
Abstract
A laser wavelength meter was calibrated to ±1 MHz 1\(\sigma\) using transitions in molecular iodine at around 700 nm, where precise experimental measurements of iodine lines are sparse. Two saturation absorption spectroscopy systems were used to measure and characterize the R(118)(2-8) a10, R(54)(3-9) a1, and P(48)(3-9) a15 hyperfine lines of molecular iodine near 700 nm. The transition frequencies were measured using a frequency comb and used to calibrate the wavelength meter. Additionally, the full hyperfine spectrum of the R(54)(3-9) transition was obtained and fitted using a theoretical model allowing for hyperfine coupling constants to be deduced, which agreed with theoretical values.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Wavelength meters (wavemeters) have become a workhorse for stabilization and frequency determination for precision laser spectroscopic measurements. One application of interest is the laser probing of rare isotopes at radioactive beam facilities. The collinear laser spectroscopy and collinear resonance-ionization laser spectroscopy techniques are commonly used for nuclear structure studies [1,2,3], for example electromagnetic moments from the hyperfine (hf) coupling constants and the charge radius from the isotope shifts of the hf spectra. Such laser spectroscopic studies of many elements have been carried out for more than 40 years at radioactive beam facilities including ISOLDE/CERN [4], JYFL/IGISOL [5], RIBF/RIKEN [6] and NSCL/MSU [7]. The accessible range of radioactive elements and their isotopes will be even further extended at the Facility for Rare Isotope Beams (FRIB) [8]. There is strong motivation to carry out laser-probing studies on isotopes with extreme proton-to-neutron ratios as peculiar behaviors are observed for nuclei at their existence limits. Studies on such nuclei provide critical experimental data to test the predictive power of state-of-the-art nuclear theories [9, 10].
A dominant contribution to the systematic uncertainty of the deduced nuclear properties is the laser frequency measurement that can also exceed the statistical contribution. To reduce the systematic error, precise knowledge in the laser frequency, to a level of 1 MHz, is necessary. With such high requirements of accuracy, there are few options for frequency measurement calibration that are robust enough for the wide range of laser frequencies used. While modern wavemeters can have very high absolute accuracy if used very close to the calibration wavelength, there is a systematic uncertainty introduced as the frequency being measured moves further from the calibration frequency [11]. In the case of an isotope shift measurement, for example, the unique resonance frequencies of the two isotopes are measured, and the laser frequency calibration system used here must be widely tunable to match the range of the laser frequencies used for each isotope. Furthermore, studies of atomic and ionic species over a wide range of elements and their isotopes require the use of wavelengths from the near-UV to near-IR (NIR) portion of the electromagnetic spectrum.
One means to overcome this challenge is the measurement of well-known absorption features of atomic or molecular species to calibrate the wavemeter near the desired wavelength. As an example, molecular iodine has thousands of hyperfine transitions in the visible and NIR wavelength range [12]. Transition frequencies of abundant lines in the visible range are well studied and aid to formulate theoretical models, which are capable of predicting the resonance frequency of iodine lines to a 1\(\sigma\)Footnote 1 accuracy of less than 1 MHz [13].
For these reasons, a Doppler-free saturation laser spectroscopy system with molecular iodine has been set up for use at a laser spectroscopy facility for nuclear structure studies (BECOLA facility) at FRIB at Michigan State University (MSU). Measurements of iodine resonance frequencies made at MSU using a wavemeter were then compared to measurements of the same iodine lines performed with a frequency-comb at TU Darmstadt. The differences between these measured resonance frequencies were then used to calibrate the wavemeter at MSU. The wavemeter at MSU has a 1\(\sigma\) accuracy limit of 10 MHz [14], whereas the frequency-comb system at TU Darmstadt has an absolute accuracy limit of less than 1 MHz.
In the present work, the absolute accuracy of the laser frequency measurements using the wavemeter at MSU was improved by an order of magnitude to less than 1 MHz for a recent study of the charge radii of stable and radioactive nickel isotopes. Three iodine hyperfine lines, R(118)(2-8) a\(_{10}\), R(54)(3-9) a\(_{1}\), and P(48)(3-9) a\(_{15}\), were selected to act as calibration lines based on their proximity in frequency to the lab frame resonance frequencies of \(^{54}\hbox {Ni}\), \(^{58}\hbox {Ni}\), and \(^{60}\hbox {Ni}\), respectively. The three iodine lines happen to be near 700 nm and within the region of 680-755 nm, where there are significantly fewer iodine lines whose transition frequencies are well studied (or precisely known) than lines in the visible regime, due to much smaller thermal population of the rovibrational states associated with the iodine line at room temperature. The shortage of reference lines causes the accuracy of the theoretical models to suffer in this region, with a typical prediction uncertainty of 15 MHz [13] compared to that of 1 MHz for the visible regime. In addition to the MSU wavemeter calibration, the absolute frequencies of the iodine lines and hyperfine coupling constants determined in the present study aid to benchmark theoretical models and allow for more confident applications of iodine hyperfine components, which are part of a complicated multiplet in the regime of 700 nm.
