
Abstract
To study the effect of the conjugated structural configuration on the two-photon absorption (TPA) properties of V-shaped compounds, two 1,3,5-triazine-based compounds with the same electron donor (D) and acceptor (A) connected in a reverse-conjugated structural configuration (T02: D-\(\pi\)-A-\(\pi\)-D; R02: A-\(\pi\)-D-\(\pi\)-A) were systematically investigated using steady-state and transient absorption spectroscopy, open-aperture Z-scan measurements, and two-photon fluorescence measurements. The TPA cross-section of compound R02 connected in a A-\(\pi\)-D-\(\pi\)-A-conjugated structural configuration with triphenylamine as the central core was 203 GM, which showed a 2.3-fold enhancement compared with compound T02 connected in a reverse D-\(\pi\)-A-\(\pi\)-D-conjugated structural configuration (90 GM, with 1,3,5-triazine as the central core). This result indicates that the conjugated structural configuration plays an important role in the TPA properties. A two-color pump–probe experiment was used to investigate the effect of the conjugated structural configuration on the excited state and intra-molecular charge transfer (ICT) properties of these V-shaped compounds. The formation and relaxation lifetimes of the ICT state were determined. The results indicate that the electron-donating/accepting strength of the central group, which serves as a communal group for two D-\(\pi\)-A subunits, was confirmed to be a key role to the overall effect of the ICT for V-shaped compounds. These ultrafast dynamic results are in agreement with the TPA properties.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Two-photon absorption (TPA) materials have promising applications in various fields, such as optical limiting [1], stereo-lithography [2,3,4,5], optical information storage [6], photodynamic therapy [7], and two-photon patterning [8,9,10]. Some TPA compounds emit an intense fluorescence under two-photon excitation. Two-photon excited fluorescence (TPF) has several advantages compared with one-photon microscopy for imaging cells or tissues. The wavelength of the excited beam is in the near-infrared region. In this special spectral region, background fluorescence can be effectively avoided, and absorption of the biological sample is effectively reduced due to the transparent property, thus leading to deeper probing and less photodamage under the excitation of intense radiation. Moreover, the two-photon excitation is confined in a small focal volume on the order of ~ \({\lambda ^3}\) and provides 3D image resolution. Thus, TPF dyes are frequently employed as molecular probes in laser scanning fluorescence microscopy imaging or in the up-conversion lasing field [11,12,13].
However, for practical imaging applications, a large TPA cross-section is usually required [14, 15]. After the observation of large TPA cross-sections of some dyes, organic dyes with unique TPA properties attracted great attention, and many dipolar, quadrupolar, multi-branched, or macrocyclic molecules were synthesized [16,17,18,19,20,21]. Usually, a dipolar compound exhibits better solubility and are easy to dye due to the small molecular size and cross-section; however, the relative small TPA cross-section limits the application of the dipolar compound. Meanwhile, a multi-branched compound exhibits a larger TPA cross-section and a larger steric hindrance, which results in a relative worse dyeing property. A V-shaped compound exhibits both a relative larger TPA cross-section and nearly the same cross-section as the dipolar molecular compound, making it a unique candidate for TPF imaging applications.
To design and optimize new organic materials with even larger TPA cross-sections, a deep understanding of the excited state and the ultrafast responses of TPA compounds is very important. In previous works, we reported the nonlinear optical properties and ultrafast responses of two V-shaped compounds [22, 23]. These compounds exhibited both relative large TPA cross-sections and good fluorescent properties. In this paper, we present the ultrafast responses, fluorescence and TPA properties of two V-shaped compounds with the same electron donor (triphenylamine) and acceptor (1,3,5-triazine) connected in a reverse-conjugated structural configuration (T02: D-\(\pi\)-A-\(\pi\)-D; R02: A-\(\pi\)-D-\(\pi\)-A). The V-shaped compound, R02, with triphenylamine as the central core and two 1,3,5-triazine groups as the terminal end was found to have a larger TPA cross-section of 203 GM, showing a 2.3-fold enhancement compared with T02 connected in a reverse-conjugated configuration. The ultrafast response study indicated the effect of the charge transfer direction on the intramolecular charge transfer (ICT) and TPA properties of the TPA compounds. Our results provide insight into the relation between the ICT direction and the TPA property.
