
Abstract
Jet-cooled laser-induced fluorescence is a spectroscopic method, specifically developed for the study of PAHs formed in flames. This technique has already been used to measure different aromatic species in sooting low-pressure methane flames such as benzene, naphthalene, and pyrene. The use of the LIF technique to excite PAHs drastically cooled down inside a supersonic jet offers the possibility to get selective and quantitative profiles of PAHs sampled from sooting flames. In this paper, we demonstrate the ability of this experimental method to separate the contribution of two mass isomers generated in sooting flames which are the pyrene and the fluoranthene. The selectivity of the method is demonstrated by studying the spectral properties of these species. The method is then applied to the measurement of both these species in two sooting flames with different equivalence ratios and stabilized at 200 torr (26.65 kPa). The sensitivity of the technique has been found to reach a few ppb in the case of fluoranthene measurements.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
The understanding of the formation of polycyclic aromatic hydrocarbons (PAHs) and soot particles in flames has considerably increased over the last few years. Nevertheless, large uncertainties still remain, especially concerning the nucleation step of the soot particles. Notably, the identity and the role of the gas-phase species at the origin of the formation of the molecular entities, leading to the soot nuclei, are still highly speculative. Soot formation has been shown to be correlated with PAHs which are considered as their main precursors. Currently, the envisaged nucleation mechanisms are expected to be either related to the dimerization of large PAHs forming stack clusters or to the contribution of cross-linked three-dimensional aromatic structures involving the combination of smaller PAHs [1, 2]. However, none of these pathways could have been clearly confirmed. The major inconsistency concerning the first envisaged pathway is that at flame temperature only dimers of very large PAHs as ovalene (C32H14) or circumcoronene (C56H18) could potentially be formed. Binding energies of dimers of lower size are not strong enough to provide stable species at flame temperature [3]. However, larger PAHs than coronene (C24H12) are generally not measured in flames and expected to be in too low concentrations to lead to the formation of the first nuclei. The other pathway, involving smaller PAHs, could seem more pertinent because of their much higher concentration in rich flames. However, according to Wang [3], this kind of mechanisms could only be plausible close to the flame front as it requires H atoms which are in too low concentration in the postflame zone to explain the persistent nucleation far above the burner. Experimental investigations are therefore still needed to provide information to answer this challenging question of the nucleation of soot particles in flames.
Pyrene and fluoranthene are assumed to be mainly formed via the HACA mechanism [4]. Pyrene has been considered during a long time as a potential species leading directly by dimerization to the nascent soot particles. It is now clearly established that the pyrene dimer cannot survive at flame temperatures due to its too weak binding energy [3, 5, 6]. Nonetheless, pyrene dimerization is still envisaged in many soot nucleation mechanisms as the nucleation step [7]. Fluoranthene, which is the main mass isomer (202 a.m.u) of pyrene, is another potential interesting species for soot nucleation. In contrast to pyrene, constituted only six-membered carbon rings, the presence of a five-membered carbon ring in fluoranthene allows the curvature of the molecule, possibly leading to the formation of three-dimensional molecular edifices. In a previous study carried out by LIF in situ in sooting flames at atmospheric pressure [8], we highlighted that the LIF emission signal coming from the flame upon various excitation wavelengths led to a broadband and poorly resolved spectrum in the UV–visible range. We showed that fluoranthene and potentially larger derivatives as rubicene (C26H14) could be good candidates to explain part of these spectral features in the visible range because of the five-membered ring units in their molecular structure. This hypothesis is supported by the fact that the fluorescence spectra of such PAHs are generally wider and more redshifted than spectra of PAHs of equivalent size containing only six-membered rings [9, 10].
Measurements of PAHs in flames can be done with different techniques. MBMS (molecular beam mass spectrometry) is one of the experimental methods used to detect moderate molecular weighted species such as pyrene and fluoranthene. However, it does not allow by itself to separate isomers and the mass 202 is generally associated to the single pyrene molecule [11, 12]. To become selective, mass spectrometry requires the use of a selective ionization method such as resonance-enhanced multiphoton ionization (REMPI) or synchrotron radiation. Reported REMPI measurements in the literature are usually performed in a molecular beam obtained after direct probe sampling of the species from the flame with conical quartz probe followed by a second expansion through a copper skimmer. In these conditions, the rotational temperature of the species cooled down in the molecular beam is only around 400 K [13, 14]. At that temperature, aromatic compounds exhibit broad overlapping absorption bands which can limit seriously their selective ionization. In order to enhance the selectivity, mass spectrometry can be associated with synchrotron ionization which enables the measurement of the PIE (photoionization efficiency spectrum), allowing a more selective identification of the species and likely the distinction between pyrene and fluoranthene. This last method is certainly the most powerful tool to selectively detect PAHs in flames. Its main limitation is maybe its moderate sensitivity. According to the literature, its sensitivity and measurement uncertainties are similar to the one of the REMPI–MBMS techniques. Hence, the sensitivity threshold is estimated around 1 ppm (±(25–40) % uncertainty) for the measurements of species with well-known ionization cross sections, while an uncertainty factor of 2–4 is reported for species with unknown cross sections [15–18]. Concentration profiles of various PAHs have been measured with this method in different non-sooting [15, 19] and sooting [18] low-pressure premixed flames. In these studies, the mass 202 is exclusively attributed to pyrene and none of these works report the measurement of fluoranthene. Maybe this lack of data comes from the too low concentration of fluoranthene in these flame conditions.
Mass isomer distinction can also be done by gas chromatography associated with mass spectrometry (GCMS) [20–23]. However, the measurement of such large species as pyrene and fluoranthene is very likely to be affected by condensation problems during their introduction in the capillary of the setup.
As mentioned earlier, in situ LIF excitation of PAHs leads to unstructured spectra preventing any selectivity for the measurement of individual PAHs [24]. To address this problem, it is necessary to drastically cool down the species. In this way, only the lowest energy levels of the molecule are populated. Fewer transitions to the excited states are possible, and the spectra are simplified at lower temperature allowing the selective measurement of PAHs. The jet-cooled laser-induced fluorescence (JCLIF) method has been developed by our group to this end. This technique is based on the sampling of the PAHs from the flame by a thin microprobe. PAHs are then directly cooled down inside an expanded free jet where they can be selectively excited by a laser beam. This technique has a very good sensitivity, enabling the detection of PAHs at concentrations of a few ppb. Moreover, the species sampled from the flame are introduced at reduced pressure (a few mbar) in the JCLIF chamber and do not require the compression of the analyzed species as commonly done with GC setups. Thus, the samples are not impacted by condensation problems. The experimental setup we used for this study is nearly the same as the one previously described in [25–27] for PAHs measurements in low-pressure methane flames.
In this paper, we present in detail the spectroscopic study we made concerning the fluoranthene in order to measure selectively this species in sooting flames. Excitation and emission spectra have been recorded for various experimental conditions as well as fluorescence lifetimes in order to bring evidences of the ability of the method to distinguish mass isomers (fluoranthene and pyrene). The experimental setup is described, notably the few modifications we made to improve its sensitivity and selectivity compared with previous versions. Finally, we report the experimental application we made by measuring quantitatively the fluoranthene and pyrene in sooting flames with two different equivalence ratios. The calibration procedure as well as the concentration profiles determined in both flames are presented and discussed.
2 Experimental setup
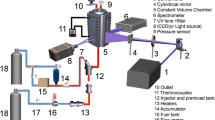
The JCLIF setup is presented in Fig. 1. Samples are extracted from the low-pressure flame with a microprobe and introduced in a high-vacuum analysis chamber. The experimental parameters leading to the conditions of free jet expansion are detailed in [25]. The position of the laser beam in the jet is optimized to probe an area which is sufficiently cooled and dense in molecules. These conditions are necessary to obtain a good signal-to-noise ratio while maintaining a sufficient cooling to insure the selectivity of the method. The laser system consists of a Quantel Nd:YAG laser, pumping a dye laser (TDL70 Quantel). The second harmonic at 532 nm is used to pump the dye laser. For pyrene measurements, DCM solved in ethanol was used to generate by frequency doubling a laser pulse around 321 nm, whereas for the fluoranthene excitation, we utilized Rhodamine 590 to generate a laser pulse around 282 nm. The beam was sent into the analysis chamber unfocused and spatially reduced (diameter around 2 mm) by using two lenses and pinholes. The laser fluence has been adjusted around 2.10−2 J/cm2 to be in the linear regime of fluorescence. Fluorescence light was collected thanks to two lenses and focused into a bundle of optical fibers connected to an Acton 2500i spectrometer (500 mm focal length–300 g/mm grating). The spectrometer was coupled to a 16-bit intensified charge-coupled device camera (Roper Pimax II) for the recording of the fluorescence spectra or to a photomultiplier (Photonis XP2020Q, spectral range 150–650 nm and maximum sensitivity around 420 nm) for the measurement of the LIF temporal decays and the determination of the concentration profiles of the species over the flame height. Temporal LIF signals were measured with a spectral bandwidth of 20 nm provided by the spectrometer. This setup is similar to the one we used in a previous study [27] except for the collection setup described above which has been improved. By this way, we increased the sensitivity of the detection by a factor of ten in comparison with our previous system. In these conditions, it was possible to use a spectrometer instead of a bandpass filter for the pyrene and fluoranthene measurements.

