
Abstract
A correction for the undesirable effects of direct and indirect cross-interference from water vapour on ammonia (NH3) measurements was developed using an optical laser sensor based on cavity ring-down spectroscopy. This correction relied on new measurements of the collisional broadening due to water vapour of two NH3 spectral lines in the near infra-red (6548.6 and 6548.8 cm−1), and on the development of novel stable primary standard gas mixtures (PSMs) of ammonia prepared by gravimetry in passivated gas cylinders at 100 μmol mol−1. The PSMs were diluted dynamically to provide calibration mixtures of dry and humidified ammonia atmospheres of known composition in the nmol mol−1 range and were employed as part of establishing a metrological traceability chain to improve the reliability and accuracy of ambient ammonia measurements. The successful implementation of this correction will allow the extension of this rapid on-line spectroscopic technique to exposure chamber validation tests under controlled conditions and ambient monitoring in the field.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Over the last century modern intensive farming practices, the increased use of nitrogen-based fertilisers, and certain industrial processes are believed to be responsible for increases in the ambient amount fraction of ammonia (NH3) found in Europe [1, 2]. Emission and deposition of NH3 both contribute to eutrophication and acidification of land and freshwater and may lead to a loss of biodiversity and undesirable changes to the ecosystem. Ammonia also affects the long-range transportation of acidic pollutants such as sulphur dioxide and the oxides of nitrogen and contributes to the production of secondary particulate matter (PM).
In the European Union (EU), NH3 emissions are regulated by legislation on national emissions ceilings [3–5], which also set emission targets for individual member states. Abatement of NH3 emissions in the pig and poultry industry is covered in the EU by Integrated Pollution Prevention and Control (IPPC), and in some European countries, including the UK, there are dedicated NH3 monitoring networks. In Germany, the Federal Immission Control Act [6] provides guidance and technical instructions on air quality control and recommends that at any assessment point the concentration of ammonia should not exceed 10 µg m−3 (equivalent to approximately 14 nmol mol−1 at ground level), thereby limiting damage to plants and ecosystems. Controlling ammonia is also important for reducing particle emissions of PM2.5 and PM10. A recent study [7] employing three chemical transport models found an underestimation of the formation of ammonium particles and concluded that the role of NH3 on PM is larger than originally thought.
Monitoring ammonia, however, poses a number of challenges: there is a lack of regulation on which analytical techniques to employ, the required uncertainties for the measurements, the establishment of agreed quality control and quality assurance (QC/QA) procedures, or the implementation of a suitable traceability infrastructure to underpin the measurements. Measurements of NH3 are often carried out using low-cost diffusive samplers and active sampling with denuders [8]. Despite currently being considered an “unofficial” reference method, denuders suffer from a number of limitations: not only do they not provide rapid measurements in real time, but they also require complex post-exposure analysis by wet chemical techniques, must be deployed over extended periods to achieve adequate sensitivity and suffer from low accuracy. These devices deliver only average amount fraction data, and their validation by traceable methods is not presently extensive.
In recent years, a number of spectroscopic techniques have been developed to measure trace gases in the atmosphere. These rapid on-line methods, such as cavity ring-down spectroscopy (CRDS) [9, 10], have the potential to overcome the limitation of the techniques currently used in the field. Ammonia sensors based on CRDS [11] and on multiple-pass cells with quantum-cascade lasers (QCL) [12–14] have been reported in the literature; however, before these sensors can be deployed routinely in the field, their potential drawbacks (such as spectral cross-interference and effects of collisional broadening on the NH3 lines of interest) must be addressed. One of the goals of this work is to extend the CRDS technology to enable more accurate measurements of ambient NH3 where the sampled atmosphere contains many species over a wide range of concentrations and is also humid. Water vapour influences the measurements through the presence of absorption features close to those of ammonia and through differences in matrix broadening effects. Conventional moisture removal devices such as Nafion™ dryers are known to introduce biases in the measurements as they also remove ammonia from the sampled air and are therefore not a practical sampling option. In this paper, we describe the development of a correction for the effects of water vapour and its application to ammonia measurements using a CRDS instrument. The correction is based on a new determination of the collisional broadening of the ammonia absorption lines by H2O, which are compared with those reported previously by Schilt [15], Owen et al. [16] and Sur et al. [17]. Crucially, the correction derived in the present study is underpinned by the establishment of metrological traceability of the measurements through the development, at NPL, of new stable ammonia primary standard gas mixtures (PSMs) prepared by gravimetry. This represents a step forward in the application of spectroscopic measurements and gas standard development towards improved quantification of ambient ammonia.