Below, the systems at each institution are described and results of the precise determination of absolute frequencies of the iodine lines are presented. Additionally, a mathematical description of the iodine hyperfine spectrum is outlined for fitting of full hyperfine spectra.
2 Experiment
2.1 Saturation spectroscopy at FRIB/MSU

Diagrams of the experimental setups at MSU (a) and at TU Darmstadt (b) used in this paper. In both setups, the laser light was split into a probe beam (solid line) and a pump beam (dashed line) which counter-propagated through the iodine cell with the pump beam modulated by an acousto-optical modulator (AOM). In (b), the probe beam was additionally frequency modulated by an electro-optical modulator (EOM). The other acronyms represent half-wave plate (\(\lambda\)/2), polarizing beam-splitter (PBS), photo-diode (PD) and collinear laser spectroscopy (CLS)
A saturation laser spectroscopy system with molecular iodine was constructed. A schematic is shown in Fig. 1a. Laser light at 704 nm was produced by a continuous-wave titanium-sapphire laser (Matisse TS, Sirah Lasertechnik) pumped by a Nd-YAG solid-state laser (Millennia eV, Spectra Physics). The main part of the laser light was used for radioactive-beam spectroscopy. A fraction of the fundamental laser light was sampled and sent to a wavemeter (WSU30, HighFinesse) for frequency measurement and stabilization; the wavemeter was calibrated against a frequency-stabilized helium-neon laser (SL 03, SIOS Meßtechnik).
Another fraction of the primary red laser light was sampled and sent to the iodine spectroscopy setup via an optical fiber (Fig. 1a). The laser light was split into a probe and a pump beam, counterpropagating through the 60-cm long iodine cell (see below). Both beams were focused at the center of the cell. The pump beam was chopped at 10 kHz by an acousto-optical modulator (AOM, Model 1205C-1, ISOMET) which in turn modulated the absorption of the probe beam. This was observed by detecting the probe-beam intensity after the iodine cell with a 125-MHz bandwidth, low-noise photoreceiver (1801, New Focus). The detector output was fed into a lock-in amplifier (LIA-MCD-200-L, Femto) for demodulation at the AOM’s frequency providing the probe beam absorption signal.
The iodine cell (custom construction, Precision Glassblowing) body of 60-cm length, 1.9-cm outer diameter and 1.6-cm inner diameter was made of quartz glass, to have a maximum operating temperature of approximately \({1000}\,^{\circ }\hbox {C}\). A schematic of the layout is shown in Fig. 2. The windows on either end of the cell were made from fused silica, angled by 11 degrees and wedged by 2 degrees to prevent reflections along the beam path and etalon effects by multiple reflections, respectively. A 10-cm long, 1-cm diameter cold-finger, located 4 cm from the one end of the cell, acted as a reservoir for approximately 200 mg of solid iodine.
The iodine cell was placed inside a temperature-controlled furnace (VST 300, Carbolite Gero). The central 30 cm of the iodine cell was held at \({600}\,^{\circ }\hbox {C}\) to maximize the population of desired rovibrational states. The ends of the cell were wrapped in a heating tape to prevent condensation of iodine on the windows. The cold-finger was placed in a 5-cm long copper heat sink with thermal paste to aid in thermal conduction. This copper block was then in contact with a thermoelectric cooler (TEC) (TECF2S, Thorlabs), operated by a temperature controller (TED200C, Thorlabs). The temperature of the TEC was regulated with the Thorlabs temperature controller and had a water-cooling loop with a \(1\text{-l}\) water reservoir as a heatsink. By adjusting the current supplied to the TEC, heat was supplied to or taken away from the cold-finger giving precise control over the cold-finger temperature from 25 up to \({50}\,^{\circ }\hbox {C}\) and, thus, to the vapor pressure of the iodine sample. The temperature of the cold-finger was measured with a 10-k\(\Omega\) thermistor (TH10K Thermistor, Thorlabs).

