11b where the SO2 conversions first increase to a peak of 90% at around 800 K and then decrease to lower than 70% at around 1000 K. The measured SO3 concentrations have larger uncertainties than those in Fig. 11a, especially at around 700 K due to the adsorption of SO3 and SO2 on the catalysts which could not be completely removed by flowing dry air even after several hours, which leads to a small amount of SO3 appearing from the oxidation of the SO2 from the last experimental case when increasing temperature.

a Measured SO2 (blue) /SO3 (green) concentrations and SO2 conversions (red) after the high-T catalytic oxidation reaction at T = 797 K and various pressures from 3 to 20 kPa. The yellow data represents the residual SO2 concentrations measured in the room-T gas cell as reference. b Measured SO2/SO3 concentrations and SO2 conversions for various temperatures from 600 to 1000 K at P = 17.5 kPa
The measured SO2 conversions are plotted as contours for all the investigated temperatures and pressures in Fig. 12a in which the largest conversions are concentrated around 750–800 K and 20 kPa. The pressure and temperature dependencies of all the measured conversions are consistent with the representative cases shown in Fig. 11. The SO2 conversions were also measured at two additional experimental conditions of 742 K, 4.89 kPa and 908 K, 15.0 kPa and are shown as the hollow symbols in Fig. 12b, c to verify the correlations (dotted lines) that are based only on the data represented by the solid symbols taken from Fig. 12a. These additional measured data points agree well with the correlations to further validate the measurement accuracy.

a Dependence of the measured catalytic conversions on temperature and pressure for 2.5% SO2, 50% O2 and 47.5% N2. b The pressure dependence of the conversions at T = 742 K and 908 K. The dotted lines are correlations of the data represented by the solid symbols while the hollow symbols are two additional measured conversions that verify the correlation. c The temperature dependence of the conversions at P = 4.89 kPa and 15.0 kPa
5.3 Comparison with thermodynamic model
The SO2 conversions were also calculated theoretically based on a thermodynamic model for the same temperatures and pressures as those used in the experiments with the predictions in Fig. 13a for comparison with the measured results. The thermodynamic SO2 conversion can be calculated from the chemical equilibrium constant with the known gas (SO2, O2, N2) composition and equilibrium temperature based on equilibrium thermodynamics and S/O mass balances [51, 52]. The chemical equilibrium constant for the catalytic reaction SO2 (g) + 0.5O2 (g) → SO3 (g) was calculated from the Gibbs free energy change for SO2 oxidation at a desired equilibrium temperature.

a Predicted dependence of the conversions of the SO2 catalytic oxidation reaction on pressure and temperature based on a thermodynamic model. b Comparison of the pressure dependence between the measured and calculated conversions at 907 K. c Comparison of the temperature dependence between the measured and calculated conversions at 4.89 kPa
In comparison between the experimental conversions and the thermodynamic conversions, the assumptions and preconditions of the two conversions should be noted. First, the experimental conversions were measured from a heterogeneous (gas and solid) chemical reaction with vanadium catalysts involved. However, thermodynamic conversions were calculated only from the thermodynamic properties (entropy and enthalpy) of SO2(g) and O2(g) based on the Gibbs free energy in a homogeneous reaction without catalysts involved. Second, the reaction time-scale in the experimental measurements was in seconds or minutes while the time-scale in the thermodynamic model is infinite since it is assumed that all the chemical reactions must go to equilibrium in the thermodynamic equations even if the chemical reaction rate is so slow that it will take infinite time to reach equilibrium.
For the temperature dependence of the two conversions, the thermodynamic conversion is always decreasing with increasing temperature with an initial value approaching 100% at around 600 K while the measured heterogeneous conversion first increases from 600 to 800 K with a peak of around 92% and then decreases to the lowest value of only 3% at 1000 K. The thermodynamic conversions (blue) in Fig. 13c show the similar temperature dependence to the results presented in [7]. Although the reaction rate is very slow at lower temperatures from 600 to 800 K, the thermodynamic model shows high conversions since the model demonstrates the results when reaching chemical equilibrium even if it takes infinite time, which likely cannot be achieved in reality. It also explains why there was no SO3 produced when SO2/O2 mixtures flowed through the empty cavity with no catalysts inside in our experiments. However, the heterogeneous conversions can occur due to the vanadium catalysts which can improve the SO3 formation rates compared with the cases in the empty cavity. As temperature increases from 600 to 800 K, the heterogeneous reaction rates and the conversions increase but with a short residence time [7, 53] in a gas mixture (SO2/N2/O2) flow rate of 400 mL/min. In addition, the temperature dependence of the adsorption capacity of the catalysts [55] may also contribute to the SO2 conversions since the heterogeneous reactions occurs on the catalytic surface.