Experimental setup
The ability of the JCLIF method to detect fluoranthene has been tested in flames stabilized on a 6-cm Holthuis flat flame burner. Two flames were stabilized at 200 torr (26.65 kPa) for two different equivalent ratios (ϕ = 2.05 and ϕ = 2.32). The composition of these flames is shown in Table 1.
A microprobe with a diameter aperture of 200 µm was used for the sampling of the species from the flame. The stainless steel line used for the transfer of the species from the probe to the nozzle of the analysis chamber was heated at 368 K, and the pressure inside the line was kept constant at 10 torr (1.33 kPa) to avoid condensation. The generator system of vapors of pure PAHs consisted of a stainless steel cell containing a porous filter (0.5 µm) and the PAH powder. The cell was heated at a fixed temperature controlled by an electronic regulation system in order to generate a constant vapor pressure of PAHs. A controlled flow of nitrogen was sent into this cell to generate a flow of pyrene or fluoranthene vapors, the concentrations of which could be adjusted by mixing this flow with a second nitrogen flow. This system was used to get reference spectra of pure PAHs and also for the calibration of the LIF signals of the PAHs sampled from the flame. The calibration of the LIF profiles into mole fraction profiles has been made according to the standard addition method described elsewhere [27], which relies on the addition of small amounts of a pure PAH with known concentrations to the PAH to be measured sampled from the flame. In our work, the temperature of the cell containing pure PAHs was fixed to 323 K for both the pyrene and fluoranthene studies, leading to respective vapor pressures of 9.67 × 10−5 torr (12.88 × 10−3 Pa) [28] and 1.18 × 10−4 torr (15.72 × 10−3 Pa) [29]. The stainless steel lines, driving the species from the cell to the nozzle of the analysis chamber, were gradually heated from 333 to 368 K to avoid their condensation along the line until the nozzle aperture used for the free jet expansion. The pressure inside these lines was kept constant to 10 torr (1.33 kPa) upstream the nozzle aperture. This led to a pressure around 10−3 torr (0.133 Pa) inside the free expansion where LIF measurements were made. Note that in comparison with our previous works [25–27, 30, 31], we used a new porous burner and a new microprobe for this study. The stabilized flame is therefore slightly different from the one studied in previous works. This explains the slight differences in the concentration profiles of pyrene and naphthalene reported in this study in comparison with previous ones.
3 Experimental results
In this part, we describe in detail the experimental procedure we followed to define the conditions, in terms of excitation wavelength and collection of the fluorescence signal, to obtain selective measurements of fluoranthene by JCLIF in flames. To this aim, we report and discuss previous spectroscopic studies published in the literature. As explained below, we preferentially chose the S 0 → S 4 transition to carry out our experiments. Excitation and emission spectra recorded with our free jet setup are then presented as well as the fluorescence lifetimes of this transition. The second part of this section is devoted to the use of the JCLIF method to the detection of fluoranthene in sooting flames. A discussion about the selectivity and sensitivity of the method is finally proposed. This work is concluded by the measurements of concentration profiles of fluoranthene and pyrene in two different sooting flames.
3.1 Spectroscopy of fluoranthene
Fluoranthene has been the subject of a few studies in the last decades. The interpretation of its fluorescence spectrum has long been a controversial issue [32–34] because of the complexity of its spectroscopy and also due to some misinterpretation of the measurements perturbed by the presence of impurities in fluoranthene samples. Fluoranthene is one of the rare PAHs with pyrene that does not follow the Kasha’s rule which normally governs the PAHs fluorescence process. According to this rule, after their excitation to some vibrational levels of a state S n (n > 1), PAHs first relax following non-radiative processes, governed by rapid intern conversion (IC) as well as intravibrational and intervibrational relaxation (IVR), to the lowest vibrational levels of the first electronic state S 1. In free collision regime, as in free jet experiments, only isoenergetic internal conversion and intravibrational relaxation can take place after the excitation of the molecule. As discussed for the pyrene molecule, which also highlight an anomalous fluorescence from the second excited state [35], the fact that in a supersonic expansion, the fluorescence emission of fluoranthene, upon S 4 excitation, can occur from the S 1 and S 2 states might indicate the possibility of reverse internal conversion as shown for pyrene. Relaxation processes of the fluoranthene vapor in free collision regime might therefore correspond to the scheme reported in Fig. 2.