Layout of the cell and temperature control system used at MSU. The cold-finger was set in a copper heatsink attached to a thermoelectric cooler (TEC) with a water reservoir to provide thermal mass. This setup allows for stable and precise control of the cold-finger temperature between 25 and 50 \(^{\circ }\)C
Absorption spectroscopy was performed and a typical spectrum of the P(48)(3-9) a\(_{15}\) iodine hyperfine component is shown in Fig. 3. The power of the probe and pump beams were set to 1 and \({20}\,\hbox {mW}\), respectively, and they had a beam waist of approximately \({400}\,\upmu \hbox {m}\) at the focus around the middle of the cell. The wavemeter reading and associated feedback loop were used to step the primary laser frequency in 1-MHz increments across the iodine hyperfine line, with a typical total scan range of 100 MHz. At each frequency step, the absorption signal was measured 100 times in quick succession with a 10-ms data acquisition, and averaged for the final value at each step. The standard deviation of the 100 measurements was used as the uncertainty of the absorption signal. The absorption spectra were fit with a Lorentzian lineshape with the absorption signal uncertainties used as weights in the least-squares fitting. The centroid value of the absorption spectra was extracted, and a typical line width was about 10 MHz (full width at half maximum, FWHM).

A typical absorption signal for the P(48)(3-9) a\(_{15}\) hyperfine component with a Lorentzian fit. There was a sidepeak present from the R(148)(6-10) a\(_{1}\) line. This sidepeak was also fit with a Lorentzian lineshape with the width fixed to that of the main peak and a free peak center
2.2 Saturation spectroscopy at TU Darmstadt
A similar system was setup at TU Darmstadt to determine the transition frequencies using a frequency comb which was referenced to a GPS-disciplined quartz oscillator. In the first step, absorption spectra were recorded in the same way as performed at MSU using a Matisse 2 ring laser. The chopping frequency of the acousto-optical modulator was \({20}\,\hbox {kHz}\), and different types of the photodetector (Thorlabs Si Switchable Gain Detector PDA36A-EC) and lock-in amplifier (EG&G Model 5209) were used. The iodine cell produced by Opthos Inc. had the same dimensions as the one used at MSU described in Sect. 2.1, but instead of thermal paste, the contact material between the copper block and the cold-finger was water, while no additional external water circuit was employed. To obtain absorption spectra, the probe and the pump beam usually had a power of 7 and 12 mW focused to 250 and \({150}\,\upmu \hbox {m}\), respectively, in the middle of the cell, optimized to obtain a good signal-to-noise ratio. A typical scan spanned \({100}\,\hbox {MHz}\) with a step size of \({1}\,\hbox {MHz}\). As the frequency comb (Menlo Systems FC1500-250-WG) had a sample rate of \({1}\,\hbox {Hz}\), five frequency and signal measurements were made at each step. The standard deviation of the frequency and signal measurements were used as the statistical uncertainty of the frequency and the signal amplitude, respectively. Lorentzian lineshapes were fitted to the observed signals to extract the centroid frequency of the hyperfine component under investigation.
2.3 FM-saturation spectroscopy
In the second step, the setup at TU Darmstadt was slightly modified to allow for frequency-modulation (FM)-saturation spectroscopy [15]. A schematic is shown in Fig. 1b. An electro-optical modulator (EOM) (New Focus Inc. Model 4002 Broadband Phase Modulator) was placed in the probe beam path operated at a modulation frequency of \(\Omega =\) \({15}\,\hbox {MHz}\) to create two sidebands of opposite phase at \(\pm \Omega\) of the laser frequency. On the photodiode, these sidebands generate two beat signals at the modulation frequency due to their interference with the carrier frequency. These signals cancel each other, due to their opposite phase, as long as both sidebands have the same intensity [16]. By scanning across the hyperfine lines, the relative intensity of the sidebands change as soon as one of them interacts with the iodine molecules. This creates (after demodulation) a signal proportional to the derivative of the saturation signal as it is shown in Fig. 4 for the case of the P(48)(3-9) a\(_{15}\) hyperfine transition, the same line as depicted for absorption spectroscopy in Fig. 3. Again, the scan had a range of \({100}\,\hbox {MHz}\) with \({1}\,\hbox {MHz}\) steps. A derivative of a Voigt profile was fitted to the observed data with the uncertainties as weights using the non-linear least-square fitting. In this case, the zero crossing represents the center frequency of the line.

A typical signal of the FM-saturation spectroscopy of the P(48)(3-9) a\(_{15}\) hyperfine line with a fit of a derivative of a Voigt profile. There is a recognizable sidepeak (see also Fig. 3), also fit with a derivative of a Voigt profile with the width fixed to that of the main peak, and a free peak center
2.4 Frequency stabilization
The form of the FM-saturation-spectroscopy signal with the zero crossing at the center frequency is ideally suited for frequency stabilization. The signal was used to control the length of the Matisse reference cavity with a digital feedback loop. The frequency of the stabilized laser was then determined with the frequency comb.