The experimental and thermodynamic conversions show the similar temperature dependence and values from 800 to 1000 K as shown in Fig. 13c likely due to the following two reasons. First, since the reaction rate increases with increasing temperature, it is easier to reach chemical equilibrium with the help of the catalysts so that the deviations between the measured values and the thermodynamic values reduced at higher temperatures. Second, the reaction SO2 (g) + 0.5O2 (g) → SO3 (g) is an exothermic reversible reaction with the equilibrium shifting to SO2 formation with increasing temperatures, which accounts for the reduction of the conversion at higher temperatures in both the thermodynamic and heterogeneous conversions.
For the pressure dependence of the heterogeneous and thermodynamic conversions, they both increase logarithmically with increasing pressures as shown in Fig. 13b since the collision frequency of the molecules (SO2/O2) increases with increasing pressures and the reaction equilibrium shifts to SO3 formation at higher pressures based on the law of chemical equilibrium. However, the heterogeneous conversions grow faster than the thermodynamic conversions at the same pressure range mainly because the adsorption capacity of the catalyst enhances with increasing pressure so that larger amount of SO2 reacted with O2 on the catalytic surface at higher pressures.
The SO3 formation from SO2 in heterogeneous catalytic reactions has also been investigated in previous studies [7, 53] in which the conversion was negligible at 773 K and maximum conversion was reached at around 973 K before falling until 1173 K, which is similar to the temperature-dependent trend of the measured conversions in our work. The heterogeneous conversions of SO2 were also measured in [27] by using an infrared multi-gas analyzer to measure the inlet and outlet SO2 concentration but only at temperatures lower than 723 K. Their conversions increased with increasing temperature to a peak around 100% at 723 K, which is similar to the trend measured in the same temperature range in our work.
SO3 can also be formed from a homogeneous gas-phase reaction but the homogeneous SO2 conversion to SO3 was not observed when the gas mixtures flowed through the empty cavity at temperatures lower than 1000 K in our work. The homogeneous conversions of SO2 to SO3 were previously studied by [7, 8, 56] in which a conversion increase was noted (less than 6%) when the temperature increased above 973 K and temperatures above 1173 K were required for obvious SO3 formation with a residence time of 1 s. In addition, the homogeneous conversions increased exponentially in the temperature range from 973 to 1273 K in [7]. Thus, it is reasonable for no SO3 formation in the empty cavity (homogeneous reactor) in our work and higher temperatures are required to convert SO2 to SO3 in homogeneous gas-phase reactions.
6 Conclusion
This work demonstrated the feasibility of using 7.16 µm QCL absorption spectroscopy to simultaneously measure both SO2 and SO3 concentrations during heterogeneous catalytic oxidation of SO2 at elevated temperatures. High-temperature individual SO2 and SO3 absorbance were measured and the overlapped spectra of the gas mixtures were resolved by combining the measured high-temperature spectra with the room-temperature spectra in the HITRAN database. The heterogeneous conversions were determined not only from the simultaneous detection of SO2 and SO3 concentrations at high temperatures but also by measuring the inlet and outlet SO2 concentrations at room temperature. The measured conversions from these two methods agreed well with each other within 7% uncertainties with the differences mainly due to the adsorption of SO2 in the tubing. The temperature and pressure dependence of the heterogeneous conversions were investigated at pressures of 3–20 kPa and temperatures of 600–1000 K and were also compared with the theoretical conversions calculated using a thermodynamic model. The heterogeneous and thermodynamic conversions both show logarithmic growths with increasing pressure. In addition, the heterogeneous conversions initially increase to about 92% at 800 K and then decrease to the conversion less than 3% at 1014 K, which differs from the continual decrease of the conversions with increasing temperature predicted by the thermodynamic model.