2 Materials and experiments
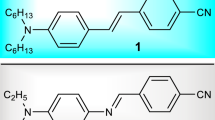
The synthesis of V-shaped compound T02 has been reported in detail [23]. The synthesis of R02 connected in reverse conjugated mode was synthesized as shown in Fig. 1. Bromination of 2,4,6-tri(p-tolyl)-1,3,5-triazine afforded 2,4,6-tris(4-(bromomethyl)phenyl)-1,3,5-triazine, followed by reaction with trimethyl phosphate, to yield the triazine derivative: dimethyl 4-(4,6-di(p-tolyl)-1,3,5-triazin-2-yl) benzylphosphonate (5). R02 was achieved by the condensation reaction of 4,4′-diformyl triphenylamine with 5. In a 100-mL round-bottom flask, 2,4,6-Tri(p-tolyl)-1,3,5-triazine (0.01 mol), NBS (0.03 mol) and BPO (1.2 mmol) were dissolved in 50 mL of chlorobenzene and heated at 110 °C for 7 h. The mixture was filtered, and the solvent was removed under vacuum. The residue was dissolved in trimethylphosphite (10 mL) and refluxed for 9 h. Excessive trimethyl phosphate was removed under vacuum. The residue was purified by column chromatography on silica to afford the product as a white power (5). In a 250-mL round-bottom flask, we added 5 (0.5 mmol), 4,4′-diformyl triphenylamine (0.2 mmol), potassium tert-butoxide (3.0 mmol), 18-crown-6 (0.08 mmol) and 100 mL of DCM under an argon atmosphere. After stirring at 45 °C for 4 h, the mixture was poured into distilled water and extracted with dichloromethane and water. The combined organic phases were dried over anhydrous MgSO4 and concentrated using a rotary evaporator. The final compounds were purified by column chromatography and recrystallization.

Structures of V-shaped compound T02 and synthetic routes to compound R02 connected in a reverse conjugation configuration
As shown in Fig. 1, the V-shaped compounds, T02 and R02, utilize 1,3,5-triazine as the electron acceptor, triphenylamine as the electron donor, and the styryl group as the conjugated bridge. The same electron donor and acceptor groups were connected in reverse-conjugated structural configuration for these two compounds (T02: D-\(\pi\)-A-\(\pi\)-D; R02: A-\(\pi\)-D-\(\pi\)-A). Different TPA properties were observed. In the experimental test, chloroform (CHCl3) was used without further distillation.
The UV–visible absorption spectra of the compounds in CHCl3 solutions were measured by a Hitachi spectrophotometer with a spectral resolution of 2 nm, and the fluorescence spectra were measured in dilute solutions (\({10^{ - 5}}\) M) using an Edinburgh FLS 920 spectrometer with a spectral resolution of 1 nm.
The TPA cross-sections were measured using an open-aperture Z-scan method [24]. The two-photon fluorescence (TPF) spectra were recorded using a TRISTAN light spectrometer. The excitation fluence in the TPF measurement was approximately 1–5 GW/cm2, and a high concentration ~ 0.01 mol/L was used for the TPF measurements. The ultrafast dynamic responses of these compounds were investigated via a femtosecond (fs) pump–probe experiment, as described in previous references [25, 26]. The fs pulses from the fs laser system (800 nm) were divided into two parts by a beam splitter. To efficiently pump the compounds into the excited state, a portion of the beam was frequency-doubled to 400 nm by a 0.5-mm-thick \(\beta\)-barium metaborate (BBO) crystal. The excitation power in transient absorption was approximately 0.7 GW/cm2. The other portion of the beam was focused on a 5-mm-thick cell with flowing water to generate a supercontinuum to sample the photo-induced excited state at various wavelengths. A monochromator after the sample cell selected the probe wavelength. To detect the dynamics without interference effects, the polarization of the pump beam was set perpendicular to that of the probe beam [27, 28]. The fs pulses were generated by the amplification stage of the fs laser system (Spitfire, Spectra-Physics). The average output power from the Spitfire was approximately 300 mW. The pulse duration was 140 fs, the wavelength was 800 nm, and the repetition rate was 1 kHz. All the measurements were performed at room temperature.
3 Results and discussion
3.1 Linear absorption and fluorescence spectra
Shown in Fig. 2 are the normalized linear absorption and one-photon fluorescence spectra of T02 and R02 in CHCl3 at a dilute concentration of approximately 10−5 M, and the obtained optical parameters are summarized in Table 1. For the linear absorption spectra, all compounds exhibited two major prominent bands. In some literatures, multi-branched triphenylamine derivatives of similar structure exhibited charge transfer state absorption peak shifts in more polar solvent [29, 30]. The absorption observed at 400–450 nm is due to the charge transfer, and the absorption bands located in the shorter wavelength region at approximately 300 nm are ascribed to the absorption of the triphenylamine group [31, 32]. The peak wavelength of the charge transfer (CT) band of dipolar compound T02 is located at 423 nm. The fluorescence maximum of V-shaped compound T02 is located at 553 nm. However, for R02 with the same electron donor and acceptor connected in reverse configuration, the peak wavelength of the CT band is located at 431 nm, showing an 8-nm red shift compared with T02. The redshifts may be due to an enhanced ICT effect, which possibly affected the TPA response. There was no apparent absorption observed at wavelengths longer than 500 nm for both V-shaped compounds. The fluorescence peak of R02 is located at 530 nm. The Stokes shift of R02 (99 nm) is shorter than that of T02 (130 nm) that is connected in the reverse conjugated mode. A similar effect was observed in our previous works [22, 33]: the Stokes shift may be related to the ICT of the compounds with similar structures. In the ultrafast response section, we will further discuss this point.