3 Frequency determination
3.1 Pressure shift
Iodine lines above \({700}\,\hbox {nm}\) originate from higher lying rovibrational states that are not populated at room temperature. Spectroscopy in this region requires heating of the iodine vapor to several \({100}\,^{\circ }\hbox {C}\). The pressure inside the cell affects the width and centroid of the iodine resonance lines [17] by intermolecular collisions. Therefore, the pressure dependence of the centroid frequency was measured for the three molecular iodine lines R(118)(2-8) a\(_{10}\), R(54)(3-9) a\(_1\), and P(48)(3-9) a\(_{15}\) by changing the cold-finger temperature. The iodine vapor pressure \(p\,[\hbox {Pa}]\) is related to the cold-finger temperature \(T\,[^{\circ }\hbox {C}]\) by the relation [18]

Pressure shift of the R(118)(2-8) a\(_{10}\) hyperfine component measured at MSU (left) and at TU Darmstadt (right) over several days using Doppler-free saturation spectroscopy. The y-intercept represents the transition frequency extrapolated to zero pressure. The averaged slope was used at MSU to determine the zero-pressure temperature from measurements performed at a single cold-finger temperature. Please note that the data point at 200 Pa of the TU Darmstadt measurements for ”23.Sep” was not taken into account in the fitting. At this day the laser was unstable, which might explain this deviation
The pressure dependence was measured on several days and the expected linear relationship between the cell pressure of the iodine cell and the iodine resonance frequency [17] was observed. Examples are shown in Fig. 5. The obtained resonance frequencies show the expected linear dependence from the vapor pressure. However, a day-to-day variation is observed, which is illustrated for the measurements at MSU (Fig. 5a) and TU Darmstadt (Fig. 5b). The stronger variation in the linear dependence seen at MSU might be caused by ambient temperature and pressure changes that slightly affect the wavemeter interferometers during the measurement periods which were several hours for each full pressure-shift measurement.
The numerical value of the (linear) pressure shift is given by the slope of the lines shown Fig. 5. To perform these fits for the MSU measurements, an uncertainty of \({0.5}\,^{\circ }\hbox {C}\) was assigned to the cold-finger temperature measured with a thermocouple and propagated to the calculated vapor pressure. The uncertainty of the centroid at each temperature was obtained from the statistical fit uncertainty of the respective absorption spectrum. The linear fit was performed by orthogonal distance regression as described in [19], taking into account x and y uncertainties. At TU Darmstadt, a cold-finger temperature uncertainty of \({1}\,^{\circ }\hbox {C}\) was estimated, based on a reference measurement where the cold-finger was replaced by an arduino temperature sensor and its reading compared to the readout from the temperature sensor of the controller. The difference between the sensors varied by typically 0.3–\({0.7}\,^{\circ }\hbox {C}\). The statistical uncertainty of each measured frequency was taken as the statistical fit uncertainty for the saturation spectroscopy and the FM spectroscopy, whereas the standard deviation of ten subsequent frequency measurements was used for the stabilization technique. Linear fitting with x and y uncertainties was performed with the linear fit algorithm by York et al. [20].
At TU Darmstadt, the pressure-shift measurement was repeated using each of the three spectroscopic methods described in Sects. 2.2–2.4. In Fig. 6a, pressure-shift results from the three different spectroscopic methods employed at TU Darmstadt are shown. An excellent linear relationship is obtained with the stabilization technique, while there is some scatter when using the methods that scan across the complete resonance. This different behavior is ascribed to temperature fluctuations during the longer time intervals required for performing the complete scan at each temperature setting as well as statistical fluctuations in the determination of the peak center in the fitting procedure.
The pressure shifts measured for all three lines at both locations on several days and with different techniques are summarized in Table 1. The average values of slopes as well as their respective weighted standard deviation are also included. All values are in a typical range of approximately \({6}\,\hbox {kHz}\,\hbox {Pa}^{-1}\), with the exception of the R(54)(3-9) a\(_1\) transition measured at the MSU cell, which is about twice as large but has also a comparatively large uncertainty. The pressure shift measured for this transition at TU Darmstadt is an order of magnitude more precise and lies within \(2\sigma\) of the MSU result. Moreover, the TU Darmstadt results are very similar to the shifts observed previously with the same cell for other lines [22].
3.2 Transition frequencies
To compare measurements made with different cells, the measured resonance frequencies are extrapolated to zero pressure, at which the resonance line is not affected anymore by collisions. This is necessary since the pressure shift depends on the composition of possible cell impurities.
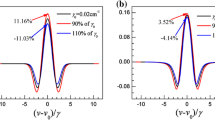
The exemplary pressure-shift measurements of the R(118)(2-8) a\(_{10}\) transition in Fig. 6a demonstrate, however, that despite the excellent linearity, the lines determined with the different techniques do not converge for \(p\rightarrow 0\). To investigate this closer, additional measurements with the simple saturation technique and the frequency-stabilization technique were repeated on several days for the P(48)(3-9) a\(_{15}\) and the R(54)(3-9) a\(_{1}\) hyperfine components, respectively. The resulting zero-pressure frequencies for all measurements were deduced and the results are shown in Fig. 6b. Each circle represents the extrapolation of a pressure-dependence measurement to the y-axis. Plotted is the difference between the individual measurement and the mean value of all measurements with all techniques for the respective transition. It is obvious that the measurements scatter much more than the estimated uncertainty from the fit or the standard deviation of a single measurement in the stabilization technique. The average and the standard deviation of the measurements created on several days are depicted by the squares and their error bars. The size of the error bar is very similar (252 and \({229}\,\hbox {kHz}\)) in both cases and represents the scatter of all measurements quite well. Moreover, there is no systematic deviation between the different techniques.