References
C. Zheng, Y. Wang, Y. Liu, Z. Yang, R. Qu, D. Ye, C. Liang, S. Liu, X. Gao, Formation, transformation, measurement, and control of SO3 in coal-fired power plants. Fuel 241(1), 327–346 (2019). https://doi.org/10.1016/j.fuel.2018.12.039
D. Bongartz, A.F. Ghoniem, Chemical kinetics mechanism for oxy-fuel combustion of mixtures of hydrogen sulfide and methane. Combustion Flame 162(3), 544–553 (2015). https://doi.org/10.1016/j.combustflame.2014.08.019
X. Zheng, E.M. Fisher, F.C. Gouldin, J.W. Bozzelli, Pyrolysis and oxidation of ethyl methyl sulfide in a flow reactor. Combustion Flame 158(6), 1049–1058 (2011). https://doi.org/10.1016/j.combustflame.2010.10.018
D. Bongartz, A.F. Ghoniem, Impact of sour gas composition on ignition delay and burning velocity in air and oxy-fuel combustion. Combustion Flame 162(7), 2749–2757 (2015). https://doi.org/10.1016/j.combustflame.2015.04.014
J. Mu, D.D. Perlmutter, Thermal decomposition of inorganic sulfates and their hydrates. Ind. Eng. Chem. Process Design Dev. 20(4), 640–646 (1981). https://doi.org/10.1021/i200015a010
J. Paulik, F. Paulik, M. Arnold, Simultaneous TG, DTG, DTA and EGA technique for the determination of carbonate, sulphate, pyrite and organic material in minerals, soil and rocks-III Operation of the thermo-gas-titrimetric equipment and the examination procedure in special cases. J Thermal Anal. 29(2), 345–351 (1984). https://doi.org/10.1007/BF02720070
L.P. Belo, L.K. Elliott, R.J. Stanger, R. Spörl, K.V. Shah, J. Maier, T.F. Wall, High-temperature conversion of SO2 to SO3: Homogeneous experiments and catalytic effect of fly ash from air and oxy-fuel firing. Energy Fuels 28(11), 7243–7251 (2014). https://doi.org/10.1021/ef5020346
D. Fleig, M.U. Alzueta, F. Normann, M. Abián, K. Andersson, F. Johnsson, Measurement and modeling of sulfur trioxide formation in a flow reactor under post-flame conditions. Combustion Flame 160(6), 1142–1151 (2013). https://doi.org/10.1016/j.combustflame.2013.02.002
L.P. Belo, R. Spörl, K.V. Shah, L.K. Elliott, R.J. Stanger, J. Maier, T.F. Wall, Sulfur capture by fly ash in air and oxy-fuel pulverized fuel combustion. Energy Fuels 28(8), 5472–5479 (2014). https://doi.org/10.1021/ef500855w
R.K. Srivastava, C.A. Miller, C. Erickson, R. Jambhekar, Emissions of sulfur trioxide from coal-fired power plants. J. Air. Waste Manag. Assoc. 54(6), 750–762 (2004). https://doi.org/10.1080/10473289.2004.10470943
R. Spörl, J. Walker, L. Belo et al., SO3 emissions and removal by ash in coal-fired oxy-fuel combustion. Energy Fuels 28(8), 5296–5306 (2014). https://doi.org/10.1021/ef500806p
A.W. Hodgson, P. Jacquinot, P.C. Hauser, Electrochemical sensor for the detection of SO2 in the low-ppb range. Anal. Chem. 71(14), 2831–2837 (1999). https://doi.org/10.1021/ac9812429
D. Weidmann, A. Hamdouni, D. Courtois, CH4/air/SO2 premixed flame spectroscopy with a 7.5-µm diode laser. Appl. Phys. B 73(1), 85–91 (2001). https://doi.org/10.1007/s003400100613
L. Joly, V. Zéninari, B. Parvitte, D. Weidmann, D. Courtois, Y. Bonetti, T. Aellen, M. Beck, J. Faist, D. Hofstetter, Spectroscopic study of the ν1 band of SO2 using a continuous-wave DFB QCL at 9.1 µm. Appl. Phys. B 77(6–7), 703–706 (2003). https://doi.org/10.1007/s00340-003-1310-8