Normalized one-photon absorption spectra and fluorescence spectra of T02 and R02 dissolved in CHCl3
3.2 Two-photon fluorescence and two-photon absorption
All the two V-shaped compounds exhibited unique fluorescent properties. The fluorescence emission can be clearly seen by the naked eye even under excitation of unfocused fs laser pulses (800 nm) with a pulse energy of several microjoules, indicating high fluorescence quantum yields and large TPA cross-sections, which is a very important factor for TPF imaging or up-conversion lasing applications. The fluorescence spectra of these compounds excited by 800 nm fs pulses of various powers were measured by a spectrometer and are shown in Fig. 3a. The spectral shape is independent of the excitation light power. The fluorescence peaks of T02 and R02 are located at 577 nm and 536 nm, respectively. Shown in Fig. 3b is the linear dependence of the fluorescence on the square of the pump intensity, which confirms that the fluorescence is generated by a TPA process [34]. The peak wavelengths of the TPF exhibit redshifts of approximately 6–28 nm, which are possibly due to the reabsorption effect caused by a much higher concentration in the TPF measurement [28]. The fluorescence quantum yields of T02 and R02 were 0.47 and 0.48, respectively [23]. The TPA cross-section is an important parameter to quantify the TPA/TPF abilities of TPA compounds. The TPA cross-sections of T02 and R02 were measured by an open-aperture Z-scan technique at fs pulses at 800 nm, which is an optimum wavelength for TPF imaging applications [35]. The TPA cross-section of V-shaped compound T02 with the D-\(\pi\)-A-\(\pi\)-D structure was 90 GM (1 GM=\(1 \times {10^{ - 50}}{\text{c}}{{\text{m}}^4}\,{\text{s}}\,{\text{photo}}{{\text{n}}^{ - 1}}\)). However, for V-shaped compound R02 with the same electron donor/acceptor connected in a reverse conjugated A-\(\pi\)-D-\(\pi\)-A mode structure, the TPA cross-section was 203 GM, which was a 2.3-fold enhancement compared with T02. The nonlinear optical results indicate the conjugated structure and the direction of ICT are important parameters to enhance the TPA responses. The TPA cross-section of R02 is comparable with other V-shaped compounds [22, 36, 37]. In the following section, we will further discuss the enhancement mechanism.

a TPF intensities of compound R02 under different excitation power densities. b Normalized TPF intensities of T02 and R02 dissolved in CHCl3. c The TPF intensity versus the square of the excitation power density
3.3 Ultrafast excited state dynamics
The transient absorption spectra of T02 and R02 dissolved in CHCl3 solutions were measured using a two-color pump–probe experiment with a perpendicular polarization configuration. The transient absorption spectra of the V-shaped D-\(\pi\)-A-\(\pi\)-D compound, T02, are shown in Fig. 4a, and the dynamic traces probed at representative probe wavelengths are shown in Fig. 5a. At an initial delay time of 0 ps, the entire region shows photo-induced absorption, except for a weak positive signal region located at ~ 500 nm. At a relative long delay time (> 5 ps), the positive region disappears, and the entire region from 400 to 800 nm shows photo-induced absorption. The observed photo-induced absorption regions are attributed to the excited state absorption, and the positive region is attributed to the stimulated emission of the fluorescence emission state or due to the absorption cross-section difference between the excited and ground states [38, 39]. For R02 with a reverse-conjugated structure, the intensity of the stimulated emission signal is relatively stronger, and the wavelength region is much wider for compound T02. The result is in agreement with our previous work [22]. The transient dynamic signal will be affected by the excited absorption and the stimulated emission, and thus, it is affected by the fluorescence quantum yield. In our previous work, V-shaped compound P-2 exhibited a weaker stimulated emission signal mainly due to the relatively lower fluorescence quantum yield. Compounds T02 and R02 with similar V-shaped-conjugated structures also exhibit relatively weaker stimulated emission bands. However, the wavelength region of R02 with the reverse conjugated structure exhibits a wider stimulated emission band. As shown by the linear absorption spectra (Fig. 2), the compound R02 exhibits a much wider overlapping band between the linear absorption and the fluorescence spectra, which results in a wider stimulated emission band. A 70-nm blue-shifted transient absorption band for the T02 compound was observed within the first 20 ps. The blue shift of excited state absorption peaks could arise from vibrational relaxation, solvation or stimulated fluorescence emission [40]. The dynamic traces were fitted by a combination of two exponential decay processes:

Femtosecond transient absorption spectra of compounds T02 and R02 in CHCl3 solutions with a delay time range of 0–90 ps

Dynamic decay traces for compounds a T02 and b R02 at representative probe wavelengths
