
Abstract
An advanced in situ diode laser hygrometer for simultaneous, sampling-free detection of interstitial H2 16O and H2 18O vapor was developed and tested in the aerosol interaction and dynamics in atmosphere (AIDA) cloud chamber during dynamic cloud formation processes. The spectrometer to measure isotope-resolved water vapor concentrations comprises two rapidly time-multiplexed DFB lasers near 1.4 and 2.7 µm and an open-path White cell with 227-m absorption path length and 4-m mirror separation. A dynamic water concentration range from 2.6 ppb to 87 ppm for H2 16O and 87 ppt to 3.6 ppm for H2 18O could be achieved and was used to enable a fast and direct detection of dynamic isotope ratio changes during ice cloud formation in the AIDA chamber at temperatures between 190 and 230 K. Relative changes in the H2 18O/H2 16O isotope ratio of 1 % could be detected and resolved with a signal-to-noise ratio of 7. This converts to an isotope ratio resolution limit of 0.15 % at 1-s time resolution.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
High-altitude cirrus clouds play a major role in earth’s climate. Cirrus clouds influence the radiation balance of the earth by infrared radiation trapping as well as reflection of incoming solar radiation [1]. To better understand cirrus cloud formation and persistent supersaturation, as recently detected in cold cirrus clouds [2–6], it is of great interest to investigate ice cloud formation in more detail on a microphysical level. Phase transitions, particularly of water, are often investigated using isotopic tracers [7–10]. For ice clouds, water (= H2 16O) and its rare isotopes provide new information about cloud formation processes and their dependencies on gas conditions or aerosol availability [11, 12]. In the past, airborne instruments were applied to measure the absolute isotope depletion of H2 18O and HDO in the atmosphere, which ranges from almost 0 to −200 ‰ [13–16]. However, fractionation factors of water isotopes for the upper troposphere and lower stratosphere (UT/LS) are only available as extrapolations of measurements as low as 230 K, yielding a fractionation of about 3 % [17–20]. Therefore, new studies of water isotope fractionation with single digit per mill precision are necessary to validate the applicability of these extrapolations to analyses of atmospheric observations in the UT/LS region [8, 17–20].
The “isotopic water in clouds” (ISOCLOUD) project is an international cooperation between the University of Chicago (UC), Karlsruhe Institute of Technology (KIT) and Physikalisch-Technische Bundesanstalt (PTB, the German national metrology institute) and was initiated to develop new instrumentation for dynamic isotopically resolved water vapor measurements, to perform isotope-resolved measurements in clouds and to gain new insights on cloud formation and isotope fractionation within these clouds [21]. In a first step, the project focused on the validation of isotope fractionation models and the direct measurement of equilibrium fractionation of HDO and H2 18O. In this context, it is of great importance to detect small relative changes in the isotopic composition. For this purpose, suitable instruments had to be developed to allow a sampling-free open-path detection of isotopic water species. This paper focusses on the technological and instrumentation advances by PTB and KIT (both funded by the German Science Foundation (DFG)) with respect to the H2 16O and H2 18O detection and takes advantage of preliminary experimental data taken during ISOCLOUD measurement campaigns. In-depth microphysical analyses and additional data for testing and/or development of new atmospheric models and theories related to isotopic water vapor and nucleation processes will be the subject of future publications.
The detection of interstitial water vapor within clouds is difficult, particularly given the risk of changing the phase equilibrium of ice and vapor during an extractive sampling process. Furthermore, cold ice clouds can show water vapor mixing ratios of only a few ppm for stratospheric temperatures and pressure levels. In addition, isotope-resolved studies are hindered by the low natural abundance of heavy isotope H2 18O, which is suppressed by 1:498 with respect to H2 16O. As a result, the sensitivity requirements for isotope-resolved measurements are very high. All these issues are already present at static conditions, but when capturing dynamic changes of isotope signatures in cold ice clouds, a time response of 1 s or faster, and an effective compensation of the unavoidable, spectrally broadband transmission fluctuations caused by the ice crystals, is required. Under these boundary conditions, isotope ratio mass spectrometry (IRMS) [22, 23] is very difficult to apply. Consequently, there is a need for an isotope-resolving instrument with a high temporal resolution and the ability to measure water vapor in equilibrium with the surrounding ice cloud without changing the phase equilibrium. By simultaneously measuring at least two isotopes, additional inter-spectrometer uncertainties and error amplification can be avoided.
Recently, laser-based isotope diagnostics including integrated cavity output spectroscopy (ICOS) [24, 25] and optical feedback cavity-enhanced absorption spectroscopy (OF-CEAS) [26, 27] have become popular [28]. However, both methods are greatly limited due to necessary gas sampling and the risk of shifting the phase equilibrium as well as surface adsorption effects from the gas handling system, resulting in systematic errors in the isotope ratio. Compared with various other laser-based techniques, direct tunable diode laser absorption spectroscopy (dTDLAS) has advantageous features for in situ cloud measurements. With dTDLAS, it is possible to obtain absolute and calibration-free vapor concentration values even with strong transmission fluctuations by broadband absorbers such as particles present on the signal [29–32]. In contrast to extractive instruments, in situ instruments are able to observe a continuously changing isotope ratio over the complete condensation process, without interference of ice particles or the risk of changing the phase equilibrium.
The investigation of cloud formation processes needs well-defined and repeatable environmental conditions, as well as the possibility to control parameters such as supersaturation, cooling rate and the type of condensation nuclei. This is almost impossible to achieve in the free atmosphere, but such conditions can be recreated in the aerosol interactions and dynamics in atmosphere chamber (AIDA) at the Karlsruhe Institute of Technology (KIT). Within AIDA, it is possible to generate atmospheric conditions with temperatures ranging from 183 K to 333 K and pressures ranging from 1 to 1000 hPa with uncertainties (1σ) of ±0.3 K and ±1 hPa, respectively). It is also possible to add different aerosols, such as H2SO4, dessert dust or organic aerosols. Furthermore, the AIDA simulation chamber is equipped with reliable and validated hygrometers capable of measuring H2 16O inside clouds (APicT using TDLAS) as well as total water concentration (APeT using TDLAS, dew point mirror 373 from MBW Calibration Ltd.) and condensed water content (FTIR) [33–37].