Results of the pressure calibration for the R(118)(2-8) a\(_{10}\) line comparing different spectroscopic methods are shown in a. The frequency-comb-related frequency measurements at TU Darmstadt obtained using the Doppler-free saturation spectroscopy, the FM-saturation spectroscopy and the frequency-stabilization technique are shown in b. Results for the different transitions are color coded. Filled circles represent individual measurements scattered over several days, while the squares represent their average and standard deviation. The y-axis shows the deviation of the individual value from the respective average across all three techniques that are obtained for each transition. All measurements in a and b are plotted as deviation from the average value obtained from averaging the results from the three methods (see text). The grey band represents the standard deviation of all individual measurements of all transitions
The final frequency for each transition listed in Table 2 is calculated as the average of the three frequencies obtained with the different techniques. Besides the statistical uncertainty, we have to include a systematic uncertainty due to possible red shifts of the transition caused by differences in the partial pressures of impurities in each cell. This has been estimated in [22] based on the comparison of two cells. For the measurements at TU Darmstadt, one of these cells (’GSI cell’) was used again to confirm that there is no significant aging during these years, we remeasured the R(114)(2-11) a\(_1\) hyperfine component and found the extrapolated frequency shifted roughly \({300}\,\hbox {kHz}\) to higher frequencies than in [22] but still within the uncertainties of both values. To be conservative, we indicate the difference between the two cells from [22] as an additional systematic uncertainty of the determined transition frequency in brackets in Table 2. Other sources of uncertainty such as the misalignment of the counter-propagating pump and probe beam, Zeemann effect, etc. are considered to be much smaller than these dominating uncertainties.
3.3 Evaluation of the wavemeter uncertainty
The resonance frequencies observed with the wavemeter at MSU were first determined from the extrapolation to zero pressure. The difference to the frequencies obtained with the frequency comb at TU Darmstadt, will then provide a correction offset for the wavemeter at the current laser frequency. Since a full measurement of the pressure dependence and the extrapolation to zero pressure is a time-consuming effort for a wavemeter calibration that has to be performed every day, a different approach was used: For each calibration measurement, the resonance frequency was measured with the wavemeter at a given cold-finger temperature. Then the previously measured slopes, listed in Table 1, were used to extrapolate to the zero-pressure frequency from which the wavemeter offset is extracted as the difference to the frequency-comb assisted iodine spectroscopy performed at TU Darmstadt.

The offset frequency of the wavemeter reading relative to that of the frequency comb was measured over several weeks. The offset moved in unison and \(\sim\) 2 MHz drift was observed
The obtained zero-pressure resonance frequencies are summarized in the second column of Table 2, where only statistical uncertainties are shown. The wavemeter used to measure the laser frequency is known to have a 10-MHz accuracy [14]. Approximately 11.5 MHz systematic offset was observed, which varies \(\pm 1\) MHz. The fluctuation is consistent with the observation for a similar wavemeter discussed in [11]. While the offsets for R(118)(2-8) a\(_{10}\) and R(54)(3-9) a\(_{1}\) were within \({100}\,\hbox {kHz}\) of each other, the offset for P(48)(3-9) a\(_{15}\) was approximately \({2}\,\hbox {MHz}\) smaller.
Understanding the stability and systematic trends of the wavemeter over an experimental period is critical for the reliability of the data and the data analysis. Repeated measurements of the iodine resonance frequencies were performed over the course of several weeks between June and August 2020 and the results are shown in Fig. 7. For each of these measurements the zero-pressure resonance frequency was determined as discussed above, and the offset between the observed resonance frequency and the resonance frequency measured at TU Darmstadt was obtained. The offsets moved in unison and showed a systematic trend of drifting over time. While the wavemeter has an uncertainty of 10 MHz, observations of the drift have shown that over several weeks the offset frequency did not drift more than 2 MHz, which is within the specifications of the frequency stabilized He:Ne laser used to calibrate the wavemeter readings (SL 03, SIOS Metechnik).
4 Hyperfine structure
Investigations of the rovibrational structure within the well-known electronic transition between the \(X ^{1}\Sigma _g^{+}\) ground state to the \(B ^{3}\Pi _{0u}^{+}\) excited state of molecular iodine have been well-documented over the last 50 years. Results have been reported in atlases [23,24,25,26,27], and spectra can be simulated using the IodineSpec5 program which is based on empirical model descriptions of the hyperfine and rovibrational structures [13, 28,29,30].