Relaxation processes of fluoranthene vapor in free collision regime (IC internal conversion, RIC reverse internal conversion)
In the case of fluoranthene, fluorescence emission may come from both S 2 and S 1 states. Absorption and excitation spectra of fluoranthene have been measured in various experimental conditions, as in gas phase [36], solvents at ambient temperature [37] or around 20 K [38], polymer matrix [34] at 77 K, and supersonic jets [39, 40]. Table 2 shows a list of previous spectroscopic works and the corresponding values determined for the energies of the first four electronic states.
The first study on fluoranthene was performed in the 60s by Pesteil et al. [37]. They analyzed this compound in a solution of n-pentane at very low temperature (20 K) and determined 19 vibrational modes issued from the \(S_{1} \leftarrow S_{0}\) transition. Those modes were then identified as mode combinations by Chan and Dantus [39] in supersonic jet conditions. Among the few teams who analyzed the first electronic transitions, Ruth and Wick [40] studied the \(S_{1} \to S_{0}\) fluorescence emission band upon the excitation of the S 4 electronic level in a continuous supersonic jet. Their main objective was to characterize the vibronic coupling between the S 3 and S 4 states. The excitation spectrum was obtained by collecting the fluorescence signal over the spectral range 401.5–424.5 nm. They found that the strongest band corresponding to the \(S_{4} \leftarrow S_{0}\) transition was located around 280.4 nm (35,664 cm−1), i.e., at a slightly higher energy level in comparison with previous works [34, 36, 38]. The authors also found that there was a contribution from the \(S_{3} \leftarrow S_{0}\) around this wavelength overlapping with the \(S_{4} \leftarrow S_{0}\) transition.
Concerning the fluorescence emission of fluoranthene, Berlman et al. [32] first showed that it could be emitted upon laser excitation at 355 nm between 16,000 and 25,000 cm−1, i.e., in the range 400–625 nm. They demonstrated that this fluorescence signal came from the S 1 state, as also observed by Karcher et al. and reported in [41]. Philen and Heges [42] were one of the first teams to demonstrate the dual fluorescence of fluoranthene, in low-temperature experiments (PAH trapped in EPA ethanol/diethyl ether/isopentane matrix) at 77 K. They showed that the excitation of fluoranthene at 365 nm (\(S_{2} \leftarrow S_{0}\)) led to an emission fluorescence signal coming from the \(S_{1} \to S_{0}\) between 400 and 540 nm while the use of a shorter excitation wavelength generated a dual fluorescence emission spectrum from S 1 (400–540 nm) and \(S_{2 }\) (370–395 nm). Spectra were recorded for two different temporal conditions (emission collected with and without a temporal delay of 120 ns after the laser pulse). The corresponding measured spectra enabled to highlight a longer fluorescence lifetime from the S 2 state compared to the S 1 state. These results are consistent with the more recent study made by Bark and Forcé [43] in the gas phase. In their work, they attributed the signal emitted in the (402–571 nm) range to the fluorescence coming from the S 1 state and the one issued from 353 to 393 nm to the \(S_{2} \to S_{0}\) emission. The authors attributed the dual fluorescence phenomena to the small energetic gap between the \(S_{1}\) and S 2 electronic states (3450 cm−1), which can facilitate \(S_{1}\)–S 2 internal conversion.
We have chosen to exploit the \(S_{4} \leftarrow S_{0}\) transition for our experiments because of the higher sensitivity it provided for the measurement of low concentrations of fluoranthene in flames. To this end, known concentrations of fluoranthene were introduced in the free jet as follows. A cell containing the pure PAH was heated at 323 K, and the flow rates of nitrogen were adjusted in order to generate a concentration of fluoranthene around 100 ppm at a constant pressure of 10 torr (1.33 kPa) upstream the supersonic expansion. In order to make reliable comparisons with the experimental studies issued from the literature, all our spectra were corrected from the transmission function of the collecting setup which has been determined using an integrating sphere (SphereOptics CSTM-LR-6-M).
Figure 3 presents the comparison of the fluorescence excitation spectrum of pure fluoranthene we get and the spectrum obtained by Ruth and Wick [40] in supersonic jet conditions. A good concordance in shape and position of the band observed around 282 nm is observed. Note that Ruth and Wick’s work was specifically dedicated to the spectroscopic investigation of fluoranthene. The better quality of their spectra is attributed to the different experimental system they used. Their spectrum has been obtained with a initial pressure (before the free jet expansion) of 517 torr (6.9 kPa) and using Argon as carrier gas to optimize the signal of the cold molecules whereas our free jet experiments were done with nitrogen, known to be less efficient for the cooling of the molecules, and with only 10 torr (1.3 kPa) as initial pressure. Moreover, their injection system allowed them to work with concentration of fluoranthene much higher than ours. The limitation in terms of pressure and therefore cooling of the species in our case is directly dependent on the sampling of the species from the flame limited to 10 torr to avoid condensation problem in the line. The corresponding fluorescence emission spectrum of fluoranthene upon laser excitation wavelength around 282 nm is reported in Fig. 4. A comparison of our spectrum with the one obtained by Philen and Hedges [42] is also plotted in this figure.