Unfortunately, for isotope-resolved cloud studies, the direct simultaneous detection of main and rare water isotopes H2 16O and H2 18O is not possible. Such a measurement would be possible by combining the data of different instruments for different isotopes. However, such a combination suffers a greatly enhanced risk of inter-instrumental differences caused by different evaluation principles as well as by different time bases or by differences in the spatial overlap through different in situ paths. The combination of these effects can lead to large systematic errors and additional noise on the isotope ratio, which then consequently would require a calibration. To minimize these risks and drawbacks, the instrument design focussed on developing the time-multiplexed dual-isotope spectrometer described below.
Within the ISOCLOUD project, two isotope-selective instruments were developed. The first instrument was designed to measure HDO fractionation and will be described elsewhere. The second instrument, presented in this paper, is designed to simultaneously measure H2 16O and H2 18O. This spectrometer is based upon the well-validated APicT (AIDA PCI in-cloud TDL) instrument [33, 34, 37], but now allows for the first time a simultaneous open-path detection of H2 18O and H2 16O. The multi-channel isotope-selective AIDA PTB in-cloud TDL spectrometer (acronym: MC-IsoAPicT) employs for this a rapidly time-multiplexed, dual-wavelength TDLAS approach to detecting H2 16O and H2 18O quasi-simultaneously along the same optical path using an identical data evaluation process. By utilizing an AIDA internal, open multi-pass White cell high sensitivity, even at 190 K corresponding to a water concentration below 1 ppm, on both isotope channels is achieved.
2 Measurement principle
2.1 TDLAS
The instrument presented here is based on the principal of direct tunable diode laser absorptions spectroscopy (dTLDAS) [38], which in turn is based on the Beer–Lambert law and continuous tuning of the diode laser wavelength via modulation of the laser current. In its basic setup, the light emitted by the laser is guided along an “open” measurement path inside AIDA. After passing through the absorption volume, the light is focused on a photodiode. The resulting photocurrent is directly digitalized including all offsets and disturbances. This system can be described by an extended version of the Beer–Lambert law [39, 40]:
where the detected intensity I(λ) is given by the emitted intensity I 0(λ), the transmission function Tr(t) and the absorption depending on the temperature-dependent line strength S(T), the absorber number density N, the absorption path length L, the normalized line shape function g(λ – λ 0) centered around λ 0 and additional broadband incident light E(t). From Eq. 1, the molecular concentration can be determined based on the ideal gas law
where the integral is the area A of the Voigt profile fitted to the absorption line. The influences of broadband transmission loss Tr(t) versus narrowband molecular absorption caused by absorber molecules such as H2O can be seen in Fig. 1. To summarize, the parameters k B, L and S(T) are either constant or calculated based on spectral databases, while temperature and pressure are measured. Since the dynamic laser-tuning coefficient ∂λ/∂t has previously been experimentally determined for the intended application, the measurement of the incident intensity, and therefore dTDLAS, allows an absolute molecule detection at ideal gas conditions without any calibration [29, 30, 34]. Additionally, the robustness of dTDLAS regarding transmission fluctuations is a great advantage in environments with highly scattering particles such as ice crystals. Detailed explanations about TDLAS can be found in [38] or [41, 42].

Schematic of the relation between the emitted and received laser intensity (I 0 (λ), I(λ)), the transmission function Tr(t) and additional broadband emissions E(t), used for direct tunable diode laser absorption spectroscopy (dTDLAS)
2.2 Isotope ratio calculation
For ultra-high resolution detection of isotopic signatures (e.g., with mass spectrometry, IRMS), it is necessary to reference very small isotope ratio changes to a stable reference material with very well-defined isotope ratio. For water isotopes, one of these reference materials is Vienna Standard Mean Ocean Water (VSMOW). All measurements of isotope ratio changes are based on this reference, whose absolute isotopic composition is not defined at the high resolution of the IRMS technique due to the lack of a suitable absolute measurement technique with an adequate high absolute accuracy. IRMS measurements are therefore ultra-high resolution relative measurements.
When using absorption spectroscopy to measure the isotopic composition of a gas sample, the physics of the absorption process allows for the differentiation of molecules of identical total masses but different isotopic composition. Therefore, we define the isotope ratio on molecular differences acc. Equation 3.
where n x is the number density of the rare isotope and n a is the number density of the main isotope of the molecule.
For isotope ratio measurements, individual absorption lines for the different isotopes are needed, so that the isotope ratio can be directly calculated. Based on Eq. 2, the line area A and the line strength S(T) are the only isotope-specific parameters. Therefore, the isotope-resolved absorber density ratio can be calculated based solely on the measurement of two different absorption lines (Eq. 4).
When calculating the isotope ratio acc. Equation 4 utilizing the line strengths taken from the HITRAN2008 database [43], it is important to consider that HITRAN line strength data are scaled to fixed values of the natural abundance [44], which coincides with the isotope abundance of VSMOW. An isotope ratio R x–a = 1 corresponds therefore to the isotope abundance of HITRAN/VSMOW rather than the absolute isotope ratio.
Molecules of different isotopic composition also differ in the saturation vapor pressure. This difference leads to an isotope-dependent fractionation during phase change. For H2 18O/H2 16O, this difference can be expressed through a fractionation factor α. For the phase change between vapor and ice, the following formula is applied:
Since isotopic fractionation is a complex process, we refer to [20, 45, 46] for additional information about isotopic fractionation and to [17, 21, 47–49] for measurements of the equilibrium fractionation.
2.3 Line selection
In recent years, laser-based water isotope measurements (H2 16O, HDO, H2 18O, H2 17O) were mostly utilizing absorption lines in the 1.4 µm ν1 + ν3 overtone band of water [8, 26, 28, 45], since highly developed and cost-efficient telecommunications hardware can be used in this spectral region. As mentioned earlier, measurement methods included CRDS and CEAS, as well as ICOS. However, for measurements of the rare water isotope H2 18O with direct absorption spectroscopy (dTDLAS), stronger absorption lines are needed which can be found in the ν1, ν3 band around 2.7 µm. Therefore, we decided to employ the well-known and previously used 211-101-line at 7299.63 cm−1 for the detection of H2 16O [29, 41, 50] and the 3786.93 cm−1 absorption line for the rare isotopes H2 18O, which was agreed upon within the ISOCLOUD project. Important criteria for the line selection were the temperature dependence of the line strength, the spectral separation to other gas species as well as other water isotope lines and the availability of lasers and optical components for the specific wavelength range. With more than 100,000 available water lines, this process is time consuming and difficult and will be described in detail in future publications. Since the H2 16O absorption line has been characterized well in previous publications [50], the uncertainties for this line are below 5 %. For the 2.7-µm line, we used the information from the HITRAN2008 database [43], where the uncertainties are significantly higher and only ranges for the uncertainties are given. For the broadening coefficients, the listed uncertainty is 2–10 % and the uncertainty of the temperature dependence is 10–20 %. For the line strength, the uncertainty in HITRAN2008 is specified to be within the 5–10 % category. It is clearly visible that for future measurements the line data accuracy has to be improved significantly.