where A1 and A2 are the contributions and t1 and t2 are the lifetimes of the two corresponding components, and C0 is a constant. The fitting time constants are listed in Table 2. At a probe wavelength of 400 nm, the V-shaped D-\(\pi\)-A-\(\pi\)-D T02 exhibits a long photo-induced absorption process of > 300 ps. When probed at a wavelength of 500 nm, a new positive signal that is approximately 8 ps was observed along with a long photo-induced absorption process with a lifetime of > 300 ps. When the probe wavelength was tuned from 425 to 650 nm, the dynamic curves changed gradually, indicating a competition between processes with different photo-induced absorption or stimulated emission properties. When the probe wavelength was tuned to > 650 nm, the dynamic curve exhibited a fast photo-induced absorption process of approximately 7–8 ps along with a long decay process of > 300 ps. The fitting time constants of the dynamics are also listed in Table 2. The fast process of approximately 7–8 ps is mainly attributed to the internal conversion from the excited state to the ICT state, and the long decay process is ascribed to the evolution of the ICT state.
The V-shaped compound R02 with the same electron donor/acceptor connected in a reverse-conjugated structural A-\(\pi\)-D-\(\pi\)-A mode exhibited a 2.3-fold enhancement compared with T02. To investigate the excited state and the ultrafast response mechanism, transient absorption spectra measurements were also performed. The corresponding evolution of the transient absorption spectra and the dynamic curves probed at representative wavelengths are shown in Figs. 4b and 5b. At an initial delay time of 0 ps, the broad region of 400–800 nm exhibits photo-induced absorption, except for a positive signal region located at 425–520 nm. The absorption region can be ascribed to the excited state absorption; however, the positive signal region may be caused by the stimulated emission from the ICT state or due to the absorption cross-section difference between the excited and ground states. However, the stimulated emission region is much wider than that of T02, and the region is also well coincident with the overlap of the absorption and fluorescence spectra. At delay times of > 5 ps, the entire wavelength region exhibits only photo-induced absorption.
For these two V-shaped compounds, the photo-induced absorption bands in the range of 550–750 nm are spectrally structured with peaks at approximately 710 and 590 nm. In addition to the signal intensities, the relaxation dynamics are very important. Figure 5b shows the dynamic traces of R02 probed at representative probe wavelengths. At a probe wavelength of 400 nm, the dynamic curve only exhibits a long photo-induced absorption process, which indicates that the absorption area of the excited state is smaller than that of the ground state. When a probe wavelength was tuned to 450 nm, the dynamics first only show a fast positive signal process for approximately 6 ps. When the probe wavelength is tuned from 451 to 600 nm, the decay curves gradually change. The fast process properties change from positive to photo-induced absorption, and after the long photo-induced absorption process of > 300 ps was observed, the intensity of the long process was also enhanced. When the probe wavelength was tuned to > 650 nm, the dynamic curve exhibited a fast photo-induced absorption process of approximately 6–7 ps along with a long decay process of > 300 ps. The dynamic trace located at 590 nm, which is one of the peak wavelengths of the transient photo-induced absorption band, was also fitted, and the obtained optical constants are listed in Table 2. The dynamic curve exhibits a fast process of approximately 6.7 ps and a long decay process of > 300 ps. A similar dynamic trace was also observed for compound T02. The fast process was fitted to be approximately 4.0 ps, which is shorter than that of R02. When the wavelength is in the range of the fluorescence emission, the dynamic trace will also be affected by the fluorescence emission. A wavelength of 750 nm is distant from the linear absorption and fluorescence emission regions of these V-shaped compounds and was chosen as a specific probe wavelength to directly compare the time constants of the two compounds. The dynamic curve probed at 750 nm for R02 showed a fast photo-induced absorption process (6.4 ps) and a long photo-induced absorption process (> 300 ps). The fast process is ascribed to the formation of the ICT state, and the subsequently long decay process can be ascribed to the evolution of the ICT state. The formation of the ICT state is a competition between the electron–phonon interaction and the internal conversion processes, and the lifetimes of some processes can be modulated by the molecular structure or surface recombination [41]. The formation of the ICT state is usually related to the electron transmission in the compound, which is an important factor that is closely related to the TPA response [42]. The ICT state formation time of V-shaped A-\(\pi\)-D-\(\pi\)-A compound R02 (6.4 ps) was