Fluorescence excitation spectra of pure fluoranthene. Current work (red line), Ruth and Wick (black line) [40]

Fluorescence emission spectra of pure fluoranthene. Current work (red line), Philen and Hedges (black line) [42]
Although we note a good concordance in shape and position between the spectra, we observe slight differences in the range 390–430 nm which certainly result from the different experimental conditions. Indeed, both fluorescence spectra have not been acquired upon the same excitation wavelength which was 282 nm in our case (\(S_{4} \leftarrow S_{0}\) transition) and 337.1 nm for the study of Philen and Hedges (\(S_{2} \leftarrow S_{0}\) transition). It is well known that the structure and shape of PAHs fluorescence spectra are highly dependent on the energy of the excited vibrational levels. Typically, the excitation of vibronic states 2000 cm−1 or more above the S 1 origin lead to the broadening of the fluorescence spectrum and the emergence of additional bands due to intramolecular vibrational energy redistribution (IVR) process. These additional broadbands are due to the emission of a large number of combination states populated through the IVR process. Concerning the fluoranthene, Chan and Dantus [39] have shown that the excitation of levels 1345 cm−1 above the S 1 origin would be sufficient to start the IVR process. Furthermore, the spectrum reported by Philen and Hedges, used for the comparison with ours in Fig. 4, has been recorded with fluoranthene cooled down in an EPA matrix at 77 K. It is also known that such a medium modifies the fluorescence spectrum and quantum yield as well as the fluorescence lifetime [10] because of radiative migration that takes place in crystals. Hence, this comparison also highlights the interest, from a spectroscopic point of view, to work under jet-cooled expansion conditions which enable the study of such large molecules without spectral artifacts or perturbations of the measured spectra.
The choice of the excitation wavelength (\(S_{4} \leftarrow S_{0}\) transition) for our measurements has motivated a specific spectroscopic work dedicated to the dual fluorescence of the fluoranthene associated with our excitation wavelength. Hence, fluorescence emission spectra of fluoranthene have been recorded according to various temporal delays after the laser pulse. For this specific work and in order to improve the sensitivity of the experimental setup, measurements were performed with a lower dilution of the PAH vapor. By this way, the pressure inside the heating cell and the line upstream the nozzle of the jet expansion was kept constant to 1.3 torr instead of 10 torr in previous experiments. The mole fraction of the fluoranthene was evaluated to 780 ppm in these conditions. With this lower initial pressure, the cooling of the fluoranthene is certainly of lesser quality than with 10 torr; however, we noted an increase of the signal-to-noise ratio of approximately a factor of 2.5. Figure 5 shows the fluorescence emission spectra of fluoranthene recorded after different temporal delays from the laser pulse and arbitrarily normalized in intensity at 350 nm.

Fluorescence emission spectra of fluoranthene recorded for different delays after the laser pulse. Fluorescence spectra were normalized at 350 nm, and initial pressure before supersonic expansion fixed at 1.3 torr. Emission due to \(S_{2} \to S_{0}\) transition (350 nm) shows a lower decrease of intensity with delay compared to \(S_{1} \to S_{0}\) emission (450 nm)
We observe that the decrease in the part of the spectrum located between 390 and 540 nm coming from the S 1 state is less important than for the part of the spectrum positioned between 340 and 390 nm issued from \(S_{2}\). These results are in good agreement with the one obtained by Philen and Hedges [42] and confirm the longer fluorescence lifetime of the signal issued from the S 2 state in comparison with the S 1 state.
The temporal fluorescence signals measured with the spectrometer centered at 440 and 350 nm, both with a spectral bandpass of 20 nm, are reported in Figs. 6 and 7 . The analysis of the fluorescence temporal decays for the two collected wavelength ranges has been performed according to a convolution procedure carried out with the help of the data analysis software DecayFit [44]. This procedure enabled the analysis of the fluorescence signals for both emission wavelength ranges. We plot in Figs. 6 and 7 , the measured LIF temporal signals (red line) as well as the laser pulse (pulse width estimated to 5.5 ns—green line).

Experimental and calculated LIF temporal decay recorded at 440 nm (±10 nm). The black line corresponds to the residual between the two decay curves

Experimental and calculated LIF temporal decay recorded at 350 nm (±10 nm). The black line corresponds to the residual between the two decay curves

Comparison of the emission fluorescence spectra of pure pyrene diluted in nitrogen (black line) and pyrene sampled from the flame (red line) after excitation at 321 nm
The convolution function (blue dash line) corresponding to the best fit of our experimental LIF signal has been obtained by considering a single exponential decay with a fluorescence lifetime of 16.9 ns in the spectral range 430–450 nm. Hence, the fluorescence lifetime value obtained in the \((S_{1} \to S_{0} )\) emission range confirms the very short duration of the process and is found close to the natural lifetime of 16.4 ns of this state determined by Berlman et al. [32]. We applied the same procedure for the signal measured in the range 340–360 nm. In this case, it was necessary to consider a bi-exponential decrease in order to get a satisfying agreement between the convolution function and the measured fluorescence decay. The values of the two decay times providing the best agreement with our experimental signal are 10.2 ns and a longer one of 36.5 ns.
3.2 Spectral assessment of the selectivity
The second step of this work concerned the selectivity of the method for the measurement of fluoranthene in flame conditions. This verification has been carried out by transposing the previous procedure to the fluoranthene sampled from a sooting flame. To this aim, we chose to sample species from the richest flame (ϕ = 2.32) 40 mm above the burner, i.e., in a zone rich in various PAHs [31] and where spectral interferences are expected to occur.
As the aim of this work was to highlight the ability of the method for the selective measurement of two mass isomers, we first checked that the excitation and emission fluorescence spectra of pyrene sampled from the flame were free from spectral interference. The comparison of these spectra with the ones obtained with pure pyrene is reported in Figs. 8 and 9. As expected from previous work, we did not observe any difference between spectra of pure pyrene and pyrene sampled from the flame.