Using the HITRAN parameters, the temperature dependence of the line strength in the relevant temperature range is calculated to be below 0.7 %/K. However, in addition to the single-isotope uncertainties, the temperature dependence is also important for accurate isotope ratio measurements. As shown in Fig. 2, the similar ground-state energy of both lines leads to a temperature dependence of the isotope ratio of below 4.5 × 10−5/K, thus achieving a sufficient stability. Alternatively, using an additional H2 16O line in the 2.7 µm range (instead of in the 1.4 µm range) would not be beneficial for the accuracy of isotope ratio measurements. Although the temperature dependence is in the same magnitude as for the actually chosen lines, the ground-state energy is significantly different, which would lead to a temperature dependence for the isotope ratio H2 18O2.7µm/H2 16O2.7µm of 40 %/K.

Temperature dependence of the line strength ratio is shown for the selected absorption lines for H2 18O (2.7 µm) and H2 16O (1.4 µm). This characterizes the gas temperature-induced uncertainty on the isotope ratio based on HITRAN uncertainties
3 Experimental design
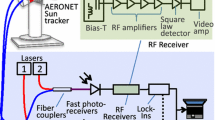
The schematic setup of the MC-IsoAPicT spectrometer is shown in Fig. 3. The spectrometer optics are directly attached to the AIDA chamber [33–36, 51] with the open-path multi-pass White cell mounted inside the chamber. The 84 m3 chamber (4 m inner diameter and 7.5 m height) is designed to allow pressure and temperature variations similar to the typical dynamic updrafts of humid air masses in the atmosphere [52–55]. Through adiabatic expansion, cooling rates up to 3 K/min can be achieved. It is even possible to generate cold cirrus cloud conditions between 190 and 230 K with the option to manipulate the freezing point of the water vapor by adding different ice nuclei (aerosols) [52–55]. Thermocouples are mounted inside the AIDA chamber along several vertical and horizontal axes to monitor the temperature homogeneity of the chamber (±0.3 K during cloud expansion), while the pressure can be stabilized to ±1 hPa. For isotope studies, the ADIA facility also allows enrichment of the water isotope abundances, which is quite advantageous compared with atmospheric studies, because it reduces the sensitivity requirements for the detection of the heavier isotopes. Additionally, a mixing fan at the bottom center ensures constant mixing of the air masses.

Setup of the multi-channel isotope-selective AIDA PTB in-cloud TDL spectrometer (MC-IsoAPicT) mounted to and inside the AIDA cloud chamber (large circle)
Directly connected to AIDA is the optics box of the H2 18O and H2 16O instrument. The box is separated from AIDA with two wedged CaF2 windows and purged with dry air. Placed inside this box are a 2.7-µm DFB laser (Nanoplus GmbH), a fiber-coupled collimator for the 1.4-µm DFB laser, whose light is supplied from outside the box via a vacuum-sealed fiber feed-through, and a detector (InAs, Hamamatsu). Besides containing the optical components of the spectrometer, the laser box also aids in avoiding additional water absorption outside the chamber. Since dTDLAS is a line of sight technique, there is always the possibility of parasitic absorption in the laser itself, the “outside” optics and detector system [56]. To avoid this, the laser box is placed on the outside of the AIDA chamber, but within the AIDA thermostatic housing. Consequently, the temperature of the laser box is about the same as the temperature of AIDA and the water vapor pressure in the box cannot be larger than the saturation vapor pressure at the AIDA chamber temperature. Since the laser box is constantly purged with dry air, the value is typically significantly lower. An 1.4-µm DFB laser (NEL photonics), as known from the APicT instrument [34], is placed outside the thermostatic housing in a separately purged box. Both lasers are controlled using a laser-driver control unit (Thorlabs) with a laser current driver module and peltier controller module each.
The light emitted by the 1.4-µm laser is transmitted via a single-mode fiber (SMF-28e) to a collimator inside the laser box, while the 2.7-µm laser is mounted in a custom laser mount and collimated by a custom PTB-designed aspheric ZnSe lens. The optics within the laser box superimpose the 2.7-µm laser and the 1.4-µm laser onto the same optical path using a custom-coated dichroic beam splitter. The dual-wavelength composite laser beam is then launched on a free-space path into the multi-pass White cell [57–60] mounted on the inside of AIDA. After passing through the White cell, both lasers are focused onto the same detector (InAs, Hamamatsu). With no spatial separation (both lasers are on the same optical path), they have to be separated in time [61–63] to avoid interference. This is realized via a time-multiplexed modulation scheme, which is shown in Fig. 4. By modulating each laser with a 2 kHz sawtooth function, which is switched off every other period and phase shifted by 180° relatively, the combined laser signal is generated avoiding interference between the two lasers. Given a combined repetition rate of 1 kHz (1000 µs), the measurements can be seen as quasi-simultaneous with respect to the time scales of the mixture and transmission fluctuations inside AIDA [52–55]. Hence, it is possible to detect both isotopes at the same time with different wavelengths within the same volume.

Time-multiplexed modulation scheme. The dashed line represents the signal of the 2.7-µm laser on the detector, while the continuous line shows the detected signal of 1.4-µm laser
The drawback of dTDLAS compared with other measurement techniques such as wavelength modulation spectroscopy (WMS) [64–66] or cavity-enhanced techniques such as CEAS, CRDS and ICOS [24, 26, 67] is the relatively low sensitivity. This is compensated by the implementation of a multi-pass White cell inside of AIDA. This White cell allows an adjustable path length of 24–257 m. With its refocusing behavior, the White cell ensures a stable alignment during the experiments despite the long absorptions paths. In addition, an off-axis parabolic mirror (OAP, effective focal length 2″) on the detection side of the White cell increases the stability of the focus onto the detector independent of alignment changes and vibrations. The detector current is converted and amplified with a trans-impedance amplifier (FEMTO) and directly digitalized with a 4 MS/s DAQ board (National Instruments) outside of the thermostatic AIDA housing ensuring proper operating conditions for these components.