2 Experimental method
2.1 Description of the standard cavity ring-down spectrometer
At the heart of the apparatus is a high finesse optical cavity that can be brought into resonance with a laser light source, allowing an intra-cavity optical field to build up [9, 10]. The light source is then abruptly turned off and the intra-cavity power decays exponentially (“rings down”). The decay time constant, τ, is measured by monitoring the intensity of the light that leaks through one of the cavity mirrors. The total cavity loss, L, is related to the ring-down time constant and the round-trip intra-cavity optical path, ℓ, by the expression:
where c is the speed of light. If a gas sample containing absorbing molecules is introduced into the cavity, the molecular absorption increases the total cavity loss by the amount αℓ, where α is the molecular absorption coefficient at the frequency that is resonant with the cavity (assuming linear absorption, which is valid for the conditions of the measurements described here, and also assuming αℓ ≪ 1). Sequential ring-down measurements at different optical frequencies generate a spectrogram of cavity loss as a function of frequency, and subtracting the empty-cavity loss gives the contribution from molecular absorption, which is proportional to the amount fraction of the absorbing species.
The work reported here was carried out using a standard commercial CRDS spectrometer (Picarro: model G2103) originally designed for the detection of trace amounts of ammonia in dry air. The light source in this instrument is a single-frequency semiconductor laser, which can be tuned over a small wavelength range by changing its temperature and drive current. The laser wavelength is measured by a wavelength monitor (WLM) that operates on the principle of a solid etalon [18, 19]. The WLM achieves a precision of better than 10−4 cm−1 for laser frequency, but has no absolute frequency reference, and consequently spectroscopic feedback is used to maintain long-term frequency accuracy.
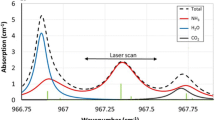
The G2103 analyser measures absorption in the spectral interval from 6548.5 to 6549.2 cm−1. Figure 1 shows the pertinent molecular spectra for the conditions of our measurements, computed from the HITRAN 2012 database [20] for a gas sample containing 10 nmol mol−1 ammonia [where 1 nmol mol−1 is equivalent to 1 part per billion (ppb)], 400 µmol mol−1 CO2 [where 1 µmol mol−1 is equivalent to 1 part per million (ppm)], and 10 mmol mol−1 water vapour (where 1 mmol mol−1 is equivalent to 0.1 %).

HITRAN simulation of absorption spectra for 10 nmol mol−1 ammonia (green), 400 µmol mol−1 carbon dioxide (red), and 10 mmol mol−1 (1 %) water vapour (blue) at 45 °C and 187 hPa
The strongly absorbing line pair at 6548.6 and 6548.8 cm−1 is measured to determine the ammonia amount fraction, whilst the water and CO2 lines around 6549.1 cm−1 provide a frequency reference for long-term stabilisation of the WLM. The water lines also deliver the measurement of the water vapour amount fraction in the sample, which is used to correct for systematic effects of water vapour on the ammonia measurement.
The ammonia amount fractions reported by the standard analyser are derived from least-squares fitting of measured absorption spectrograms to a spectral model of absorption versus frequency for the three molecular species that are measured. Molecular absorption is modelled as a sum of discrete spectral lines, each of which is described by a Galatry profile [21] because the Voigt profile is noticeably inadequate.
The Galatry line shape function, G(x), is specified by four parameters: the line centre ν 0 (in units of cm−1), the Doppler width σ (in units of cm−1), and two dimensionless shape parameters, y and z. Physically, the parameter y has the same meaning as for the Voigt profile, namely the ratio of the rate of transition dipole dephasing collisions to the Doppler width, whilst the parameter z can be described as the rate of velocity-changing collisions to the Doppler width.
The independent variable x is the dimensionless detuning and is defined by Eq. (2):
and the Doppler width is given explicitly by:
where k is Boltzmann’s constant, T is the sample temperature, and M is the molecular mass. The Galatry function also obeys a normalisation condition:
With this parameterisation, we may write the absorption coefficient as:
where the subscript i runs over all the lines in the spectrum and the coefficients A i (expressed in units of cm−1) relate the dimensionless Galatry functions to the observed absorption coefficient. Here the dimensionless detunings, x i , are given by Eq. (2) evaluated with the appropriate centre frequency for each individual line.
The spectral model used in the G2103 CRDS instrument is based on high-resolution spectra of the known gases, from which the line shape parameters [22] (i.e. line centres and shape parameters y and z, for the lines in Fig. 1) were determined. The Doppler widths are not free parameters since they are determined by the centre frequencies, sample temperature, and molecular mass.
Carbon dioxide was modelled by a single absorption line, and ammonia was modelled by three lines. Water vapour is a more complex case, requiring six lines for an adequate description. In addition to the lines at 6549.1 and 6549.2 cm−1, there are two much weaker lines at 6548.6 and 6548.8 cm−1 that interfere with the ammonia spectral features (barely discernible in Fig. 1) and two very strong lines at 6547.2 and 6549.8 cm−1 (not shown in Fig. 1), the off-resonant “tails” of which contribute measurably to the absorption in the ammonia region. For water and ammonia, which have more than one absorption line in the spectral model, the coefficients A i in Eq. (5) have fixed proportions, which are also determined from the high-resolution spectra: therefore, only one A coefficient is needed to specify the magnitude of the molecular absorption.