The hyperfine splitting of a rovibrational level of molecular iodine is described by the effective hyperfine Hamiltonian [31], \(H_\mathrm{hfs}\),
where \(H_\mathrm{EQ}\), \(H_\mathrm{SR}\), \(H_\mathrm{SSS}\) and \(H_\mathrm{TSS}\) correspond to the dominant contributions from the electric quadrupole, spin-rotation, scalar spin–spin, and tensor spin–spin interactions, respectively [32]. Four additional terms, arising from second order interactions may be included [31, 32] but are disregarded here due to the negligible contribution. The matrix elements of each term in the effective hyperfine Hamiltonian are the product of a geometrical factor and a hyperfine coupling constant. The geometrical factors are functions of the appropriate quantum numbers I, J, and F, which represent the total nuclear spin, molecular angular momentum, and total angular momentum of the electronic state respectively, for a specified state and are calculated with spherical tensor algebra to give expressions that are well-defined in literature [32, 33]. Algebraic expressions for the matrix elements of \(H_\mathrm{EQ}\), \(H_\mathrm{SR}\), \(H_\mathrm{SSS}\) and \(H_\mathrm{TSS}\) are provided in Eqs. (3–8) where eqQ(J,J'), C, A, and d are the electric quadrupole, spin-rotation, scalar spin-spin, and tensorial spin-spin interaction coupling constants [32], respectively. The coupling constants can be determined experimentally by fitting the hyperfine structure.
The energy of each hyperfine state in both the X and B electronic states are calculated by constructing a hyperfine Hamiltonian matrix for each electronic level. To construct each matrix, the number of hyperfine states was first determined using the quantum numbers I, J, and F. In an iodine molecule, two \(^{127}\)I nuclei each with spin \(I_1=\frac{5}{2}\) can couple to give a total nuclear spin, I of 0, 1, 2, 3, 4, or 5. The coupling of I and J gives the hyperfine quantum number, \(F = I + J\). Due to the nuclear statistics (Pauli principle) for even J levels in the ground electronic state, the value of I must also be even; therefore, I can take on the values 0, 2, or 4. Conversely, odd values of J can have I equal to 1, 3, or 5. The coupling of I and J results in 15 hyperfine states for even J and 21 hyperfine states for odd J in the ground electronic state [34].
The electric quadrupole interaction mixes \(\Delta J = \pm 2\) states in the same electronic level. As a consequence, the coupling to neighboring rotational states must be considered. The additional interactions that need to be included are 15 (21) states for \(J' = J+2\) and 15 (21) states for \(J' = J-2\), resulting in 45 (63) coupled hyperfine energy levels for even (odd) values of J [33]. A hyperfine Hamiltonian matrix, composed of 45 (63) states with unique F, I, and J values can be constructed for each upper and lower electronic levels of an iodine transition. To take into account the energy difference between different J states, differential rotational energies were added to states where \(J' = J \pm 2\), and the matrices were subsequently diagonalized. Specifically, differential changes in rotational energy given as
were added to the relevant \(\Delta J = \pm 2\) matrix elements. The rotational constants \(B_v\) and \(D_v\) used to determine the rotational energy are known [35], and uncertainties in these constants impact calculated splittings by less than 0.5 kHz [31]. Frequencies for the 15 (21) transitions were obtained from the difference of the corresponding energies from the two separate Hamiltonians after diagonalization. Higher-order contributions from \(\Delta J = \pm 4\) are not included here and would typically impact the deduced frequencies by a maximum of 100s of Hz [31] and are negligible compared with the precision required for nuclear structure studies.
In the present work, a Python application utilizing the NumPy [36] and SciPy [37] stacks was written to populate and diagonalize hyperfine Hamiltonians described above, as well as fit hyperfine spectra presented in Sect. 4.1. The performance of the application was validated against the results and matrix elements presented in Ref. [34].
4.1 Molecular iodine hyperfine spectrum
A saturated absorption signal was measured for the R(54)(3-9) hyperfine spectrum using the MSU setup, and the result is presented in Fig. 8. The temperature of the cold-finger in the iodine cell was 24.9 \(^{\circ }\)C, and the cell was set to a temperature of 700 \(^{\circ }\)C. The hyperfine spectrum was fitted using the Python application described in Sect. 4. A Lorentizan lineshape was used, and all 15 peaks in the spectrum shared a common width and the hyperfine coupling constants for the ground state were fixed to theoretical values [28, 29] during the fit. Fitting parameters included the center-of-gravity frequency, the common line width, four hyperfine coupling constants for the excited state. During the fitting routine, the hyperfine Hamlitonian was diagonalized for each minimization step. The centroid frequency, width, and hyperfine coupling constants are presented in Table 3. The first error reported in the table is the statistical error from the fitting routine, and the second error is the estimated systematic error from a potential non-linearity of wavemeter readings over the measurement range [11]. The reported center-of-gravity frequency of 425 632 361.2(1)[5] MHz is corrected for the 12.8(5) MHz offset described in Table 2; it was assumed that the offset was constant throughout the entire spectrum, and the uncertainty from this offset is included as a systematic error. The deduced values of the hyperfine coupling constants agree well with the modeled values.