4 Results and discussion
4.1 Single-Isotope performance
The optical performance of the two MC-IsoAPicT wavelength channels was characterized at static AIDA conditions of 203 K and 230 hPa. Figure 5 shows the absorption line profile of the H2 16O line at 7299 cm−1 after baseline correction. The measurement data are displayed as circles, while the line represents the fitted absorption line profile. The fit is an average of 1000 raw scans and is fitted with a combination of a third-order polynomial and a multi-line Voigt profile array consisting of 19 absorption lines around the 211-101 main line optimized by a Levenberg–Marquardt algorithm [68]. The residual in Fig. 5 is dominated by the well-known deficiencies of the applied Voigt profile in high signal-to-noise TDLAS scenarios. These systematic effects of line shape mismatch have been recently investigated in detail under well-controlled metrological conditions [30] and were found to be highly deterministic and stable. For line ratio measurements of a single species, this effect is quite effectively canceled as both lines show the identical behavior. The remaining systematic errors are further reduced in the presented study as we mostly aim toward relative changes in the isotopic ratio during cloud formation. For single-line detection schemes (aiming at species concentrations), however, the systematic and strongly pressure-dependent effects caused by the line shape deficiencies has to be taken into account, as they can cause systematic deviation of up to a few percent [30].

Fitted absorption line profile of the H2 16O line on the 1.4-µm channel. A 19-line Voigt fit was applied to a 1000 raw scans averaged line profile at 203 K, 230 hPa and 227-m absorption path, resulting in a H2 16O mixing fraction of 10.3 ppm, while the local residual yields a SNR of 3940
The single-scan performance of the instrument can be determined as a signal-to-noise ratio (SNR) of 3940 (based on the data in Fig. 5), which is calculated using the local 1σ standard deviation of the residual (7.5 × 10−5) and the peak height of the absorbance (2.96 × 10−1). Given the fitted concentration of 10.3 ppm, the 1σ H2 16O detection limit is calculated to be 2.6 ppb or 1.8 \({\text {ppm}} \cdot {\text {m}} \cdot \sqrt{(\text {Hz})} \). The overall dynamic range for H2 16O in this configuration is defined by a maximum absorbance value of 3 as the upper detection limit and the absolute detection limit as the lower detection limit, resulting in a range from 2.6 ppb to 87 ppm at 230 hPa and 203 K.
On the 2.7-µm wavelength channel, two strong absorption lines of H2 16O and H2 18O can be detected, as well as some weaker H2 16O lines. Since the H2 16O lines are within 0.8 cm−1 of the H2 18O line, cross talk between those lines becomes highly relevant for the fitting procedure. H2 18O and H2 16O must be treated fully independent from each other to ensure a high-quality H2 18O measurement. Taking the inappropriate temperature dependence of the 2.7-µm H2 16O line into account, it is necessary to calculate the line area of the 2.7-µm H2 16O lines based on the 1.4-µm measurements, doing so it is possible to fit the H2 16O background underneath the H2 18O line isotopic independent. Given a much wider wavelength tuning range than usual, a sixth-order polynomial was used for the reconstruction of the base line, and a multi-line Voigt profile consisting of 37 lines was used to fit the complete H2 16O background. The H2 18O line at 3786.93 cm−1 was fitted with an 11-line Voigt profile array. This fitting approach results in an isotope-independent background fit for H2 16O, allowing high precision H2 18O measurements in close vicinity. An exemplary measurement of the 2.7-µm channel is shown in Fig. 6 (averaging 100 single scans at 230 hPa and 203 K), yielding a total H2 18O mixing ratio of 19 ppb. From the peak absorbance and the 1σlocal standard deviation of the residual, a SNR of 217 was calculated, which can be converted into a H2 18O detection limit for this measurement of 87 ppt or 19 \({\text {ppb}} \cdot {\text {m}} \cdot \sqrt{(\text {Hz})} \). For H2 18O at 230 hPa and 203 K, a dynamic range of 87 ppt to 3.6 ppm is estimated as described above.

Top: Measured spectrum of the H2 18O line measurement (1000 scans averaged, 230 hPa, 203 K, 227 m), with the residual between measurement and fit shown below. The Voigt profile fit yields a H2 18O mixing fraction of 19 ppb with a signal-to-noise ratio of 217
The short- to mid-term stability and precision can be analyzed using an Allan-Werle deviation (shown in Fig. 7) on the single-isotope measurements at static AIDA conditions of 300.1 hPa and 194.12 K. The gas conditions (water vapor mole fraction, isotope ratio, pressure and temperature) were kept constant for the Allan-Werle measurement while the data were recorded averaging 100 raw scans. The resulting Allan-Werle plot [69] indicates the optimum number of raw scans to be averaged. Figure 7 shows an optimal averaging time of 10 s (10,000 raw scans) and 125 s (125,000 raw scans) for H2 16O and H2 18O, respectively. To achieve a sub-second time resolution, a 1000 raw scan average was used. The Allan-Werle deviation also indicates a slope larger than negative unity on the falling slope, which indicates that noise sources other than white noise are present in the signal. At large facilities such as AIDA with significant electromagnetic noise, it is difficult to clearly identify said additional noise sources. The flat bottom of the 1.4-µm channel Allan-Werle plot is a typical behavior caused by fringes. These optical interferences are limiting the number of scans to be averaged and usually originate from the fiber collimation optics or the detector window. In many studies using relatively short White cells, strong fringing caused by the White cell was observed. In our case, however, due to the mirror separation of 4.1 m, we would expect a fringe spacing of 1.2 × 10−3 cm−1 or even less. This fringe spacing is much smaller than typical widths of the water lines and already is comparable to the spectral resolution of the instrument of about 1.8 × 10−3 cm−1. Due to this situation, we were never able to see fringes caused by the White cell or detect any effect caused by such disturbances.