notably shorter than that of T02 (8.1 ps), which was connected in a reverse D-\(\pi\)-A-\(\pi\)-D-conjugated structural configuration. Usually, a stronger electron-donating/accepting group will improve the ICT and enhance the TPA property. Besides the electron transmission efficiency of the compound under excited state, the overall ICT is another important factor that limits the TPA property. To cause an overall ICT of the compound to be as large as possible, the conjugated structure and the charge transfer direction of the V-shaped compound is very important. Because the central core serves as a communal group for two D-\(\pi\)-A subunits, the electron-donating/accepting strength of the central group should play a key role in the overall effect of the ICT. The triphenylamine group has a strong electron-donating ability, which can guarantee an effective ICT in-between two D-\(\pi\)-A subunits. However, for T02 connected in a reverse structural configuration, the electron accepting ability of the 1,3,5-triazine group is very limited and not strong enough to guarantee effective ICT in-between the two D-\(\pi\)-A subunits. Therefore, the TPA cross-section of A-\(\pi\)-D-\(\pi\)-A compound R02 with the triphenylamine as the central core is notably larger (2.3-fold) than that of T02, which is connected in a reverse D-\(\pi\)-A-\(\pi\)-D-conjugated structural configuration. Moreover, the long processes of T02 and R02 are > 300 ps. The evolution of the ICT state is always correlated with the fluorescence emission, the longer lifetime of the ICT state, and the better two-photon fluorescence property. Both T02 and R02 exhibit strong Stokes shifts of > 90 nm (130 and 99 nm for T02 and R02, respectively), and generally, the stronger ICT, the larger Stokes shift [20]. In general, many mechanisms, such as ICT, excited state intramolecular proton transfer (ESIPT) and fluorescence resonance energy transfer (FRET), may cause large Stokes shifts [43,44,45,46]. ICT usually occurs in excited states of an electron donor–acceptor compound in which a fraction of the electronic charge is transferred between the molecular entities. Because of the loss of excitation energy by the ICT during the excitation and emission cycle, fluorescence with ICT always results in large Stokes shifts in emission. One of the ICT processes is a twisted intramolecular charge transfer (TICT). TICT is an electron transfer process that occurs upon photoexcitation in molecules with intramolecular twisting between the electron donor and acceptor [47]. The TICT state usually emits dual fluorescence in emission spectra and results in large Stokes shifts. Another kind of ICT process is planar intramolecular charge transfer (PICT). With a rigid planar donor/acceptor molecule, efficient ICT occurs without large amplitude increases in the PICT [48]. This means that fast and efficient ICT is possible in planar donor/acceptor molecules. However, PICT typically has small Stokes shifts due to the very rigid ground and excited state molecular structures [49]. The third kind of ICT process is planarized intramolecular charge transfer (PLICT) [50]. With the aid of two donor portions, PLICT compounds are completely twisted in the ground states and are planar in the excited states. For a large ICT effect in the V-shaped structure, both T02 and R02 exhibit large Stokes shifts. There are two D-\(\pi\)-A subunits linked to the terminal end of the central triphenylamine due to the larger steric hindrance and the more obvious restrictive effect by the two subunits. The rigid planar property of R02 is much better than that of T02, and the PICT process mainly dominates the ICT process of R02. Thus, the Stokes shift may be affected by the geometry relaxation of the excited state, and similar phenomenon was observed by Zheng et al. [51]. An energy level diagram in Fig. 6 shows the excited state decay dynamics of the investigated compounds. The ultrafast dynamic results are in good agreement with the TPA and TPF results.

Energy level diagram showing the excited state decay dynamics of the investigated compounds
4 Conclusions
In this work, we presented the TPA properties and ultrafast responses of V-shaped compounds that connect the same electron donor (triphenylamine) and acceptor (1,3,5-triazine) in a reverse configuration: T02 (D-\(\pi\)-A-\(\pi\)-D) and R02 (A-\(\pi\)-D-\(\pi\)-A). V-shaped A-\(\pi\)-D-\(\pi\)-A compound R02 exhibited a notably larger TPA cross-section (203 GM, 2.3-fold enhancement) relative to T02, which was connected in a reverse D-\(\pi\)-A-\(\pi\)-D configuration, indicating that the direction of the ICT is closely related to the TPA response. The electron-donating/accepting ability of the central core that serves as a communal group for two D-\(\pi\)-A subunits was confirmed to play a key role in the overall effect of the ICT. The results can assist the optimization of new TPA compounds with improved TPA responses. Our study also suggests that V-shaped compounds have potential applications in biological TPF imaging or in the up-conversion lasing field.