To measure the composition of an unknown sample, the CRDS analyser acquires spectra, expressed as cavity loss per unit length versus frequency. To achieve the maximum data rate in this analysis mode, the spectrum is sampled on a grid of adjacent cavity modes spaced by the cavity free spectral range, equal to approximately 0.02 cm−1 in the case of this instrument. Therefore, no gross mechanical motion of the cavity mirrors (which would slow the data acquisition) is needed. The doublet of ammonia lines at the low-frequency end of Fig. 1 is sampled approximately every 2 s by 160 ring-down measurements on ten cavity modes. Approximately once every 30 s, the entire range of Fig. 1 is sampled by 160 ring-downs on 36 modes; this longer scan is used only to update the water concentration and the absolute frequency calibration of the wavelength monitor. In either case, the analyser performs a nonlinear least-squares fit (Levenberg–Marquardt algorithm) to determine the parameters in the spectral model which minimises the deviation of the modelled absorption from the measured data. In this fitting procedure, the line centres and shape parameters are fixed.
There are therefore only five free parameters, namely the magnitude of absorption for ammonia, carbon dioxide, and water, an offset describing the empty-cavity absorption, and a global frequency offset, due to the fact that the WLM does not provide an absolute frequency scale.
The frequency offset is used to continually update the WLM readout software, so that the reported optical frequency is always very close to the correct value. The optical absorption due to ammonia is reported as the absorption coefficient at the peaks of two strong ammonia lines that were measured at 6548.6 and 6548.8 cm−1. The two are averaged, and a linear transformation is applied to convert from absorption to amount fraction. Similarly, the absorption at the peak of the spectral line at 6549.1 cm−1 was employed for water vapour measurements, together with the other five already detailed. The slope of the linear transformation was derived from an in-house measurement of NH3 by the manufacturer, which is applied to all instruments. The intercept was determined for each individual analyser from a measurement of air that had been scrubbed of ammonia by a chemical filter, taking into account instrumental offsets, including in the absorption coefficient, due to imperfections in the empty-cavity model.
2.2 Description of the modified cavity ring-down spectral analysis
The G2103 CRDS instrument was originally conceived to measure trace ammonia in well-controlled environments, such as clean rooms. Environmental monitoring encounters a much wider range of conditions. Consequently, the effects of water vapour on the ammonia spectroscopy, which could be neglected in the original instrument, have to be addressed for the present application.
Water affects the measured ammonia spectra in two ways, which can be called direct and indirect interference. A direct interference arises from absorption by water molecules at the same optical frequency where ammonia molecules absorb. The instrument software was modified at the factory to apply an empirical, linear correction to the ammonia amount fraction, based on measurements of ammonia-free air with varying water amount fraction.
Indirect interference is a result of the contribution of water molecules to the collisional broadening of the ammonia spectral lines. The cross-section for spectral broadening is different (typically larger) for H2O than for air. Therefore, the ammonia lines are broader, and the peak absorption is smaller when measured in humid air than when measured in dry air at the same amount fraction.
Since the analyser reports amount fraction based on peak absorption, it systematically under reports the ammonia amount fraction measured in a humid environment. This indirect interference manifests itself, to lowest order, as an error in the coefficient relating peak absorption to amount fraction; this error is in general a nonlinear function of the water amount fraction. To illustrate the effect, Fig. 2 shows raw high-resolution spectra, acquired from samples of 3 µmol mol−1 NH3 in dry air, to which humidified air was added to generate water vapour amount fractions of 1.0, 10, and 25 mmol mol−1. As expected, there is a clear drop observed in NH3 peak absorption and an increase in line width at higher humidity.

Raw spectra for ammonia (3 μmol mol−1) in air with varying water vapour amount fractions: 1 mmol mol−1 (black points), 10 mmol mol−1 (green points), and 25 mmol mol−1 (blue points). Sample conditions are 45 °C and 187 hPa. Each individual spectral point is the result of one ring-down measurement, with the x-axis being the wavenumber measured by the instrument’s wavelength monitor and the y-axis being the reciprocal of the speed of light times the ring-down time constant, which we treat as an “absorption coefficient”. Between ring-downs, the cavity length was adjusted so as to change the resonant frequency in steps of 0.0005 cm−1 by moving a piezoelectric transducer, with the laser temperature and current adjusted accordingly to change the laser frequency by the same amount
Close to 100 spectra were collected for the three water vapour amount fractions listed and then individually fitted with Galatry profiles, allowing the y parameter to be adjusted to deliver the best-fit value. For completeness, an example spectrum with its fitted model and fit residuals is shown in Fig. 3. The simplified model made use of three spectroscopic lines for ammonia, neglecting several of the very weak lines. This is adequate to ensure that the agreement between the recorded ammonia absorption and the model is considerably better than ±1 % and also to quantify the variation of NH3 line width with water vapour concentration. A commonly used figure of merit for the fitting procedure is the ratio of the peak absorption to the root-mean-square residuals, designated as “signal-to-noise ratio” by Cygan et al. [23] and as “quality of fit” by Lisak et al. [24] For the data provided in Fig. 3, where the spectrum is complex because the lines are not completely isolated, this measure is approximately 160. This is sufficient for this application, but is not as high as could be achieved with very well isolated lines that can serve as much better tests of spectral line shape theory.