Allan-Werle deviation of a simultaneous mixing ratio measurement of H2 18O (scaled by natural abundance 498:1) and H2 16O under static conditions with a low mark at 10 s for H2 16O and 125 s for H2 18O
At such high sensitivities, it is important to discuss the measurement uncertainties. An overview of the different contributions to the overall measurement uncertainty of both isotopes is given in Table 1. Besides the uncertainty of the line strength, which is dominating for both H2 16O and H2 18O, the most important contribution is the temperature dependence of the line strength. It directly scales the temperature uncertainty with the mixing ratio uncertainty and depends on the ground-state energy of the chosen absorption lines. For both absorption lines used in this publication, the relative temperature coefficient is below 0.7 %/K. Including the uncertainty of the absorption length, pressure, temperature and fit accuracy, the total relative uncertainty is calculated by propagation of uncertainty for H2 16O to 3.8 % and for H2 18O to 10.1 %. Although the laser box is purged with dry air, it is difficult to completely avoid parasitic absorption and additional absorption in the laser box and the detector can occur. This leads to an offset uncertainty defined by the possible absorbance in the optical system outside AIDA. Thus, for the total instrument accuracy, it is necessary to take into account the relative uncertainty and the absolute offset uncertainty.
4.2 Isotope ratio performance
From the separate measurements of each isotope, it is possible to calculate the isotope ratio and determine the Allan-Werle deviation for the ratio itself as shown in Fig. 8 (averaging 100 raw scans for each isotope, resulting in 0.1-s time resolution). A decreasing trend flattening out around 70 s is visible. The slope of this Allan-Werle plot is comparable to the Allan-Werle plots on the individual isotope mole fractions and indicates a rather similar noise characteristic. Calculating the ratio of two mole fractions with similar temporal behavior and noise can lead to the advantageous situation of inter-dependent disturbance suppression. Thus, the Allan-Werle plot of the isotope ratio indicates greater stability than the individual isotopic mole fractions.

Allan-Werle deviation of the isotope ratio flattening around averaging time of 70 s
4.3 Dynamic isotope detection during cloud formation
Conclusively, the performance during dynamic cloud formation was tested. The experimental data shown in Fig. 9 were recorded during the experimental simulation of a convective updraft from 220 to 170 hPa with a temperature drop of 7 K from 206 to 199 K. Induced by the dust aerosol inside the chamber, ice cloud formation began 2 min (roughly 15:45–15:47) into the pump down. Due to this ice cloud formation and vibrations due to the pump down, the MC-IsoAPicT measurements experienced transmission fluctuations, which were successfully handled. Through cloud formation, the water was removed from the gas phase, which is indicated by the decreasing mole fraction of both isotopes. From the simultaneous measurement of H2 16O and H2 18O, the isotopic ratio was derived. The results imply a preference of H2 18O to condensate into the ice phase, as indicated by an earlier mole fraction decrease for H2 18O. To allow inter-instrument comparison, data during this simulation were recorded with a temporal resolution of 1 Hz. The precision of the instrument before the pump down was determined to 7 ppb for H2 16O and 40 ppt for H2 18O (230 m absorption path). The standard deviation of the noise on the isotope ratio \({\text {R}}_ {{\text {H}_{2} {^{18}{\text {O}}}}-{\text {H}_{2}\, ^{16}{\text {O}}}}\) was 0.15 % before and 0.3 % during cloud formation. Thus, additional noise from ice cloud formation and pumping does not change the precision on the isotope ratio significantly, and it is possible to resolve the experiment fractionation of H2 18O to H2 16 O through phase change in the order of 1 % with a signal-to-noise ratio of 7.

Adiabatic cloud expansion experiment to create ice clouds at 200 K. The decrease in the H2 18O to H2 16O isotope ratio is resolved to 0.15–0.3 %
The achieved precision for isotope ratio measurements is comparable to other laser-based instruments. For example, the extractive instrument from the Kerstel group in Grenoble achieves 0.44 % σ(H2 18O) at 20 ppm and 4-s time resolution [26]. MC-IsoAPicT can achieve a comparable performance (up to 0.15 %) with a better time resolution (0.96 s) and at lower total water concentrations (10 ppm). In particular, MC-IsoAPicT as an open-path in situ instrument does not suffer from sampling artefacts (e.g., due to H2O adsorption or unwanted shifts in the phase partitioning), which can be a significant and hard to identify source of uncertainty in extractive instruments.
With the achieved isotope ratio precision, it is now possible to study isotope ratio changes \(\Delta {\text {R}}_ {{\text {H}_{2} {^{18}{\text {O}}}}-{\text {H}_{2}\, ^{16}{\text {O}}}}\) to investigate the consistency of theoretical microphysical cloud formation predictions [70, 71], which will be discussed in future publications.
5 Summary and outlook
We presented the new multi-channel, isotope-selective AIDA PTB in-cloud TDL spectrometer (MC-IsoAPicT) which was designed for quasi-simultaneous open-path H2 16O and H2 18O measurements in ice clouds at temperatures between 190 and 230 K, as well as 170 and 230 hPa. The spectrometer is highly selective and sensitive and able to detect H2 16O and H2 18O down to detection limits of 2.6 ppb and 87 ppt, respectively. Due to the simultaneous dual water isotope detection within the same measurement volume, it is possible to extract changes in the isotope ratio in the interstitial gas phase water in ice clouds, with an isotope ratio resolution as low as 0.15 % (with a time resolution of 1 Hz). Measuring such low water concentration levels was made possible via a complex, isotope-independent background correction of the H2 18O channel and the effective prevention of the parasitic absorption in the laser path. The precision between 0.15 and 0.3 % was sufficient to observe isotopic effects such as equilibrium fractionation during cloud formation. We showed that MC-IsoAPicT can also maintain this performance during the formation of ice clouds within the optical path, despite transmission fluctuations through ice particles and vibrations of the optical setup.
In future measurement campaigns at the AIDA chamber within the ISOCLOUD project, we expect to gain new information with MC-IsoAPicT measurements about supersaturated cirrus clouds and water isotope fractionation coefficients.