B. Grouiez, B. Parvitte, L. Joly, D. Courtois, V. Zeninari, Comparison of a quantum cascade laser used in both cw and pulsed modes. Application to the study of SO2 lines around 9 µm. Appl. Phys. B 90(2), 177–186 (2008). https://doi.org/10.1007/s00340-007-2857-6
L. Richard, I. Ventrillard, G. Chau, K. Jaulin, E. Kerstel, D. Romanini, Optical-feedback cavity-enhanced absorption spectroscopy with an interband cascade laser: application to SO2 trace analysis. Appl. Phys. B 122(9), 1–7 (2016). https://doi.org/10.1007/s00340-016-6502-0
F. Xu, Y.G. Zhang, G. Somesfalean, Z.G. Zhang, H.S. Wang, S.H. Wu, Temperature-corrected spectroscopic evaluation method for gas concentration monitoring. Appl. Phys. B 86(2), 361–364 (2007). https://doi.org/10.1007/s00340-006-2489-2
H.S. Wang, Y.G. Zhang, S.H. Wu, X.T. Lou, Z.G. Zhang, Y.K. Qin, Using broadband absorption spectroscopy to measure concentration of sulfur dioxide. Appl. Phys. B 100(3), 637–641 (2010). https://doi.org/10.1007/s00340-010-4151-2
J. Henningsen, Measurement of free SO2 in wine with 74-µm difference frequency spectrometer. Appl. Phys. B 76(4), 451–456 (2003). https://doi.org/10.1007/s00340-003-1141-7
J. Henningsen, J. Hald, Quantitative analysis of dilute mixtures of SO2 in N2 at 7.4 µm by difference frequency spectroscopy. Appl. Phys. B 76(4), 441–449 (2003). https://doi.org/10.1007/s00340-003-1140-8
L. Wang, Y. Zhang, X. Zhou, F. Qin, Z. Zhang, Optical sulfur dioxide sensor based on broadband absorption spectroscopy in the wavelength range of 198–222 nm. Sens. Actuators, B 241, 146–150 (2017). https://doi.org/10.1016/j.snb.2016.10.055
Y. Matsumi, H. Shigemori, K. Takahashi, Laser-induced fluorescence instrument for measuring atmospheric SO2. Atmosp. Environ. 39(17), 3177–3185 (2005). https://doi.org/10.1016/j.atmosenv.2005.02.023
X.T. Lou, G. Somesfalean, Z.G. Zhang, S. Svanberg, Sulfur dioxide measurements using an ultraviolet light-emitting diode in combination with gas correlation techniques. Appl. Phys. B 94(4), 699–704 (2009). https://doi.org/10.1007/s00340-009-3399-x
Y. Cao, H. Zhou, W. Jiang et al., Studies of the fate of sulfur trioxide in coal-fired utility boilers based on modified selected condensation methods. Environ. Sci. Technol. 44(9), 3429–3434 (2010). https://doi.org/10.1021/es903661b
R.F. Maddalone, S.F. Newton, R.G. Rhudy, R.M. Statnick, Laboratory and field evaluation of the controlled condensation system for SO3 measurements in flue gas streams. J. Air Pollut. Control Assoc. 29(6), 626–631 (1979). https://doi.org/10.1080/00022470.1979.10470834
D. Fleig, E. Vainio, K. Andersson, A. Brink, F. Johnsson, M. Hupa, Evaluation of SO3 measurement techniques in air and oxy-fuel combustion. Energy Fuels 26(9), 5537–5549 (2012). https://doi.org/10.1021/ef301127x
C. Zheng, X. Li, Z. Yang, Y. Zhang, W. Wu, X. Wu, X. Wu, X. Gao, Development and experimental evaluation of a continuous monitor for SO3 measurement. Energy Fuels 31(9), 9684–9692 (2017). https://doi.org/10.1021/acs.energyfuels.7b01181
D. D. Stuart (2010) Acid dewpoint temperature measurement and its use in estimating sulfur trioxide concentration, ISA automation week 2010: Technology and Solutions Event 339–350.