References
G.S. He, G.C. Xu, P.N. Prasad, B.A. Reinhardt, J.C. Bhatt, A.G. Dillard, 2-photon absorption and optical-limiting properties of novel organic-compounds. Opt. Lett. 20, 435–437 (1995)
S. Kawata, H.B. Sun, T. Tanaka, K. Takada, Finer features for functional microdevices—micromachines can be created with higher resolution using two-photon absorption. Nature 412, 697–698 (2001)
S. Shukla, E.P. Furlani, X. Vidal, M.T. Swihart, P.N. Prasad, Two-photon lithography of sub-wavelength metallic structures in a polymer matrix. Adv. Mater. 22, 3695–3699 (2010)
J. Fischer, M. Wegener, Three-dimensional optical laser lithography beyond the diffraction limit. Laser Photonics Rev. 7, 22–44 (2013)
R.P. Chaudhary, A. Jaiswal, G. Ummethala, S.R. Hawal, S. Saxena, S. Shukla, Sub-wavelength lithography of complex 2D and 3D nanostructures without two-photon dyes. Addit. Manuf. 16, 30–34 (2017)
B.H. Cumpston, S.P. Ananthavel, S. Barlow, D.L. Dyer, J.E. Ehrlich, L.L. Erskine, A.A. Heikal, S.M. Kuebler, I.-Y.S. Lee, D. McCord-Maughon, J.G. Qin, H. Röckel, M. Rumi, X.-L. Wu, S.R. Marder, J.W. Perry, Two-photon polymerization initiators for three dimensional optical data storage and microfabrication. Nature 398, 51–54 (1999)
J.D. Bhawalkar, N.D. Kumar, C.F. Zhao, P.N. Prasad, Two-photon photodynamic therapy. J. Clin. Laser Med. Surg. 15, 201–204 (1997)
S. Shukla, X. Vidal, E.P. Furlani, M.T. Swihart, K.T. Kim, Y.K. Yoon, A. Urbas, P.N. Prasad, Subwavelength direct laser patterning of conductive gold nanostructures by simultaneous photopolymerization and photoreduction. ACS Nano 5, 1947–1957 (2011)
R.P. Chaudhary, G. Ummethala, A. Jaiswal, S. Hawal, S. Saxena, S. Shukla, One-step subwavelength patterning of plasmonic gratings in metal-polymer composites. RSC Adv. 6, 113457–113462 (2016)
G. Ummethala, A. Jaiswal, R.P. Chaudhary, S. Hawal, S. Saxena, S. Shukla, Localized polymerization using single photon photoinitiators in two photon process for fabricating subwavelength structures. Polymer 117, 364–369 (2017)
W. Denk, J. Strickler, W. Webb, 2-Photon laser scanning fluorescence microscopy. Science 248, 73–76 (1990)
J.D. Bhawalkar, G.S. He, C.K. Park, C.F. Zhao, G. Ruland, P.N. Prasad, Efficient, two-photon pumped green upconverted cavity lasing in a new dye. Opt. Comm. 124, 33–37 (1996)
G.S. He, L.X. Yuan, Y.P. Cui, M. Li, P.N. Prasad, Studies of two-photon pumped frequency-upconverted lasing properties of a new dye material. J. Appl. Phys. 81, 2529–2537 (1997)
G.S. He, L.S. Tan, Q.D. Zheng, P.N. Prasad, Multiphoton absorbing materials: molecular designs, characterizations, and applications. Chem. Rev. 108, 1245–1330 (2008)
Y.C. Wang, Y.L. Yan, B. Li, S.X. Qian, Recent progress on two-photon absorbing organic materials. Prog. Phys. 32, 135–164 (2012)
H.B. Xiao, C. Mei, N. Ding, T.T. Wei, Y.Z. Zhang, B. Li, Synthesis and photophysical properties of a novel pyridinium salt based on dipicolinate. J. Photochem. Photobiol. A Chem. 273, 29–33 (2014)
X. He, Y.Q. Liu, X. Du, Y.Q. Yang, B. Xu, W.J. Tian, Y.G. Ma, Excited-state relaxation processes of DPA-DSB: Investigation of the reason for high fluorescence quantum yield of symmetric D–D molecule. Chem. Phys. Lett. 501, 296–299 (2011)
C.K.R. Namboodiri, S.R. Bongu, P.B. Bisht, R. Mukkamala, B. Chandra, I.S. Aidhen, T.J. Kelly, J.T. Costello, Enhanced two photon absorption cross-section and optical nonlinearity of a quasi-octupolar molecule. J. Photochem. Photobiol. A Chem. 314, 60–65 (2016)
Y.C. Wang, Y.H. Jiang, D.J. Liu, Y.Z. Wang, G.Q. Wang, J.L. Hua, Ultrafast relaxation processes of multi-branched compounds based on 1,3,5-triazine: an investigation of the causes of a high fluorescence quantum yield after modification with perfluoroalkyl chains. J. Lumn. 190, 89–93 (2017)