References
D. Qin, M. Manning, Z. Chen, M. Marquis, K. B. Averyt, Report from Working Group I of the Intergovernmental Panel on Climate Change (2007)
M. Krämer, C. Schiller, A. Afchine, R. Bauer, I. Gensch, A. Mangold, S. Schlicht, N. Spelten, N. Sitnikov, S. Borrmann, M. de Reus, P. Spichtinger, Ice supersaturations and cirrus cloud crystal numbers. Atmos. Chem. Phys. 9(11), 3505–3522 (2009)
U. Burkhardt, B. Kärcher, U. Schumann, Global modeling of the contrail and contrail cirrus climate impact. Bull. Am. Meteorol. Soc. 91(4), 479–484 (2010)
K. Grerens, U. Schumann, M. Helten, H. Smit, P.-H. Wang, ice-supersaturated regions and subvisible cirrus in the northern midlatitude upper troposphere. J. Geophys. Res. 105, 22743–22753 (2000)
B. Kärcher, P. Spichtinger, Cloud-controlling factors of cirrus, in Clouds in the Perturbed Climate System, ed. by J. Heintzenberg, R.J. Charlson (MIT Press, Cambridge, 2009), p. 597
U. Lohmann, P. Spichtinger, S. Jess, T. Peter, H. Smit, Cirrus cloud formation and ice supersaturated regions in a global climate model. Environ. Res. Lett. 3(4), 045022 (2008)
M. Bolot, B. Legras, E.J. Moyer, Modelling and interpreting the isotopic composition of water vapour in convective updrafts. Atmos. Chem. Phys. 13(16), 7903–7935 (2013)
C.R. Webster, A.J. Heymsfield, Water isotope ratios D/H, 18O/16O, 17O/16O in and out of clouds map dehydration pathways. Science (New York, N.Y.) 302(5651), 1742–1745 (2003)
G. Lis, L.I. Wassenaar, M.J. Hendry, High-precision laser spectroscopy D/H and 18O/16O measurements of microliter natural water samples. Anal. Chem. 80(1), 287–293 (2008)
A. Angert, T. Weiner, S. Mazeh, M. Sternberg, Soil phosphate stable oxygen isotopes across rainfall and bedrock gradients. Environ. Sci. Technol. 46(4), 2156–2162 (2012)
O. Möhler, Effect of sulfuric acid coating on heterogeneous ice nucleation by soot aerosol particles. J. Geophys. Res. 110(D11), D11210 (2005)
A. Mangold, M. Krämer, C. Giesemann, H. Teichert, V. Ebert, Microphysics of cirrus clouds and its dependency on different types of aerosols. in Proceedings of Aviation, Atmosphere and Climate (2003), pp. 1–6
A. Gettelman, Simulations of water isotope abundances in the upper troposphere and lower stratosphere and implications for stratosphere troposphere exchange. J. Geophys. Res. 110(D17), D17301 (2005)
D.S. Sayres, L. Pfister, T.F. Hanisco, E.J. Moyer, J.B. Smith, J.M. St. Clair, A.S. O’Brien, M.F. Witinski, M. Legg, J.G. Anderson, Influence of convection on the water isotopic composition of the tropical tropopause layer and tropical stratosphere. J. Geophys. Res., 115, D00J20 (2010)
C.R. Webster, A.J. Heymsfield, Water isotope ratios D/H, 18O/16O, 17O/16O in and out of clouds map dehydration pathways. Science (New York, N.Y.) 302(5651), 1742–1745 (2003)
D.G. Johnson, K.W. Jucks, W.A. Traub, K.V. Chance, Isotopic composition of stratospheric water vapor: measurements and photochemistry. J. Geophys. Res. Atmos. 106(D11), 12211–12217 (2001)
M. Majoube, Fractionation factor of 18O between water vapour and ice. Nature 226, 1242 (1970)
M. Majoube, Fractionnnement en oxygène 18 et en Deutèrium entre l’eau et sa vapeur. J. Chem. Phys. 68, 1423–1436 (1971)
L. Merlivat, Molecular diffusivities of H216O, HDO, H218O in gases. J. Chem. Phys. 6, 2861–2871 (1978)
M.D. Ellehoj, H.C. Steen-Larsen, S.J. Johnsen, M.B. Madsen, Ice–vapor equilibrium fractionation factor of hydrogen and oxygen isotopes: experimental investigations and implications for stable water isotope studies. RCM 27(19), 2149–2158 (2013)
E. Barkan, B. Luz, Diffusivity fractionations of H2(16)O/H2(17)O and H2(16)O/H2(18)O in air and their implications for isotope hydrology. RCM 21(18), 2999–3005 (2007)
L. Berdié, I. Casals, I. Fernández, O. Jáuregui, M. Marimon, J. Perona, P. Teixidor, Basics of mass spectrometry, in Handbook of instrumental techniques for materials, chemical and biosciences research, 1st edn., ed. by J.R. Seoane, X. Llovet (Centres Cientifics i Tecnologics, Universitat Barcelona, Barcelona, 2012)
R.M. Marimon, J. Perona, P. Teixidor, Isotope ratio—mass spectrometry, in Handbook of Instrumental Techniques for Materials, Chemical and Biosciences Research, 1st edn., ed. by J.R. Seoane, X. Llovet (Centres Cientifics i Tecnologics Universitat Barcelona, Barcelona, 2012)
G.S. Engel, W.S. Drisdell, F.N. Keutsch, E.J. Moyer, J.G. Anderson, Ultrasensitive near-infrared integrated cavity output spectroscopy technique for detection of CO at 1.57 microm: new sensitivity limits for absorption measurements in passive optical cavities. Appl. Opt. 45(36), 9221–9229 (2006)
Y.A. Bakhirkin, A.A. Kosterev, C. Roller, R.F. Curl, F.K. Tittel, Mid-infrared quantum cascade laser based off-axis integrated cavity output spectroscopy for biogenic nitric oxide detection. Appl. Opt. 43(11), 2257–2266 (2004)
J. Landsberg, D. Romanini, E. Kerstel, Very high finesse optical-feedback cavity-enhanced absorption spectrometer for low concentration water vapor isotope analyses. Opt. Lett. 39(7), 1795–1798 (2014)
D.J. Hamilton, M.G.D. Nix, S.G. Baran, G. Hancock, A.J. Orr-Ewing, Optical feedback cavity-enhanced absorption spectroscopy (OF-CEAS) in a ring cavity. Appl. Phys. B 100(2), 233–242 (2009)