Comparison of the fluorescence excitation spectra of pure pyrene diluted in nitrogen (black line) and pyrene sampled from the flame (red line)
Concerning the fluoranthene, we also noted a good concordance between the excitation spectra of pure and sampled fluoranthene from the flame as reported in Fig. 10. This part of the spectrum corresponds to the \(S_{4} \leftarrow S_{0}\) transition collected with a bandwidth collection around (430–450 nm) already discussed in the paper. We note a poorer signal-to-noise ratio for the spectrum obtained in the flame because of the lower concentration of fluoranthene in the flame.

Comparison of the fluorescence excitation spectra of pure fluoranthene diluted in nitrogen (black line) and fluoranthene sampled from the flame (red line)
The emission fluorescence spectra of pure and sampled fluoranthene from the flame are reported in Fig. 11. It shows that the spectrum of the fluoranthene extracted from the flame is perturbed by a strong interference in the range (300–400 nm) coming from other species also sampled at 40 mm in the flame.

Comparison of the fluorescence emission spectra upon 282 nm excitation of pure fluoranthene diluted in nitrogen (black line) and fluoranthene sampled from the flame (red line)
The likely species responsible for this interference should be compounds absorbing at 282 nm. Considering the much higher intensity of the fluorescence signal in the range (300–400 nm) than at 450 nm, the most probable candidates are expected to be smaller PAHs than the fluoranthene. Hence, we speculated the possible contribution of the most relevant PAHs fulfilling these requirements which are naphthalene, anthracene, phenanthrene, and pyrene. In order to validate this hypothesis, we have simulated the fluorescence spectrum generated by a mixture of these compounds with fluoranthene. For this purpose, we followed a similar procedure as reported in [45] and considered that the intensities of the LIF signals of the different species were only function of their concentrations and molar extinction coefficients. The molar extinction coefficients were issued from the work published by Karcher et al. [41] except for naphthalene (not reported in [41]) for which we considered the value given by Berlman [9]. All these values have been determined at ambient temperature and in cyclohexane. This solvent effect induces a slight spectral shift of 6 nm with our spectra recorded in the gas phase. This value has been determined by comparing the peak position of the transition measured at 282 nm for fluoranthene in the gas phase (our conditions) which corresponds to the spectral feature determined at 288 nm in cyclohexane [41]. Hence, all the molar extinction coefficients considered for the simulation (see Table 3) correspond to the molar extinction coefficients determined at 288 nm by Karsher et al. [41] and Berlman [9] for each PAHs.
The concentrations of naphthalene, pyrene, and fluoranthene at 40 mm above the burner have been measured by using the JCLIF method [46]. The entire procedure used for pyrene and fluoranthene measurement is described in the Sect. 3.3. For naphthalene, we used the same procedure as described in [26]. As mentioned in Sect. 2, the slight difference in the concentrations of pyrene and naphthalene between this work and previous ones is due the use of a new burner as well as new pressure regulation system which led to slightly different concentration profiles. Anthracene and phenanthrene mole fractions (not studied in this work) have been estimated in-between the mole fraction values of naphthalene and pyrene according to flame modeling [31]. All these values are reported in Table 3.
By this way, we could define the relative intensities of the emission fluorescence spectrum of each PAH by multiplying their normalized spectra by the product of the corresponding mole fraction and molar extinction coefficient. Reference fluorescence spectra of naphthalene, pyrene, and fluoranthene have been measured with our experimental setup whereas we used the anthracene and phenanthrene emission spectra reported by Karcher et al. [41]. We finally summed the individual spectra of each species and obtained a simulation of the global fluorescence emission spectrum coming from the flame. In Fig. 12, we have plotted the spectral contributions of the considered PAHs and the corresponding global spectrum.

Simulation of the fluorescence spectra upon 282 nm excitation issued from the main PAHs sampled from the flame with the concentrations measured or estimated around 40 mm above the burner. The purple spectrum corresponds to the sum of the different spectral contributions for each PAH
Figure 13 shows the comparison of the simulated spectrum with the experimental spectrum obtained with species sampled at 40 mm from the flame. Despite the uncertainties affecting the spectroscopic data used for the simulation and the fact that the fluorescence quantum yield has not been taken into account, the good concordance between the simulated and experimental spectra gives confidence to our approach. We deliberately chose to not consider the quantum efficiencies of the different species in our calculation as there are many parameters that can influence their values which are difficult to take into account correctly. Quantum efficiency depends indeed on the radiative and non-radiative relaxation processes which are not all well known for these compounds. Especially, the efficiency of internal conversion (IC) and intersystem conversion (ISC) which govern the relaxation processes in free collision regime are function of the excitation energy and so difficult to predict. Quantum efficiencies provided in the literature are generally given for PAHs in solution [10] and so, not necessarily representative of the gas phase. This is why we did quantum yield that was not considered in the simulation.