G. Bhaskar, Z.K. Ramakrishna, R. Lu, J.M. Twieg, D.J. Hales, E.V. Hagan, T. Stryland, Goodson, III, Investigation of two-photon absorption properties in branched alkene and alkyne chromophores. J. Am. Chem. Soc. 128, 11840–11849 (2006)
M. Williams-Harry, A. Bhaskar, G. Ramakrishna, T. Goodson, I.I.I.M. Imamura, A. Mawatari, K. Nakao, H. Enozawa, T. Nishinaga, M. Iyoda, Giant thienylene-acetylene-ethylene macrocycles with large two-photon absorption cross-section and semishape-persistence. J. Am. Chem. Soc. 130, 3252–3253 (2008)
Y.C. Wang, S.Y. Liu, D.J. Liu, G.Q. Wang, H.B. Xiao, Ultrafast responses of dipolar and V-shaped dipicolinate derivatives with potential applications in the labeling of biomolecules. AIP Adv. 6, 025016 (2016)
F.S. Meng, B. Li, S.X. Qian, K.C. Chen, H. Tian, Enhanced two-photon properties of tri-branched styryl derivatives based on 1,3,5-triazine. Chem. Lett. 33, 470–471 (2004)
M. Sheik-Bahae, A. Said, T. Wei, D. Hagan, V. Stryland, Sensitive measurement of optical nonlinearities using single beam. IEEE J. Quantum Electron. 26, 760–769 (1990)
J. Mi, L. Guo, Y. Liu, W. Liu, G. You, S. Qian, Excited-state dynamics of magnesium phthalocyanine thin film. Phys. Lett. A 310, 486–492 (2003)
Y. Wang, Y. Jiang, J. Hua, H. Tian, S. Qian, Optical limiting properties and ultrafast dynamics of six-branched styryl derivatives based on 1,3,5-triazine. J. Appl. Phys. 110, 033518 (2011)
J. Mi, B. Li, R. Zhu, W. Liu, S. Qian, F. Meng, H. Tian, Femtosecond response from two copolymers with intense two-photon absorption. Appl. Phys. B Lasers Opt. 80, 541–545 (2005)
J.F. Ge, Y.T. Lu, Q.F. Xu, W. Liu, N.J. Li, R. Sun, Y.L. Song, J.M. Lu, Third-order nonlinear optical properties of a new type of D–π–D unsymmetrical phenoxazinium chloride with resonance structures. Chem. Phys. 382, 74–79 (2011)
B. Li, R. Tong, R.Y. Zhu, F.S. Meng, H. Tian, S.X. Qian, The ultrafast dynamics and nonlinear optical properties of tribranched styryl derivatives based on 1,3,5-triazine. J. Phys. Chem. B 109, 10705–10710 (2005)
H.B. Xiao, C. Mei, Y.C. Wang, H. Li, S.X. Qian, H.Y. Yin, Z.S. Xu, Novel triphenylamine-cored two-photon absorbing dyes for labeling of biomolecules. Mater. Chem. Phys. 130, 897–902 (2011)
H.J. Xia, J.T. He, P. Peng, Y.H. Zhou, Y.W. Li, W.J. Tian, Synthesis and photophysical properties of triphenylamine-based dendrimers with 1,3,5-triphenylbenzene cores. Tetrahedron Lett. 48, 5877–5881 (2007)
H.J. Lee, J. Sohn, J. Hwang, S.Y. Park, H. Choi, M. Cha, Triphenylamine-cored bifunctional organic molecules for two-photon absorption and photorefraction. Chem. Mater. 16, 456–465 (2004)
Y.C. Wang, Y.H. Jiang, Y.Z. Wang, G.Q. Wang, D.J. Liu, J.L. Hua, Ultrafast responses of multi-branched compounds based on 1,3,5-triazine: investigation of the reason for enhanced two-photon absorption property. Appl. Phys. A 123, 516 (2017)
Y.C. Wang, Y.L. Yan, D.J. Liu, G.Q. Wang, S.Z. Pu, Photochromism induced nonlinear optical absorption enhancement and ultrafast responses of several dithienylethene compounds. J. Appl. Phys. 118, 183104 (2015)
Y.C. Wang, D.K. Zhang, H. Zhou, J.L. Ding, Q. Chen, Y. Xiao, S.X. Qian, Nonlinear optical properties and ultrafast dynamics of three novel boradiazaindacene derivatives. J. Appl. Phys. 108, 033520 (2010)
Y. Wang, G.S. He, P.N. Prasad, T. Goodson, III, Ultrafast dynamics in multibranched structures with enhanced two-photon absorption. J. Am. Chem. Soc. 127, 10128–10129 (2005)