E. Kerstel, L. Gianfrani, Advances in laser-based isotope ratio measurements: selected applications. Appl. Phys. B 92(3), 439–449 (2008)
B. Buchholz, B. Kühnreich, H.G.J. Smit, V. Ebert, Validation of an extractive, airborne, compact TDL spectrometer for atmospheric humidity sensing by blind intercomparison. Appl. Phys. B 110(2), 249–262 (2012)
B. Buchholz, N. Böse, V. Ebert, Absolute validation of a diode laser hygrometer via intercomparison with the German national primary water vapor standard. Appl. Phys. B 116(4), 883–899 (2014)
H. Teichert, T. Fernholz, V. Ebert, Simultaneous in situ measurement of CO, H2O, and gas temperatures in a full-sized coal-fired power plant by near-infrared diode lasers. Appl. Opt. 42(12), 2043–2051 (2003)
W. Gurlit, R. Zimmermann, C. Giesemann, T. Fernholz, V. Ebert, J. Wolfrum, U. Platt, J.P. Burrows, Lightweight diode laser spectrometer CHILD (Compact High-altitude In-situ Laser Diode) for balloonborne measurements of water vapor and methane. Appl. Opt. 44(1), 91–102 (2005)
J. Skrotzki, P. Connolly, M. Schnaiter, H. Saathoff, O. Möhler, R. Wagner, M. Niemand, V. Ebert, T. Leisner, The accommodation coefficient of water molecules on ice–cirrus cloud studies at the AIDA simulation chamber. Atmos. Chem. Phys. 13(8), 4451–4466 (2013)
D.W. Fahey, R.-S. Gao, O. Möhler, H. Saathoff, C. Schiller, V. Ebert, M. Krämer, T. Peter, N. Amarouche, L.M. Avallone, R. Bauer, Z. Bozóki, L.E. Christensen, S.M. Davis, G. Durry, C. Dyroff, R.L. Herman, S. Hunsmann, S.M. Khaykin, P. Mackrodt, J. Meyer, J.B. Smith, N. Spelten, R.F. Troy, H. Vömel, S. Wagner, F.G. Wienhold, The AquaVIT-1 intercomparison of atmospheric water vapor measurement techniques. Atmos. Meas. Tech. Discuss. 7(4), 3159–3251 (2014)
B.J. Murray, T.W. Wilson, V. Ebert, S. Dobbie, Z. Cui, S.M.R.K. Al-Jumur, O. Möhler, M. Schnaiter, R. Wagner, S. Benz, M. Niemand, H. Saathoff, S. Wagner, B. Kärcher, Heterogeneous nucleation of ice particles on glassy aerosols under cirrus conditions. Nat. Geosci. 3(4), 233–237 (2010)
V. Ebert, H. Teichert, C. Giesemann, H. Saathoff, U. Schurath, Fibre-coupled in situ laser absorption spectrometer for the selective detection of water vapour traces down to the ppb-level. Tech. Mess. 72(1–2005), 23–30 (2005)
V. Ebert, C. Lauer, H. Saathoff, S. Hunsmann, Simultaneous, absolute gas-phase and total water detection during cloud formation studies in the AIDA chamber using a dual 1.37 µm TDL-spectrometer. Geophys. Res. Abstr. p. Vol. 10, EGU2008–A–10066 (2008)
V. Ebert, J. Wolfrum, Absorption, in Optical Measurements—Techniques and Applications, 2nd edn., ed. by F. Mayinger, O. Feldmann (Springer, Heidelberg, 2000), pp. 273–312
J.H. Lambert, Photometria sive de mensura et gradibus luminis colorum et umbrae (Eberhard Klett, Augsburg, 1790)
A. Beer, Determination of the absorption of red light in colored liquids (Bestimmung der Absorption des rothen Lichts in farbigen Flüssigkeiten), in Annalen der Physik und Chemie, 3rd edn., ed. by J.C. Poggendorff (Verlag von Johann Ambrosius Barth, Leibzig, 1852), pp. 78–888
S. Hunsmann, K. Wunderle, S. Wagner, U. Rascher, U. Schurr, V. Ebert, Absolute, high resolution water transpiration rate measurements on single plant leaves via tunable diode laser absorption spectroscopy (TDLAS) at 1.37 μm. Appl. Phys. B 92(3), 393–401 (2008)
A. Pogány, O. Ott, O. Werhahn, V. Ebert, Towards traceability in CO2 line strength measurements by TDLAS at 2.7µm. J. Quant. Spectrosc. Radiat. Transfer 130, 147–157 (2013)
L.S. Rothman, I.E. Gordon, A. Barbe, D.C. Benner, P.F. Bernath, M. Birk, V. Boudon, L.R. Brown, A. Campargue, J.-P. Champion, The HITRAN 2008 molecular spectroscopic database. J. Quant. Spectrosc. Radiat. Transf. 110(9–10), 533–572 (2009)
L.S. Rothman, D. Jacquemart, A. Barbe, D. Chrisbenner, M. Birk, L. Brown, M. Carleer, C. Chackerianjr, K. Chance, L. Coudert, The HITRAN 2004 molecular spectroscopic database. J. Quant. Spectrosc. Radiat. Transfer 96(2), 139–204 (2005)
G.D. Farquhar, L.A. Cernusak, B. Barnes, Heavy water fractionation during transpiration. Plant Physiol. 143(1), 11–18 (2007)
R.D. Deshpande, A.S. Maurya, B. Kumar, A. Sarkar, S.K. Gupta, Kinetic fractionation of water isotopes during liquid condensation under super-saturated condition. Geochim. Cosmochim. Acta 100, 60–72 (2013)
C.D. Cappa, Isotopic fractionation of water during evaporation. J. Geophys. Res. 108(D16) (2003)
M. Majoube, Fractionnnement en 18O entre la glace et la vapeur d’eau. J. Chem. Phys. 68(4), 625–636 (1971)
G. Jakli, D. Staschewski, Vapour Pressure of H218O Ice (−50 to 0°C) and H218O Water (0 to 170°C). J. Chem. Soc., Faraday Trans. 73(1), 1505–1509 (1977)
S. Hunsmann, S. Wagner, H. Saathoff, O. Möhler, U. Schurath, V. Ebert, Messung der Linienstärken und Druckverbreiterungskoeffizienten von H2O-Absorptionslinien im 1,4 µm - Band. VDI Berichte 1959, 149–164 (2006)
T. Klostermann, A. Afchine, J. Barthel, M. Höh, S. Wagner, O. Witzel, H. Saathoff, C. Schiller, V. Ebert, HAI: a new TDLAS hygrometer for the HALO research aircraft. Geophys. Res. Abstr. 12, 9485 (2010)
O. Möhler, S. Benz, H. Saathoff, M. Schnaiter, R. Wagner, J. Schneider, S. Walter, V. Ebert, S. Wagner, The effect of organic coating on the heterogeneous ice nucleation efficiency of mineral dust aerosols. Environ. Res. Lett. 3(2), 025007 (2008)