Comparison of the fluorescence emission spectrum from the flame after excitation at 282 nm (red line) and the simulated spectrum issued from the sum of pure PAHs spectra (purple line) after excitation at 282 nm
In this simulation, we attribute the range 430–450 nm to the fluorescence emission of fluoranthene. This attribution is supported by different evidences which are discussed below. First, we can eliminate all the PAHs that cannot provide emission signal in the range 430–450 nm. This is the case for all six-membered rings consisting of only two or three aromatic rings [9], except anthracene which can likely highlight some small spectral features in this wavelength range, as can be seen in Fig. 12. We can also eliminate the case of smaller species as fluorene or acenaphthene (expected in higher concentration than fluoranthene). Indeed, they do not provide emission bands above 370 nm [9]. We also envisaged the case of substituted PAHs of naphthalene or anthracene with side chains. However, in addition to the fact that they are always expected in considerably lower concentration than the corresponding based compounds [47], these kinds of PAHs do not have emission bands enough shifted to the red to provide significant signal in the range 430–450 nm [9]. Pyrene could potentially interfere in this range. This is why we took it into account in our simulation. However, we showed that the absorption coefficient of this species and substituted derivatives upon 282 nm excitation is not high enough to lead to a measureable emission contribution in the range 430–450 nm. Other higher molecular weight PAHs, with more than four aromatic rings, providing they have strong enough absorption bands around 282 nm, are in too low concentrations to induce a measureable emission signal in the current condition of our setup.
The dependence of the fluorescence signal to a specific excitation wavelength at 282 nm is also very important and has to be kept in mind to evaluate the selectivity of the setup. Indeed, not only likely interfering PAHs should have an emission band in the range 430–450 nm but above all they should possess an absorption band around 282 nm. The comparison of the excitation spectrum recorded in the flame 40 mm above the burner (i.e., in a region rich and full of various PAHs) and the one recorded from the pure fluoranthene sample (with a collection bandwidth centered on 440 ± 10 nm) did not highlight any significant difference. Hence, even if the spectral feature measured in the flame is much noisier because of the lower concentration, the position and linewidth of the absorption band look very similar for both excitation spectra. In case of interference with other PAHs excited from the gas sampled from the flame, we would expect differences in the line shape as enlargement or change in the spectral feature or shift of the peak position. This is clearly not the case here, which reinforces our confidence that we selectively excite fluoranthene.
The last argument supporting the selective excitation of fluoranthene concerns the temporal fluorescence signal. This last parameter appears indeed strongly correlated to the excited species. It is not only a question of structure of the molecule but it depends on numerous factors as the radiative lifetimes and various deactivation processes (IC, IVR, or intersystem crossing ISC), which are all very specific of the PAHs. The probability that two excited PAHs at a same wavelength provide an emission signal in the same wavelength range characterized by an identical temporal fluorescence signal, even if possible, is somehow very low. This last point has been verified by recording the temporal fluorescence signal upon 282 nm excitation wavelength and collected in the wavelength range (430–450 nm). As can be seen in Fig. 14, the fluorescence signal is very short in comparison with the one of naphthalene and pyrene. Now, if we compare the fluorescence signal obtained for pure fluoranthene and the one measured from species sampled from the flame, as reported in Fig. 15, an excellent agreement is obtained.

Comparison of the LIF temporal signals of naphthalene, pyrene, and fluoranthene measured in the conditions of the free jet expansion experiment

Comparison of LIF temporal decay signals of pure fluoranthene (black line) and fluoranthene sampled from the flame (red line). Signals are collected between 430 and 450 nm
In conclusion, even if some uncertainties can remain concerning the selectivity of our measurements, the signal emitted in the range 430–450 nm upon 282 nm excitation can be mainly assigned to the fluoranthene fluorescence and provide a reasonable confidence of the selectivity of the JCLIF method for the measurements of the two mass isomers, pyrene and fluoranthene, in sooting flames.
3.3 Application of the method for the quantitative measurement of pyrene and fluoranthene profiles in sooting flames
The LIF experimental profiles of pyrene and fluoranthene have been measured in two different flames (ϕ = 2.05 and ϕ = 2.32) and calibrated according to the standard addition method. This method relies on the addition of small amounts of known concentrations of a pure PAH to the flow of PAHs sampled from the flame. This calibration is done at fixed position of the microprobe in the flame, generally at the peak concentration of the profile (or in a plateau). By varying the concentration of the added PAH to the PAH sampled from the flame (which is constant), the corresponding variations of the LIF signals measured in the jet are thus directly proportional to the concentration of the added PAH. Doing this, we determine a calibration curve related to the variations of the LIF signal coming from both the PAHs sampled from the flame and the added one. From this curve, we can show that the absolute value of the x intercept (LIF signal equal to zero) corresponds to the value of the concentration of the PAH sampled from the flame at the height of sampling. All the details concerning the method have been published in a previous work [27]. In the case of the linear regime of excitation, the LIF signal can be expressed as:
Here, the PAH mole fraction \(X_{{{\text{PAH}}, {\text{flame}}}}\) is affected by the added flow of nitrogen used to carry the pure PAH. To obtain the initial mole fraction of the PAH sampled from the flame \(X_{{{\text{PAH, flame}},0}}\), the experimentally determined mole fraction \(X_{\text{PAH, flame}}\) has to be corrected for the flow rate (FR) dilution:
In these experiments, the flow rate of the sampled gases was measured to 130 ml/min while the added flow rate of the pure PAHs diluted in nitrogen was 20 ml/min, leading to a flow rate dilution of 1.15. This method offers the benefit to calibrate the LIF signal within the same gaseous environment as for the determination of the relative concentration profiles of the PAHs formed in the flame. As all additional gas flows required for the calibration method are accurately regulated, the main uncertainty concerning the concentration measurement with this method comes from the possible fluctuations of the flow rate of the gases sampled from the flame [i.e., the FRtotal/FRsampling ratio in Eq. (2)]. The observed variation of the FRtotal/FRsampling ratio was determined to 5 %, leading to uncertainties about 10 % on the concentration measurements.
The Fig. 16 shows the calibration curves obtained using the standard addition method for pyrene and fluoranthene.