CRDS spectrum of ammonia (3 μmol mol−1) in air with 1 mmol mol−1 water vapour (black points), together with the best-fit model (green curve), as described in the text. The lower panel shows the residuals of the fit
The use of a Galatry profile led to an improvement (+35 %) in the quality of the spectral fit compared to using a Voigt profile; in addition, the quality of fit was essentially found to be the same for all three humidity conditions considered here. These validation tests enabled us to establish the systematic effect of water vapour on line width, in the selected spectral window, well enough to make accurate absorption-based measurements of ammonia in both dry and humidified atmospheres.
The average values of the y parameters show a linear dependence with water amount fraction, as illustrated in Fig. 4. Converting the water amount fraction to partial pressure, and multiplying the y parameter by the ammonia Doppler width, the slope of the fitted lines can be converted to a broadening parameter of the form tabulated in the HITRAN database [20], indicated here as γ water. The average value obtained this way is γ water = (0.31 ± 0.04) cm−1 atm−1, which is intermediate between the HITRAN values for air- and self-broadening, 0.0957 and 0.486 cm−1 atm−1, respectively. The only other study of ammonia lines broadened by water vapour in the near infra-red is that of Schilt [15], who found γ water = 0.148 cm−1 atm−1 for an absorption feature at 6612.7 cm−1 and γ water = 0.24 cm−1 atm−1 for a feature at 6596.4 cm−1. It should be noted, however, that Schilt measured absorption from multiple, unresolved lines at atmospheric pressure, so that the Lorentzian line width reported in their study arises from a combination of collisional broadening and the smearing of unresolved lines with different centre frequencies and is therefore not directly comparable to our measurement of the widths of resolved lines. Owen et al. [16] measured an absorption feature consisting of six partially resolved transitions near 1103.46 cm−1 and reported broadening coefficients for all of them. The reported values for γ water range from 0.276 to 0.336 cm−1 atm−1, depending on the transition and the choice of line shape model (both Voigt or Galatry were considered), in fairly close agreement with our observations. Similarly, Sur et al. [17] reported values for γ water for nine ammonia lines in the frequency range 961.5–965.5 cm−1: they observed broadening coefficients ranging from 0.257 to 0.486 cm−1 atm−1, in good agreement with the findings of Owen et al. [16] and those from this study. However, it must be noted that Sur et al. [17] only used Voigt line shapes in their analysis.

Dependence of the line broadening parameter y on water amount fraction. The upper points (black circles) refer to the ammonia line at 6548.6 cm−1, and the lower points (red squares) refer to the line at 6548.8 cm−1. The points are average values from fitting of experimental spectra, with error bars corresponding to the standard error of the mean, and the straight lines are linear fits
Starting from the observed linear dependence of the y parameter on water concentration shown in Fig. 4, it is possible to derive a correction for the effect of water vapour on the reported ammonia amount fraction. The sample temperature and pressure are stabilised in the instrument cell to known measured values that are constant, and consequently the ammonia absorption is proportional to a dimensionless Galatry function, G, as described in Eq. (5). Since the ammonia in the sample is quantified using the absorption at the line centre (i.e. when the dimensionless detuning x is 0), and as the instrument is calibrated with dry gas (i.e. [H2O] = 0 mol mol−1), then a Galatry function in the form G(y = y 0; x = 0) is employed in Eq. (5), where the expression y = y 0 indicates the value of the broadening parameter y in the absence of water, and x = 0 is the detuning at the line centre. When the same instrument is used to measure a sample with water vapour amount fraction [H2O] ≠ 0 mol mol−1, the plot in Fig. 4 is used to determine the y parameter of the humid sample, \(y_{{{\text{H}}_{2} {\text{O}}}}\), as follows:
where s is the average slope of the linear dependence in Fig. 4. Therefore, whilst the standard fitting routine fixes the y parameter to y 0, the absorption of the humid sample relative to a dry sample is obtained by G(\(y = y_{{{\text{H}}_{2} {\text{O}}}}\); x = 0)/G(y = y 0; x = 0), which can be re-written using Eq. (6) as G(\(y = y_{0} + s \times \left[ {{\text{H}}_{2} {\text{O}}} \right]\); x = 0)/G(y = y 0; x = 0). This ratio of Galatry functions can be thought of as the systematic change in line shape introduced by the presence of water vapour. To make the evaluation of the water correction more rapid during operation, the ratio of Galatry functions was numerically evaluated for water amount fractions from zero to 0.1 mol mol−1 and then approximated (with relative accuracy better than 5 × 10−4) by a power series of the form 1 + a[H2O] + b[H2O]2. In addition, we implemented a small correction for the direct interference of water vapour with the ammonia lines that was not perfectly accounted for in the original spectral model. The final result was a modified instrument with two reported ammonia outputs, both in units of nmol mol−1, namely “NH3_raw” (no change in the original design) and “NH3_corrected” (based on the most recent near simultaneous measurement of the amount fraction of water vapour), given by:
where [H2O] is the water amount fraction in the sample in units of mol mol−1, and a and b are the constants in the power series approximation to the ratio of Galatry functions as described above, with values a = − 1.78 and b = + 2.35. The offset coefficient has the numerical value of 1.73 × 10−8, corresponding to a 0.173 nmol mol−1 correction to the ammonia amount fraction per percent amount fraction of water vapour in the sample.