O. Möhler, O. Stetzer, S. Schaefers, C. Linke, M. Schnaiter, R. Tiede, H. Saathoff, M. Krämer, A. Mangold, P. Budz, P. Zink, J. Schreiner, K. Mauersberger, W. Haag, B. Kärcher, U. Schurath, Experimental investigation of homogeneous freezing of sulphuric acid particles in the aerosol chamber AIDA. Atmos. Chem. Phys. 3(1), 211–223 (2003)
R. Wagner, H. Bunz, C. Linke, O. Möhler, K.-H. Naumann, H. Saathoff, M. Schnaiter, U. Schurath, Chamber simulations of cloud chemistry: the AIDA Chamber, in Environmental Simulation Chambers: Application to Atmospheric Chemical Processes, 62nd edn., ed. by I. Barnes, K.J. Rudzinski (Springer, Netherlands, 2006), pp. 67–82
A. Mangold, R. Wagner, H. Saathoff, U. Schurath, C. Giesemann, V. Ebert, M. Kramer, O. Mohler, Experimental investigation of ice nucleation by different types of aerosols in the aerosol chamber AIDA: implications to microphysics of cirrus clouds. Meteorol. Z. 14(4), 485–497 (2005)
B. Buchholz, V. Ebert, Offsets in fiber-coupled diode laser hygrometers caused by parasitic absorption effects and their prevention. Meas. Sci. Technol. 25(7), 075501 (2014)
J.U. White, Long optical paths of large aperture. J. Opt. Soc. Am. 32(5), 285 (1942)
R. Dominique, C. Patrick, Multiple-pass cell for very-long-path infrared spectrometry. Appl. Opt. 38(19), 4145–4150 (1999)
D. Horn, G.C. Pimentel, 2.5-km Low-temperature multiple-reflection cell. Appl. Opt. 10(8), 1892–1898 (1971)
R. Wagner, S. Benz, O. Möhler, H. Saathoff, U. Schurath, Probing ice clouds by broadband mid-infrared extinction spectroscopy: case studies from ice nucleation experiments in the AIDA aerosol and cloud chamber. Atmos. Chem. Phys. 6, 4775–4800 (2006)
V. Ebert, H. Teichert, P. Strauch, T. Kolb, H. Seifert, J. Wolfrum, Sensitive in situ detection of CO and O2 in a rotary kiln-based hazardous waste incinerator using 760 nm and new 2.3 μm diode lasers. Proc. Combust. Inst. 30(1), 1611–1618 (2005)
V. Ebert, J. Fitzer, I. Gerstenberg, K.U. Pleban, H. Pitz, J. Wolfrum, M. Jochem, J. Martin, Simultaneous laser-based in situ detection of oxygen and water in a waste incinerator for active combustion control purposes. Symp. (Int.) Combust. 27(1), 1301–1308 (1998)
E. Schlosser, T. Fernholz, H. Teichert, V. Ebert, In situ detection of potassium atoms in high-temperature coal-combustion systems using near-infrared-diode lasers. Spectrochim. Acta A Mol. Biomol. Spectrosc. 58(11), 2347–2359 (2002)
B. Lins, P. Zinn, R. Engelbrecht, B. Schmauss, Simulation-based comparison of noise effects in wavelength modulation spectroscopy and direct absorption TDLAS. Appl. Phys. B 100(2), 367–376 (2010)
P.A. Martin, Near-infrared diode laser spectroscopy in chemical process and environmental air monitoring. Chem. Soc. Rev. 31(4), 201–210 (2002)
P. Kluczynski, Å.M. Lindberg, O. Axner, Background signals in wavelength-modulation spectrometry by use of frequency-doubled diode-laser light. II. Experiment. Appl. Opt. 40(6), 794 (2001)
M.J. Hendry, B. Richman, L.I. Wassenaar, Correcting for methane interferences on δ(2)H and δ(18)O measurements in pore water using H(2)O((liquid))-H(2)O((vapor)) equilibration laser spectroscopy. Anal. Chem. 83(14), 5789–5796 (2011)
J.J. Moré, The Levenberg-Marquadt-Algorithm: implementation and theory, in Lecture notes in mathematics, ed. by G.A. Watson (Springer, Berlin, 1978), pp. 105–116
P. Werle, R. Mäcke, F. Slemr, The limits of signal averaging in atmospheric trace-gas monitoring by tunable diode-laser absorption spectroscopy (TDLAS). Appl. Phys. B Photophys. Laser Chem. 57(2), 131–139 (1993)
J.R. Gat, Oxygen and hydrogen isotopes in the hydrologic cycle. Annu. Rev. Earth Planet. Sci. 24(1), 225–262 (1996)
K. Scott, X. Lu, C. Cavanaugh, J. Liu, Optimal methods for estimating kinetic isotope effects from different forms of the Rayleigh distillation equation. Geochim. Cosmochim. Acta (2004)
Acknowledgments
This work was part of the ISOCLOUD initiative, which was funded by the Deutsche Forschungsgesellschaft (DFG) [Project Numbers DFG EB235/4-1 and DFG MO668/3-1] and the US National Science Foundation (NSF) as part of the Joint DFG/NSF 2009/2010 call on “International Collaboration in Chemistry between US Investigators and their Counterparts Abroad (ICC).” We like to thank all our cooperation partners within the ISOCLOUD project for the thoughtful discussions and the generous support we have experienced throughout the project. In particular, we want to mention the selection of water isotope transitions, which has been agreed upon by all partners within the ISOCLOUD project and will be described elsewhere. Support in the line selection for the H2 18O line by E. J. Moyer from the Department of Geophysical Sciences at the University of Chicago is acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Kühnreich, B., Wagner, S., Habig, J.C. et al. Time-multiplexed open-path TDLAS spectrometer for dynamic, sampling-free, interstitial H2 18O and H2 16O vapor detection in ice clouds. Appl. Phys. B 119, 177–187 (2015). https://doi.org/10.1007/s00340-015-6074-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00340-015-6074-4