Calibration procedure using the standard addition method for pyrene (in black) and fluoranthene (in red). Intersection of the line and the X axis gives the value of concentration of the PAH in flame, which has to be corrected for the dilution used during the experiment
Table 4 sums up the experimental conditions and results for the calibration using the standard addition method applied to the flame at ϕ = 2.32.
As expected, the calibration curves in Fig. 16 appear as straight lines whose intersections with the horizontal axis provide the values of the mole fraction of pyrene sampled from the flame. After the correction due to the dilution, we obtain the value of the mole fraction of pyrene sampled at 10 mm above the burner equal to 390 (±39) ppb for the reference flame (P = 200 Torr and ϕ = 2.32). For the fluoranthene, the concentration is 104 (±11) ppb at a height of 11 mm in the sootiest flame. By comparison of the LIF signals in the two flames, we have determined that in the 2.05 flame, the concentration of pyrene is 50 (±5) ppb. The concentration of fluoranthene is equal to 9.5 (±1) ppb in this same flame at 11 mm.
In the 2.32 flame, the uncertainties of our measurements (taking into account the reproducibility of the LIF experiments and the uncertainties due to the calibration) have been estimated around 17 and 20 % for pyrene and fluoranthene, while in the 2.05 flame, they are about 20 and 30 %. In these conditions, we estimate a sensitivity of the method to around 1 ppb, as for previous work. The difference is that the study has been done here with a spectrometer instead of bandpass filter, which provides a much better control of the selectivity of the measurements. Concerning the profiles of pyrene, which are reported in Fig. 17, we observe that they have a shape similar to the shape already observed in previous works [27]. There is a first fast increase of the concentration until a maximum value around 10 mm, followed by a decrease and a valley around 13 mm, before a strong re-increase of the concentration in the burnt gases.

Mole fraction profiles of pyrene for the two flames at equivalent ratio of ϕ = 2.32 (black squares) and ϕ = 2.05 (red squares) stabilized at 200 torr
The fluoranthene profiles, which have been plotted in Fig. 18, highlight a maximum value around 10 mm above the burner followed by a plateau of concentration, featuring equilibrium between formation and consumption reactions, before a re-increase of the concentration beyond 20 mm. As expected from the literature, the pyrene concentration is higher than the fluoranthene one. Indeed, the concentration ratio of pyrene over fluoranthene seems to be almost constant around a factor of 4 all along the profiles in the two flames.