2.3 Preparation and validation of NH3 primary standard gas mixtures
An accurate ammonia primary standard gas mixture (PSM) was necessary to be able to produce test atmospheres of known ammonia content. Due to the reactivity of ammonia, the preparation of gas mixtures is affected by a series of issues, including adsorption onto internal surfaces and reaction with impurities in the matrix gas used, as highlighted by the lack of consensus between National Metrology Institutes (NMIs) in an international key comparison in 2006–07, CCQM-K46 [25]. This exercise focused on the analysis of mixtures of 30–50 µmol mol−1 ammonia in nitrogen; the disagreement between the results (up to 5 % in some cases) was attributed to a number of reasons, including the different cylinder passivation chemistries used by the participating NMIs to produce their own reference mixtures.
In the light of these issues, a suite of seven PSMs was prepared in 10-L high-pressure aluminium gas cylinders, all of which had undergone different internal passivation treatments, which included Spectra-Seal™ (BOC plc [26]), Spectra-Seal™ with NPL’s proprietary treatment (BOC plc and NPL), and Aculife IV™ (Air Liquide/Scott [27]).
The gas mixtures were prepared by gravimetry in accordance with the standard procedures defined in ISO 6142 [28], using high-accuracy calibrated single pan balances and adding each component separately either by small transfer vessels or direct gas transfer. Prior to filling, the cylinders were evacuated to 1 × 10−7 mbar with an oil-free rotary pump and turbo-molecular pump combination. The PSMs were prepared from pure NH3 (Air Products, VLSI, 99.999 % purity) and purified BIP+ nitrogen (Air Products, BIP+, 99.99995 % purity) via a number of different dilution routes, in order to minimise any potential biases that might be introduced into the method. Figure 5 summarises the hierarchy of gas mixtures prepared in the amount fraction regime of 100 µmol mol−1 and the internal passivation treatments employed.

Hierarchy of ammonia gas mixtures prepared at NPL; mixtures prepared in Spectra-Seal™, Spectra-Seal™ with NPL’s proprietary treatment and Aculife IV™ cylinders are shown in orange, lilac, and green, respectively
Ammonia amount fractions and their stability with time were measured with a non-dispersive infra-red (NDIR) spectrometer (ABB, Uras 26), using a “standard/unknown” routine in which each mixture was in turn treated as the unknown and was certified against the other six, effectively used as standards. By this method, it was possible to establish that the ammonia mixtures were internally consistent and stable with respect to each other, irrespective of the preparation path employed.
The amount fraction of the “unknown” mixture, x u, is given by:
where R u is the analyser response for the “unknown” mixture, R 0 is the analyser response to the zero gas (purified BIP+ nitrogen), R st is the analyser response to the standard, and x st is the gravimetric amount fraction of the standard. Each mixture was therefore assigned six certified values of x u. The results obtained are summarised in Table 1 and show that, for all mixtures, the certified amount fractions are within less than 1 % from the gravimetric amount fractions (calculated only from the gravimetric preparation data). This indicated that loss of ammonia was minimal, and a conservative estimate of the expanded (k = 2) uncertainty in the amount fraction of ±2 % was used for further calculations.
The stability over time of the PSM used for the dilutions, NPL30718 (as described in Sect. 2.4 below), was monitored by recertifying it against newly made mixtures, nominally at the same amount fraction, at regular intervals. No signs of ammonia loss were observed.
2.4 Generation of trace ammonia amount fractions for spectroscopic measurements
Tests were carried out to establish the response of the CRDS spectrometer to well-characterised humidified atmospheres containing ammonia and to confirm that the changes incorporated by the manufacturer into the CRDS design were successful in removing water cross-interference effects and delivering improved values of the NH3 amount fraction.
A NH3/N2 primary standard gas mixture (Cylinder NPL 30718) was employed to carry out the main ammonia evaluation tests. The PSM contained a certified ammonia amount fraction of 99.95 ± 2.00 µmol mol−1 in a cylinder treated internally with the Aculife IV™ commercial passivation process.
The PSM was subsequently diluted with known amounts of high-purity diluent air (Peak Scientific) and water vapour to generate various ammonia atmospheres in the nmol mol−1 amount fraction regime. Comparative tests with a range of diluents showed that the scrubbed air supply was a cost-effective source of high-purity diluent gas. Mass flow controllers (MKS and Brooks Instruments) were employed to deliver the gases, and these were all calibrated with traceable high-accuracy Bios DryCal flow meters (Mesa Laboratories, base unit: ML-800 series with ML-800-44 and ML-800-3 measuring cells). Water vapour was introduced from a liquid water reservoir contained in a high-pressure cylinder and fed from a 4 bar back pressure of helium (Air Products). The liquid H2O was then directed into a low flow Coriolis mass flow controller and liquid injection vapouriser system (Quantim Series, Brooks Instruments). Here, the output was combined with the relevant dry NH3 sample in a dedicated manifold to produce the required humidified atmospheres. The liquid water employed had previously been de-ionised using a purifier (Elga DV 35 Purelab (Option S15 BP)).