5 Conclusion
A wavlength meter (WSU30, HighFinesse) at MSU was calibrated to a level of ± 1 MHz using a Doppler-free saturation laser spectroscopy system with molecular iodine in the regime of 700 nm. Hyperfine components of R(118)(2-8) a\(_{10}\), R(54)(3-9) a\(_{1}\) and P(48)(3-9) a\(_{15}\) near 700 nm were observed and the centroid frequencies measured by MSU’s wavemeter with 10-MHz accuracy were compared with the absolute transition frequencies determined using a frequency comb at TU Darmstadt. Approximately an 11-MHz offset was observed, which drifted ± 1 MHz over a three week time period. A Hamiltonian for the hyperfine interaction of molecular iodine was constructed to calculate energies of each hyperfine state and then transition frequencies of the hyperfine components. The R(54)(3-9) hyperfine spectrum was fitted using the obtained theoretical form of the hyperfine spectrum. The results allow for more confident use of the molecular iodine lines in the regime of 700 nm, where much fewer lines are precisely known than those in the visible regime, as a calibration tool for laser frequencies.
Notes
Hereafter all numbers are given in 1\(\sigma\).
References
E.W. Otten, Nuclear Radii and Moments of Unstable Isotopes, Treatise on Heavy Ion Science Vol. 8: Nuclei Far from Stability (Plenum Publishing Corp. (Springer), New York, 1989)
P. Campbell, I. Moore, M. Pearson, Prog. Part. Nucl. Phys. 86, 127 (2016)
R. Neugart, J. Billowes, M.L. Bissell, K. Blaum, B. Cheal, K.T. Flanagan, G. Neyens, W. Nörtershäuser, D.T. Yordanov, J. Phys. G Nucl. Part. Phys. 44, 064002 (2017)
K. Riisager, P. Butler, M. Huyse, R. Krücken, HIE-ISOLDE: The Scientific Opportunities, CERN Yellow Reports: Monographs (CERN, Geneva, 2007)
I. Moore, T. Eronen, D. Gorelov, J. Hakala, A. Jokinen, A. Kankainen, V. Kolhinen, J. Koponen, H. Penttilä, I. Pohjalainen, M. Reponen, J. Rissanen, A. Saastamoinen, S. Rinta-Antila, V. Sonnenschein, and J. Äystö (2013) Nucl. Inst. Methods Phys. Res. B. 317, 208 (2012)
T. Kubo, Nucl. Inst. Methods Phys. Res. B. 204, 97 (2003)
K. Minamisono, P. Mantica, A. Klose, S. Vinnikova, A. Schneider, B. Johnson, B. Barquest, Nucl. Instrum. Methods Phys. Res. Sect. A Accel. Spectrom. Detect. Assoc. Equip. 709, 85 (2013)
J. Wei, et. al, 'Progress Towards the Facility for Rare Isotope Beams' PAC2013, (Pasadena, CA, 2013
A. Gade, B.M. Sherrill, Phys. Scr. 91, 053003 (2016)
National Research Council, Nuclear Physics: Exploring the Heart of Matter (National Academies Press, Washington, DC, 2013)
K. König, P. Imgram, J. Krämer, B. Maaß, K. Mohr, T. Ratajczyk, F. Sommer, W. Nörtershäuser, Appl. Phys. B 126, 86 (2020)
H. Kato, M. Baba, S. Kasahara, K. Ishikawa, M. Misono, Y. Kimura, J. O’Reilly, H. Kuwano, T. Shimamoto, T. Shinano, C. Fujiwara, M. Ikeuchi, N. Fujita, M. H. Kabir, M. Ushino, R. Takahashi, Y. Matsunobu. Doppler-Free High Resolution Spectral Atlas of Iodine Molecule 15,000 to 19,000 cm−1 (Japan Society for the Promotion of Science, 2000)
H. Knöckel, B. Bodermann, E. Tiemann, Eur. Phys. J. D Atom. Mol. Optic. Plasma Phys. 28, 199 (2004)
HighFinesse, “Ws8-30”, https://www.highfinesse.com
J.L. Hall, L. Hollberg, T. Baer, H.G. Robinson, Appl. Phys. Lett. 39, 680 (1981)
W. Demtröder, Laser Spectroscopy: Vol. 2: Experimental Techniques, Laser Spectroscopy (Springer, Berlin, 2008)