Mole fraction profiles of fluoranthene for the two flames at equivalent ratio of ϕ = 2.32 (black squares) and ϕ = 2.05 (red squares) stabilized at 200 torr
These results are concordant with previous works [20–23], demonstrating a prominence of the pyrene concentration compared to the fluoranthene concentration. For instance, Petrarca and Marconi [21] observed in a n-heptane diffusion flame a ratio of 5, while Tregrossi et al. [22] measured a ratio of 1.5 in rich premixed benzene flames stabilized at atmospheric pressure. According to Melton et al. [23], this ratio is equal to 3 in atmospheric premixed ethane flames and to 2.5 in methane flames [20]. Sanchez et al. [48] studied the pyrolysis of acetylene between 973 and 1323 K. Their work also showed a constant ratio of the concentrations of pyrene and fluoranthene equivalent to 1.3 in this temperature range.
4 Conclusion
In this work, we studied the ability of the jet-cooled LIF technique for the measurements of two mass isomers, fluoranthene and pyrene, envisaged as key species in soot formation mechanisms. The selectivity of the method has been tested and discussed on the basis of spectroscopic and photophysics considerations. Although uncertainties remain, because it is not possible to consider all the PAHs in this analysis, we believe that we can reasonably envisage the emission signal measured in the range 430–450 nm upon 282 nm excitation as mainly coming from the fluoranthene. The selectivity of the method could be greatly enhanced with a more efficient cooling of the species. This is part of a future project which necessitates the modification of the free jet expansion system to enable to work with a gas carrier at higher pressure without condensing the sampled species. This improvement should favorably improve the sensitivity of the setup as well. This sensitivity, relying on the spectroscopic properties of the species, has been currently estimated around 1 ppb. Quantitative concentration profiles of pyrene and fluoranthene could have been measured in two low-pressure methane flames with different equivalence ratios. In both flames, the concentration of pyrene has been found higher than the concentration of fluoranthene. A constant concentration ratio around 4 has been measured which is quite concordant with the few measurements issued from the literature performed by GCMS. These results confirm the favorable formation of pyrene in comparison to its main isomer in flames. We note that the experimental profile of the two PAHs highlights a peak concentration around 10 mm above the burner. These profiles seem to indicate a formation of fluoranthene and pyrene that could be interconnected. However, the sp 2 hybridization of fluoranthene could enable the torsion of its central five-membered carbon ring and therefore the formation of tridimensional edifices while pyrene can only lead to larger planar PAHs. From these experimental results, we showed that fluoranthene, which is often neglected in soot mechanisms because of the difficulties of its measure with conventional methods, has the potential in terms of concentration to play a key role in the mechanisms leading to the formation of soot particles. An analysis of these results, thanks to kinetic modeling, may provide crucial information about the reactions potentially leading to the formation of this species and its role in the formation of soot particles.
References
A. D’Anna, Combustion Generated Fine Carbonaceous Particles (KIT Scientific Publishing, Karlsruhe, 2009)
A. D’Anna, Proc. Combust. Inst. 32, 593 (2009)
H. Wang, Proc. Combust. Inst. 33, 41 (2011)
M. Frenklach, H. Wang, Symp. Int. Combust. 23, 1559 (1991)
H. Sabbah, L. Biennier, S.J. Klippenstein, I.R. Sims, B.R. Rowe, J. Phys. Chem. Lett. 1, 2962 (2010)
J.D. Herdman, J.H. Miller, J. Phys. Chem. A 112, 6249 (2008)
J. Appel, H. Bockhorn, M. Frenklach, Combust. Flame 121, 122 (2000)
S. Bejaoui, X. Mercier, P. Desgroux, E. Therssen, Combust. Flame 161, 2479 (2014)
I.B. Berlman, Handbook of Fluorescence Spectra of Aromatic Molecules, 2nd edn. (Academic Press, New York, 1971)
J.B. Birks, Photophysics of Aromatic Molecules, 1st edn. (Wiley-Interscience, London, 1970)
H. Hepp, K. Siegmann, K. Sattler, Chem. Phys. Lett. 233, 16 (1995)
K. Siegmann, K. Sattler, H.C. Siegmann, J. Electron Spectrosc. Relat. Phenom. 126, 191 (2002)
M. Kamphus, M. Braun-Unkhoff, K. Kohse-Höinghaus, Combust. Flame 152, 28 (2008)
M. Kamphus, N.N. Liu, B. Atakan, F. Qi, A. McIlroy, Proc. Combust. Inst. 29, 2627 (2002)
Y. Li, L. Zhang, Z. Tian, T. Yuan, J. Wang, B. Yang, F. Qi, Energy Fuels 23, 1473 (2009)
T.S. Kasper, P. Obwald, M. Kamphus, K. Kohse-Höinghaus, Combust. Flame 150, 220 (2007)
N. Hansen, T. Kasper, S.J. Klippenstein, P.R. Westmoreland, M.E. Law, C.A. Taatjes, K. Kohse-Höinghaus, J. Wang, T.A. Cool, J. Phys. Chem. A 111, 4081 (2007)
Y. Li, Z. Tian, L. Zhang, T. Yuan, K. Zhang, B. Yang, F. Qi, Proc. Combust. Inst. 32, 647 (2009)
B. Yang, Y. Li, L. Wei, C. Huang, J. Wang, Z. Tian, R. Yang, L. Sheng, Y. Zhang, F. Qi, Proc. Combust. Inst. 31, 555 (2007)
T.R. Melton, A.M. Vincitore, S.M. Senkan, Symp. Int. Combust. 27, 1631 (1998)
L. Petarca, F. Marconi, Combust. Flame 78, 308 (1989)
A. Tregrossi, A. Ciajolo, R. Barbella, Combust. Flame 117, 553 (1999)
T.R. Melton, F. Inal, S.M. Senkan, Combust. Flame 121, 671 (2000)
P. Desgroux, X. Mercier, K.A. Thomson, Proc. Combust. Inst. 34, 1713 (2013)
X. Mercier, M. Wartel, J.F. Pauwels, P. Desgroux, Appl. Phys. B 91, 387 (2008)
M. Wartel, J.F. Pauwels, P. Desgroux, X. Mercier, Appl. Phys. B 100, 933 (2010)
M. Wartel, J.F. Pauwels, P. Desgroux, X. Mercier, J. Phys. Chem. A 115, 14153 (2011)
K. Ruužička, I. Mokbel, V. Majer, V. Ru̇u̇žička, J. Jose, M. Zábranský, Fluid Phase Equilib. 148, 107 (1998)
J.L. Goldfarb, E.M. Suuberg, J. Chem. Eng. Data 53, 670 (2008)
I.S. Burns, X. Mercier, M. Wartel, R.S.M. Chrystie, J. Hult, C.F. Kaminski, Proc. Combust. Inst. 33, 799 (2011)
A. El Bakali, X. Mercier, M. Wartel, F. Acevedo, I. Burns, L. Gasnot, J.F. Pauwels, P. Desgroux, Energy 43, 73 (2012)
I.B. Berlman, H.O. Wirth, O.J. Steingraber, J. Am. Chem. Soc. 90, 566 (1968)
J.W. Hofstraat, G.P. Hoornweg, C. Gooijer, N.H. Velthorst, Spectrochim Acta Part A 41, 801 (1985)
J. Kolc, E.W. Thulstrup, J. Michl, J. Am. Chem. Soc. 96, 7188 (1974)
Y. Numata, Y. Suzuki, I. Suzuka, J. Photochem. Photobiol. A 237, 49 (2012)
J. Michl, J. Mol. Spectrosc. 30, 66 (1969)
L. Pesteil, P. Pesteil, F. Laurent, Can. J. Chem. 42, 2601 (1964)
E. Heilbronner, J.-P. Weber, J. Michl, R. Zahradnik, Theoret. Chim. Acta 6, 141 (1966)
I.Y. Chan, M. Dantus, J. Chem. Phys. 82, 4771 (1985)
A.A. Ruth, M.T. Wick, Chem. Phys. Lett. 266, 206 (1997)
W. Karcher, R.J. Fordham, J.J. Dubois, P.G.J.M. Glaude, J.A.M. Ligthart, Spectral Atlas of Polycyclic Aromatic Compounds (D. Reidel Publishing Company, Dordrecht, 1985)
D.L. Philen, R.M. Hedges, Chem. Phys. Lett. 43, 358 (1976)
K.M. Bark, R.K. Force, J. Phys. Chem. 93, 7985 (1989)
S. Preus, DecayFit—Fluorescence Decay Analysis Software 1.3, FluorTools. www.fluortools.com
R. Sun, N. Zobel, Y. Neubauer, C. Cardenas Chavez, F. Behrendt, Opt Lasers Eng 48, 1231 (2010)
T. Mouton. Analyse des processus de nucléation et de croissance des particules de suie dans des flammes par fluorescence induite par laser en jet froid appliquée aux hydrocarbures aromatiques polycycliques et par incandescence induite par laser Ph.D. Thesis, University of Lille1, Villeneuve d’Ascq (France), 2014
K.H. Homann, Symp. Int. Combust. 20, 857 (1985)
N.E. Sanchez, A. Millera, R. Bilbao, M.U. Alzueta, J. Anal. Appl. Pyrolysis 103, 126 (2013)
Acknowledgments
The authors thank Sébastien Batut, Pascal Demaux and Olivier Hombert for their technical help and significant investment in this work. The PC2A participates to the Institut de Recherche en Environnement Industriel (IRENI) and to the research program CPER Climibio, which are financed by the Région Nord Pas-de-Calais, the Communauté Urbaine de Dunkerque, the Ministère de l’Enseignement Supérieur et de la Recherche, the CNRS and the European Funds for Regional Economic Development (FEDER). The CaPPA project (Chemical and Physical Properties of the Atmosphere) is funded by the French National Research Agency (ANR) through the PIA (Programme d’Investissement d’Avenir) under Contract ANR-11-LABX-0005-01 and by the Regional Council Nord-Pas de Calais and the European Funds for Regional Economic Development (FEDER).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Mouton, T., Mercier, X. & Desgroux, P. Isomer discrimination of PAHs formed in sooting flames by jet-cooled laser-induced fluorescence: application to the measurement of pyrene and fluoranthene. Appl. Phys. B 122, 123 (2016). https://doi.org/10.1007/s00340-016-6397-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00340-016-6397-9