E. Vainio, D. Fleig, A. Brink, K. Andersson, F. Johnsson, M. Hupa, Experimental evaluation and field application of a salt method for SO3 measurement in flue gases. Energy Fuels 27(5), 2767–2775 (2013). https://doi.org/10.1021/ef400271t
V. Bondybey, J. English, Infrared spectra of SO3 polymers and complexes in rare gas matrices. J. Mol. Spectrosc. 109, 221–228 (1985). https://doi.org/10.1016/0022-2852(85)90308-X
S.W. Sharpe, T.A. Blake, R.L. Sams, A. Maki, T. Masiello, J. Barber, N. Vulpanovici, J.W. Nibler, A. Weber, The ν3 and 2ν3 bands of 32S16O3, 32S18O3, 34S16O3, and 34S18O3. J. Mol. Spectrosc. 222(2), 142–152 (2003). https://doi.org/10.1016/j.jms.2003.08.001
F. Tetsuo, H. Ninomiya, SO3 concentration measurement using ultraviolet absorption spectroscopy and thermal conversion. IEEJ Trans. Fundamentals Materials 126, 977–982 (2006). https://doi.org/10.1541/IEEJFMS.126.977
J. Mellqvist, H. Axelsson, A. Rosén, Doas for flue gas monitoring - III In-situ monitoring of sulfur dioxide, nitrogen monoxide and ammonia. J. Quantitative Spectrosc. Radiative Trans. 56(2), 225–240 (1996). https://doi.org/10.1016/0022-4073(96)00044-1
D.M. Sonnenfroh, M.G. Allen, W.T. Rawlins, C.F. Gmachl, F. Capasso, A.L. Hutchinson, D.L. Sivco, A.Y. Cho, Pollutant emission monitoring using QC laser-based mid-IR sensors. Water Ground Air Pollut. Monit. Remediation 4199, 86 (2001). https://doi.org/10.1117/12.417364
O.G. Buzykin, S.V. Ivanov, A.A. Ionin, A.A. Kotkov, A.Y. Kozlov, Spectroscopic detection of sulfur oxides in the aircraft wake. J. Russian Laser Res 26(5), 402–426 (2005). https://doi.org/10.1007/s10946-005-0043-z
J. M. Hensley, W. T. Rawlins, D. B. Oakes, D. M. Sonnenfroh, M. G. Allen, A quantum cascade laser sensor for SO2 and SO3, 2005 Conference on Lasers and Electro-Optics, CLEO 2 (31) (2005) 1073–1075. doi:https://doi.org/10.1364/AO.44.006635
T. Hieta, M. Merimaa, Simultaneous detection of SO2, SO3 and H2O using QCL spectrometer for combustion applications. Appl. Phys. B 117(3), 847–854 (2014). https://doi.org/10.1007/s00340-014-5896-9
T.A. Berkoff, J.C. Wormhoudt, R.C. Miake-Lye, Measurement of SO2 and SO3 using a tunable diode laser system[C]//environmental monitoring and remediation technologies. Int. Soc. Opt. Photonics 3534, 686–693 (1999). https://doi.org/10.1117/12.339057
W.T. Rawlins, J.M. Hensley, D.M. Sonnenfroh et al., Quantum cascade laser sensor for SO2 and SO3 for application to combustor exhaust streams. Appl. Opt. 44(31), 6635–6643 (2005). https://doi.org/10.1364/AO.44.006635
A. Tokura, O. Tadanaga, T. Nishimiya, K. Muta, N. Kamiyama, M. Yonemura, S. Fujii, Y. Tsumura, M. Abe, H. Takenouchi, K. Kenmotsu, Y. Sakai, Investigation of SO3 absorption line for in situ gas detection inside combustion plants using a 4-µm band laser source. Appl. Opt. 55(25), 6887 (2016). https://doi.org/10.1364/ao.55.006887
I. E. Gordon, L. S. Rothman, C. Hill, R. V. Kochanov, Y. Tan, P. F. Bernath, M. Birk, V. Boudon, A. Campargue, K. V. Chance, B. J. Drouin, J. M. Flaud, R. R. Gamache, J. T. Hodges, D. Jacquemart, V. I. Perevalov, A. Perrin, K. P. Shine, M. A. Smith, J. Tennyson, G. C. Toon, H. Tran, V. G. Tyuterev, A. Barbe, A. G. Császár, V. M. Devi, T. Furtenbacher, J. J. Harrison, J. M. Hartmann, A. Jolly, T. J. Johnson, T. Karman, I. Kleiner, A. A. Kyuberis, J. Loos, O. M. Lyulin, S. T. Massie, S. N. Mikhailenko, N. Moazzen-Ahmadi, H. S. Müller, O. V. Naumenko, A. V. Nikitin, O. L. Polyansky, M. Rey, M. Rotger, S. W. Sharpe, K. Sung, E. Starikova, S. A. Tashkun, J. V. Auwera, G. Wagner, J. Wilzewski, P. Wcisło, S. Yu, E. J. Zak, The HITRAN2016 molecular spectroscopic database, J. Quantitative Spectrosc. Radiative Trans. 203 (2017) 3–69. https://doi.org/10.1016/j.jqsrt.2017.06.038.
R. K. Hanson, R. M. Spearrin, C. S. Goldenstein, Spectroscopy and optical diagnostics for gases, Springer International Publishing AG Switzerland (2016).