H. Margenau, Phys. Rev. 40, 387 (1932)
L.J. Gillespie, L.H.D. Fraser, J. Am. Chem. Soc. 58, 2260 (1936)
C. Wu, J.Z. Yu, Atmos. Meas. Tech. 11, 1233 (2018)
D. York, N.M. Evensen, M.L. Martinez, Delgado J. De Basabe, Am. J. Phys. 72, 367 (2004)
A.H. James, J. Filliben, DATAPLOT Reference Manual (SED, ITL, NIST, Gaithersburg, 1993)
S. Reinhardt, B. Bernhardt, C. Geppert, R. Holzwarth, G. Huber, S. Karpuk, N. Miski-Oglu, W. Nörtershäuser, C. Novotny, T. Udem, Opt. Commun. 274, 354 (2007)
S. Gerstenkorn, P. Luc, Atlas Du Spectre d’absorption de La Molécule d’iode, 15,600–20,000 Cm\(^{-1}\) (Centre National de la Recherche Scientifique, Paris, 1977)
S. Gerstenkorn, P. Luc, Atlas Du Spectre d’absorption de La Molécule d’iode, 14,800–20,000 Cm\(^{-1}\) (Centre national de la recherche scientifique, Paris, 1978)
S. Gerstenkorn, P. Luc, Rev. Phys. Appl. (Paris) 14, 791 (1979)
S. Gerstenkorn, P. Luc, Atlas Du Spectre d’absorption de La Molécule d’iode, 14,000–15,600 Cm\(^{-1}\) (Centre national de la recherche scientifique, Paris, 1980)
S. Gerstenkorn, P. Luc, Atlas Du Spectre d’absorption de La Molécule d’iode, 11,000–14,000 Cm\(^{-1}\) (Centre national de la recherche scientifique, Paris, 1982)
B. Bodermann, H. Knöckel, E. Tiemann, Eur. Phys. J. D 19, 31 (2002)
E.J. Salumbides, K.S.E. Eikema, W. Ubachs, U. Hollenstein, H. Knöckel, E. Tiemann, Mol. Phys. 104, 2641 (2006)
E.J. Salumbides, K.S.E. Eikema, W. Ubachs, U. Hollenstein, H. Knöckel, E. Tiemann, Eur. Phys. J. D 47, 171 (2008)
C.J. Bordé, G. Camy, B. Decomps, J.-P. Descoubes, J. Vigué, Journal de Physique 42, 1393 (1981)
M. Broyer, J. Vigué, J. Lehmann, Journal de Physique 39, 591 (1978)
G. Hanes, J. Lapierre, P. Bunker, K. Shotton, J. Mol. Spectrosc. 39, 506 (1971)
S. Fredin-Picard, Bureau International des Poids et Mesures Rapport BIPM-90/5 (1990)
S. Gerstenkom, P. Luc, Journal de Physique 46, 867 (1985)
C.R. Harris, K.J. Millman, S.J. vander Walt, R. Gommers, P. Virtanen, D. Cournapeau, E. Wieser, J. Taylor, S. Berg, N.J. Smith, R. Kern, M. Picus, S. Hoyer, M.H. van Kerkwijk, M. Brett, A. Haldane, J. FernándezdelRío, M. Wiebe, P. Peterson, P. Gérard-Marchant, K. Sheppard, T. Reddy, W. Weckesser, H. Abbasi, C. Gohlke, T.E. Oliphant, Nature 585, 357–362 (2020)
P. Virtanen, R. Gommers, T.E. Oliphant, M. Haberland, T. Reddy, D. Cournapeau, E. Burovski, P. Peterson, W. Weckesser, J. Bright, S.J. van der Walt, M. Brett, J. Wilson, K.J. Millman, N. Mayorov, A.R.J. Nelson, E. Jones, R. Kern, E. Larson, C.J. Carey, I. Polat, Y. Feng, E.W. Moore, J. Vander Plas, D. Laxalde, J. Perktold, R. Cimrman, I. Henriksen, E.A. Quintero, C.R. Harris, A.M. Archibald, A.H. Ribeiro, F. Pedregosa, P. van Mulbregt, SciPy 1.0 contributors. Nat. Methods 17, 261 (2020)
Acknowledgements
This work was supported by the National Science Foundation (US) (Grant nos. PHY-15-65546, and PHY-19-13509), andDeutsche Forschungsgemeinschaft (DFG, German Research Foundation)—Project-ID 279384907—SFB 1245.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Powel, R., Koble, M., Palmes, J. et al. Improved wavelength meter calibration in near infrared region via Doppler-free spectroscopy of molecular iodine. Appl. Phys. B 127, 104 (2021). https://doi.org/10.1007/s00340-021-07650-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00340-021-07650-5