The water vapour generator was calibrated by on-line measurements of the mass loss of the liquid water reservoir, using a top pan balance (Mettler Toledo SG32001). With the above method, it was possible to generate traceable humidified NH3 amount fractions in the nominal range of 10.2–215 nmol mol−1, each set at a relative humidity of 40, 50, 60, and 70 %. The various multi-component mixtures generated were then introduced into the CRDS instrument via a perfluoroalkoxy (PFA) pipe fitted with an excess flow that was also directed into a safe exhaust.
2.5 Testing for CO2 spectroscopic interference
Finally, the CRDS was checked to confirm that there was not a potential problem from cross-interference with carbon dioxide (CO2). A CO2 amount fraction of 505 µmol mol−1 was introduced into the dry atmosphere, which is well above the levels expected for normal ambient monitoring. The spectral features of this molecule were found to be too weak to be of significance to the measurements. For ambient monitoring or humidified atmospheres, Fig. 1 shows that the absorption lines of CO2 and H2O overlap, but this is taken into account by allowing the line strength of both molecules to adjust independently when fitting the measured spectra. Consequently, this method enables the relative contributions to the absorption from CO2 and H2O to be separated.
3 Results and discussion
The separate water and ammonia CRDS measurement ranges were evaluated by introducing humidified air containing several known water vapour amount fractions into the instrument up to 18 mmol mol−1 (corresponding to a relative humidity of approximately 70 % at 20 °C). The objectives here were to confirm that the factory internal water calibration was in agreement with the known delivered H2O input amount fractions generated at NPL, to check that the instrument showed a linear response to water vapour, and to measure the cross-sensitivity to water in the ammonia measurement channel.
3.1 Water vapour spectroscopic measurements
Figure 6 is an example of a lack of fit plot showing the CRDS water response to a series of known traceable H2O input amount fractions generated at NPL and is linear with r 2 effectively equal to one. The factory internal calibration was found to be in agreement to within 1.3 % with the on-line mass loss calibration method. This provided further confidence that such water vapour measurements could be employed by the manufacturer to verify the success of modifications to the instrument. The factory water vapour calibration made use of results from earlier work [29], and quadratic terms were applied to the raw data such that the reported water amount fraction from the present measurements at 6549.1 cm−1 matched the amount fraction reported by a different CRDS analyser measuring this compound at 6057.8 cm−1, which was calibrated against a chilled mirror hygrometer.

Lack of fit plot showing CRDS water response to traceable H2O input amount fractions
3.2 NH3 spectroscopic measurements
Figure 7 is an example of a lack of fit plot showing the CRDS ammonia response (“NH3_raw”) to a series of traceable NH3 input amount fractions at a known measured relative humidity of nominally 60 % at 20 °C. Figure 8 shows the corresponding modified (“NH3_corrected”) results for the same input amount fractions as transformed by Eq. (7).

Lack of fit plot showing mean CRDS response (“NH3_RAW”) to traceable humidified (RH = 60 %) ammonia input amount fractions. The solid line is the generalised least-squares (GLS) fit to the data, described in the text

Lack of fit plot showing mean CRDS response (“NH3_Corrected”) to traceable humidified (RH = 60 %) ammonia input amount fractions. The solid line is the generalised least-squares (GLS) fit to the data, described in the text
In this example, each of the amount fractions reported by the CRDS was background-subtracted using the apparent NH3 amount fraction obtained from a sample of humidified zero air at 60 % RH and scrubbed free of ammonia. A similar subtraction procedure was carried out for the other NH3 lack of fit data, using the corresponding zero gas (either dry or humidified) applicable to a relative humidity of: 0, 40, 50, and 70 %.
To generate the lack of fit plots, uncertainties were assigned to each of the NH3 input amount fractions, in preparation for analysis using XLGENLINE, which is a generalised least-squares (GLS) Microsoft Excel-based software package for low-degree polynomial fitting developed at NPL [30]. This was also used in the analysis of the water vapour data detailed above. XLGENLINE employed a user-defined input file containing values of x and u(x) (respectively, the known NH3 input amount fractions and combined standard uncertainty), y and u(y) (respectively, the reported mean zero-corrected CRDS NH3 amount fractions and repeatability standard deviation).
The software package was used to perform a first-order polynomial GLS fit, in this case forced through zero. The fitted function automatically calculated analytical results and defined uncertainties for any number of unknown samples.
Table 2 summarises the output gradients calculated by XLGENLINE for all the NH3 atmosphere lack of fit plots, each applicable to a relative humidity of: 0, 40, 50, 60, and 70 %. The column marked “NH3_raw” shows the gradients obtained for results generated with the original CRDS instrument configuration, together with their combined expanded standard uncertainties (with a coverage factor k = 2, providing a coverage probability of approximately 95 %). The column marked “NH3_corrected” shows the corresponding gradients generated using the modified CRDS instrument configuration, designed to account for the influence of water vapour.