C. S. Goldenstein, R. M. Spearrin, J. B. Jeffries, R. K. Hanson, Infrared laser-absorption sensing for combustion gases, Progress energy Combustion Sci. 60 (2017) 132–176. https://doi.org/10.1016/j.pecs.2016.12.002.
K.K. Schwarm, H.Q. Dinh, C.S. Goldenstein, D.I. Pineda, R.M. Spearrin, High-pressure and high-temperature gas cell for absorption spectroscopy studies at wavelengths up to 8 µm. J. Quantitative Spectrosc. Radiative Trans. 227, 145–151 (2019). https://doi.org/10.1016/j.jqsrt.2019.01.029
J. Li, A.P. Nair, K.K. Schwarm, D.I. Pineda, R. Mitchell Spearrin, Temperature-dependent line mixing in the R-branch of the ν3 band of methane. J. Quantitative Spectrosc. Radiative Trans. 255(19), 107271 (2020). https://doi.org/10.1016/j.jqsrt.2020.107271
H. Wang, D. Xie, Q. Chen et al., Kinetic modeling for the deactivation of TiO2 during the photocatalytic removal of low concentration SO2. Chem. Eng. J. 303, 425–432 (2016). https://doi.org/10.1016/J.CEJ.2016.06.015
H. Wang, C. You, Photocatalytic oxidation of SO2 on TiO2 and the catalyst deactivation: a kinetic study. Chem. Eng. J. 350, 268–277 (2018). https://doi.org/10.1016/J.CEJ.2018.05.152
J. Li, K.K. Schwarm, C. Wei et al., Robust cepstral analysis at variable wavelength scan depth for narrowband tunable laser absorption spectroscopy. Measure. Sci. Technol. 32(4), 045502 (2021). https://doi.org/10.1088/1361-6501/abcd6a
H. Xiao, Y. Ru, Q. Cheng et al., Homogeneous and heterogeneous formation of SO3 in flue gases burning high sulfur coal under oxy-fuel combustion conditions. Int. J. Greenhouse Gas Control 78, 420–428 (2018). https://doi.org/10.1016/j.ijggc.2018.09.009
R.F. Majkowski, R.J. Blint, J.C. Hill, Infrared absorption coefficients of gaseous H2SO4 and SO3. Appl. Opt. 17(7), 975–977 (1978). https://doi.org/10.1364/AO.17.000975
M. J. King, W. G. Davenport, M. S. Moats, Oxidation of SO2 to SO3—Equilibrium curves, Sulfuric Acid Manufacture (c) (2013) 125–133. Doi:https://doi.org/10.1016/b978-0-08-098220-5.00010-1.
M. J. King, W. G. Davenport, M. S. Moats, Catalytic oxidation of SO2 to SO3, Sulfuric Acid Manuf. (2013) 73–90. https://doi.org/10.1016/b978-0-08-098220-5.00007-1.
P. Marier, H.P. Dibbs, The catalytic conversion of SO2 to SO3 by fly ash and the capture of SO2 and SO3 by CaO and MgO. Thermochim. Acta 8(1–2), 155–165 (1974). https://doi.org/10.1016/0040-6031(74)85082-3
N.A. Burdett, W.E. Langdon, R.T. Squires, Rate coefficients for the reaction SO2+O2→SO3+O in the temperature range 900–1350 K. J. Inst. Energy 57, 373–376 (1984)
Y. Sarbassov, L. Duan, V. Manovic et al., Sulfur trioxide formation/emissions in coal-fired air-and oxy-fuel combustion processes: a review. Greenhouse Gases 8(3), 402–428 (2018). https://doi.org/10.1002/ghg.1767
D.J. Bayless, J. Jewmaidang, S. Tanneer et al., Kinetics of low-temperature homogeneous SO3 formation for use in flue gas conditioning for improved electrostatic precipitator performance. Proc. Combust. Inst. 28(2), 2499–2505 (2000). https://doi.org/10.1016/S0082-0784(00)80665-7
Acknowledgements
This work was supported by the National Key R&D Program of China (2019YFB2006002), the National Natural Science Foundation of China (11972213, 51906120) and the Tsinghua University-China Huaneng Group Co. Ltd. Joint Institute for Base Energy (HNKJ20-H50).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Li, J., Ding, Y., Li, Z. et al. Simultaneous measurements of SO2 and SO3 in the heterogeneous conversions of SO2 using QCL absorption spectroscopy. Appl. Phys. B 128, 61 (2022). https://doi.org/10.1007/s00340-022-07776-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00340-022-07776-0