S.J. Chung, K.S. Kim, T.C. Lin, G.S. He, J. Swiatkiewicz, P.N. Prasad, Cooperative enhancement of two-photon absorption in multi-branched structures. J. Phys. Chem. B 103, 10741–10745 (1999)
G. Ramakrishna, T. Goodson III, Excited-state deactivation of branched two-photon absorbing chromophores: a femtosecond transient absorption investigation. J. Phys. Chem. A 111, 993–1000 (2007)
Y. Wang, S. Yin, J. Liu, L. Yao, G. Wang, D. Liu, B. Jing, L. Cheng, H. Zhong, X. Shi, Q. Fang, S. Qian, Probing ultrafast excited state dynamics and nonlinear absorption properties of three star-shaped conjugated oligomers with 1,3,5-triazine core. RSC Adv. 4, 10960–10967 (2014)
M. Zhou, S. Long, X. Wan, Y. Li, Y.L. Niu, Q.J. Guo, Q.M. Wang, A.D. Xia, Ultrafast relaxation dynamics of phosphine-protected, rod-shaped Au20 clusters: interplay between solvation and surface trapping. Phys. Chem. Chem. Phys. 16, 18288 (2014)
H.J. Yan, B.L. An, Z.F. Fan, X.Y. Zhu, X. Lin, Z.M. Jin, G.H. Ma, Ultrafast terahertz probe of photoexcited free charge carriers in organometal CH3NH3PbI3 perovskite thin film. Appl. Phys. A 122, 414 (2016)
Y.L. Yan, B. Li, K.J. Liu, Z.W. Dong, X.M. Wang, S.X. Qian, Enhanced two-photon absorption and ultrafast dynamics of a new multibranched chromophore with a dibenzothiophene core. J. Phys. Chem. A 111, 4188–4194 (2007)
K.B. Eisenthal, Intermolecular and intramolecular excited state charge transfer. Laser Chem. 3 145–162 (1983)
S. Park, S. Kim, J. Seo, S. Park, Application of excited-state intramolecular proton transfer (ESIPT) principle to functional polymeric materials. Macromol. Res. 16, 385–395 (2008)
R.B. Sekar, A. Periasamy, Fluorescence resonance energy transfer (FRET) microscopy imaging of live cell protein localizations. J. Cell Biol. 160, 629–633 (2003)
Z.Y. Zhang, G.Q. Zhang, J.X. Wang, S.S. Sun, Z.Z. Zhang, The mechanisms of large stokes shift and fluorescence quantum yields in aniline substituted rhodamine analogue: TICT and PICT. Comput. Theor. Chem. 1095, 44–53 (2016)
J. Su, T. Xu, K. Chen, H. Tian, Electroluminescence properties of twisted dyad 1,8-naphthalic anhydride derivatives. Synth. Met. 91, 249–251 (1997)
T. Yoshihara, S.I. Druzhinin, K.A. Zachariasse, Fast intramolecular charge transfer with a planar rigidized electron/acceptor molecule. J. Am. Chem. Soc. 126, 8535–8539 (2004)
X. Liu, B. Cho., L.-Y. Chan, W.L. Kwan, C.-L.K. Lee, Development of asymmetrical near infrared squaraines with large stokes shift. RSC Adv. 5, 106868–106876 (2015)
G. Haberhauer, R. Gleiter, C. Burkhart, Planarized intramolecular charge transfer: a concept for fluorophores with both large stokes shifts and high fluorescence quantum yields. Chem. A Eur. J. 22, 971–978 (2016)
Q.D. Zheng, S.K. Gupta, G.S. He, L.-S. Tan, P.N. Prasad, Synthesis, characterization, two-photon absorption, and optical limiting properties of ladder-type oligo-p-phenylene-cored chromophores. Adv. Funct. Mater. 18, 2770–2779 (2008)
Acknowledgements
We sincerely appreciate the financial support of the National Natural Science Foundation of China (11404048, 11604038, and 11375034), the Liaoning Provincial Natural Science Foundation of China (201602061 and 201602062), the Program for Liaoning Educational Committee (L2015071), and the Fundamental Research Funds for the Central Universities (3132018236 and 3132018235).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Wang, Y., Jiang, Y., Liu, D. et al. Ultrafast responses of two V-shaped compounds with a reverse conjugated structural configuration: an investigation of the reason for the enhanced two-photon absorption cross-section. Appl. Phys. B 124, 98 (2018). https://doi.org/10.1007/s00340-018-6972-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00340-018-6972-3