As expected, the gradient results obtained for the dry ammonia data are similar in both “NH3_raw” and “NH3_corrected”. The trend for the humidified ammonia data shows a departure from the supplied amount fraction in “NH3_raw”, which within the uncertainty is rectified in “NH3_corrected” for all the conditions tested.
3.3 Error propagation and uncertainty analysis
In each case, the NH3 amount fractions delivered to the CRDS (\(C_{\text{Final}}\), in units of nmol mol−1) were calculated using Eq. (9):
where \(C_{\text{Cylinder}}\) is the ammonia amount fraction in the cylinder (in units of µmol mol−1), \(F_{\text{Span}}\) is the flow rate of NH3 span gas, \(F_{{{\text{Diluent}}1}}\) and \(F_{{{\text{Diluent}}2}}\) are the flow rates of diluent zero gas, and \(F_{{{\text{H}}_{2} {\text{O}}}}\) is the flow rate of water vapour.
The sources of error identified include the NH3 amount fraction of the parent cylinder, individual repeatability standard deviations in the mass flow rates, mass flow controller temperature dependencies, gravimetric water calibration (including balance drift), mass flow meter calibrations, and time. Errors were then allocated to each term in Eq. (9) that they influenced.
Following the method outlined in ISO 6145-7:2010 [31], a “sensitivity” was assigned to each component in Eq. (9) by differentiating the amount fraction with respect to each component and summing in quadrature in accordance with Eq. (10):
The typical combined expanded standard uncertainties calculated for the humidified NH3 atmospheres were no greater than ±2.3 %. The relevant individual values were used as inputs into XLGENLINE, along with the corresponding values for the readings recorded for the CRDS instrument.
4 Conclusion
The collisional broadening due to water vapour of two ammonia (NH3) lines in the near infra-red (6548.6 and 6548.8 cm−1) was measured over a range of relative humidity by means of cavity ring-down spectroscopy (CRDS). The average value obtained for the broadening parameter, γ water [γ water = (0.31 ± 0.04) cm−1 atm−1] is in good agreement with previous studies. The measurement of γ water allowed the implementation of a method which accounted for the cross-interference of water vapour and has delivered an improved accuracy for low amount fraction measurements of NH3 in the 10.2–215 nmol mol−1 regime with relative humidity in the range: 0, 40, 50, 60, and 70 %. The development and validation of new stable NH3 primary standard gas mixtures (PSMs) at 100 µmol mol−1 was crucial for the further collaborative development of the CRDS sensor as part of the process of establishing traceability to measurements of ambient ammonia. The correction for the effects of water vapour on the reported ammonia amount fraction has been incorporated by the manufacturer in all new CRDS ammonia sensors. The analyser has potential use as a “spectroscopic reference instrument” for monitoring ammonia in future exposure chamber tests to validate diffusive and pumped samplers, and also for field measurements.
References
M. Hornung, M.R. Ashmore, M.A. Sutton, Ammonia in the UK, Chapter 3 (DEFRA, London, 2002), pp. 24–33
C.E.R. Pitcairn, I.D. Leith, M.A. Sutton, D. Fowler, R.C. Munro, Y.S. Tang, D. Wilson, Environ. Pollut. 102, 41 (1998)
Directive 2001/81/EC of the European Parliament and of the Council of 23 October 2001 on national emission ceilings for certain atmospheric pollutants
Air Pollution and Climate Secretariat website, http://www.airclim.org/tags/clrtap. Accessed June 2016
Implementation of the National Emission Ceilings Directive website: http://ec.europa.eu/environment/air/pollutants/implem_nec_directive.htm. Accessed June 2016
Federal Ministry for Environment, Nature Conservation and Nuclear safety. First General Administrative Regulation Pertaining the Federal Immission Control Act (Technical Instructions on Air Quality Control—TA Luft) of 24 July 2002. (GMBI. [Gemaieisames Ministerialblatt-Joint Ministerial Gazette] p. 511) (Techniische Anlei-tung zur Reinhaltung der Luft-TA Luft)
B. Bessagnet, M. Beauchamp, C. Guerreiro, F. De Leeuw, S. Tsyro, A. Colette, F. Meleux, L. Rouil, P. Ruyssenaars, F. Sauter, G.J.M. Velders, V.L. Foltescu, J. Van Aardenne, Environ. Sci. Policy 44, 149 (2014)
A. Pogány, D. Balslev-Harder, C.F. Braban, N. Cassidy, V. Ebert, V. Ferracci, T. Hieta, D. Leuenberger, N. Lüttschwager, N.A. Martin, C. Pascale, C. Tiebe, M.M. Twigg, O. Vaittinen, J. van Wijk, K. Wirtz, B. Niederhauser: 17th International Congress of Metrology 07003 (2015). doi:10.1051/metrology/201507003
D.A. Anderson, J.C. Frisch, C.S. Masser, Appl. Opt. 23, 1238 (1984)
E. Crosson, B. Fidric, B. Paldus, S. Tan: Wavelength control for cavity ring-down spectrometer, US Patent 7,106,763 B2, Picarro, Inc. 2006
A.R. Awtry, J.H. Miller, Appl. Phys. B 75, 255 (2002)
J.B. Leen, X.-Y. Yu, M. Gupta, D.S. Baer, J.M. Hubbe, C.D. Kluzek, J.M. Tomlinson, M.R. Hubbell, Environ. Sci. Technol. 47, 10446 (2013)
D.J. Miller, K. Sun, L. Tao, M.A. Khan, M.A. Zondlo, Atmos. Meas. Tech. 7, 81 (2014)
K. Sun, Le Tao, D.J. Miller, M.A. Khan, M.A. Zondlo, Environ. Sci. Technol. 48, 3943 (2014)
S. Schilt, Appl. Phys. B 100, 349 (2010)
K. Owen, E. Es-sebbar, A. Farooq, J. Quant. Spectrosc. Radiat. Transf. 121, 56 (2013)
R. Sur, R.M. Spearrin, W.Y. Peng, C.L. Strand, J.B. Jeffries, G.M. Enns, R.K. Hanson, J. Quant. Spectrosc. Radiat. Transf. 175, 90 (2016)
E.R. Crosson, Appl. Phys. B 92, 403 (2008)
S. M. Tan, Wavelength measurement method based on combination of two signals in quadrature, US Patent 7,420,686 B2, Picarro, Inc. 2008
L.S. Rothman, I.E. Gordon, Y. Babikov, A. Barbe, D.C. Benner, P.F. Bernath, M. Birk, L. Bizzocchi, V. Boudon, L.R. Brown, A. Campargue, K. Chance, E.A. Cohen, L.H. Coudert, V.M. Devi, B.J. Drouin, A. Faytl, J.-M. Flaud, R.R. Gamache, J.J. Harrison, J.-M. Hartmann, C. Hill, J.T. Hodges, D. Jacquemart, A. Jolly, J. Lamouroux, R.J. Le Roy, G. Li, D.A. Long, O.M. Lyulin, C.J. Mackie, S.T. Massie, S. Mikhailenko, H.S.P. Müller, O.V. Naumenko, A.V. Nikitin, J. Orphal, V. Perevalov, A. Perrin, E.R. Polovtseva, C. Richard, M.A.H. Smith, E. Starikova, K. Sung, S. Tashkun, J. Tennyson, G.C. Toon, V.G. Tyuterev, G. Wagner, J. Quant. Spectrosc. Radiat. Transf. 130, 4 (2013)
L. Galatry, Phys. Rev. 122, 1218 (1961)
P.L. Varghese, R.K. Hanson, Appl. Opt. 23, 2376 (1984)
A. Cygan, P. Wcisło, S. Wójtewicz, P. Masłowski, J. Domysławska, R.S. Traviński, R. Ciuryło, D. Lisak, J. Phys. Conf. Ser. 548, 012015 (2014)
D. Lisak, A. Cygan, D. Bermejo, J.L. Domenech, J.T. Hodges, H. Tran, J. Quant. Sectrosc. Radiat. Transf. 164, 221 (2015)
A.M.H. van der Veen, G. Nieuwenkamp, R.M. Wessel, M. Maruyama, G.S. Heo, Y.-D. Kim, D.M. Moon, B. Niederhauser, M. Quintilii, M.J.T. Milton, M.G. Cox, P.M. Harris, F.R. Guenther, G.C. Rhoderick, L.A. Konopelko, Y.A. Kustikov, V.V. Pankratov, D.N. Selukov, V.A. Petrov, E.V. Gromova, Metrologia 47, 08023 (2010)
Spectra-Seal™, Registration Nr. 3853813, 2010, BOC Limited
Aculife™, Registration Nr. 4104439, 2012, Air Liquide America Specialty Gases LLC
International Organization for Standardization (ISO) 6142:2001. Gas analysis—Preparation of calibration gas mixtures—Gravimetric method
J. Winderlich, H. Chen, C. Gerbeig, T. Seifert, O. Kolle, J.V. Larvič, C. Kaiser, A. Höfer, M. Heimann, Atmos. Meas. Tech. 3, 1113 (2010)
I. M. Smith, Software for determining polynomial calibration functions by generalised least squares: user manual, NPL Report MS 11 (Teddington, 2010)
International Organization for Standardization (ISO) 6145-7:2010 Gas analysis: Preparation of calibration gas mixtures using dynamic volumetric methods. Part 7: Thermal mass-flow controllers
Acknowledgments
We gratefully acknowledge the funding received from the Chemical and Biological Metrology Programme of the UK Department for Business, Innovation and Skills (BIS) and the European Metrology Research Programme (EMRP) of the European Union. The EMRP is jointly funded by the EMRP participating countries within EURAMET and the European Union. © Crown copyright 2016 and reproduced by permission of the Controller of HMSO and the Queen’s Printer for Scotland.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Martin, N.A., Ferracci, V., Cassidy, N. et al. The application of a cavity ring-down spectrometer to measurements of ambient ammonia using traceable primary standard gas mixtures. Appl. Phys. B 122, 219 (2016). https://doi.org/10.1007/s00340-016-6486-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00340-016-6486-9