
Abstract
An experimental setup and data collection strategy for femtosecond transient absorption spectroscopy on thin (\(<\)1 \(\upmu \hbox {m}\)) solid polymer film samples is described. The experiment allows for parallel detection of the changes in optical density \(\Delta {\mathrm{OD}}\) via broadband supercontinuum probing in the VIS/UV range and single-color detection at an independently selected wavelength from the deep UV to the IR with a sensitivity of \(\Delta {\mathrm{OD}} \approx 10^{-3}\) per laser shot (r.m.s. standard deviation) and a time resolution below 40 fs. A fast and reproducible bi-directional translation of a two-dimensional film sample of \(20 \times 20\,\hbox {mm}^2\) in size is used to measure fresh sample spots at each detection interval. Signal readout at a 1 kHz rate enables single-shot analysis and automated signal discrimination, as well as detailed statistics on sample homogeneity, signal evolution with increasing number of pump pulses, and reproducibility. The technique was employed to study the photoisomerization of Disperse Red 1 in films of polymethylmethacrylate after \(\pi \pi ^*\) photoexcitation at \(\lambda =473\) nm. The results revealed excited-state dynamics characterized by time constants of \(\tau _1 = 0.06\) and \(\tau _2 = 1.0\,\hbox {ps}\), almost identical as in solution, but evidently enhanced vibrational excitation and slower vibrational cooling (time constant \(\tau _3 = 14\,\hbox {ps}\)) after return to the electronic ground state due to the constraining polymer environment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Photochromic molecules such as azobenzenes, fulgides or diarylethenes [1–7] have proved as successful photoswitchable building units for applications ranging from optical data storage devices to biomedical superresolution imaging [5, 8–12]. In most cases, the photoswitchable molecules need to be embedded in complex environments such as bulk polymer materials or thin films in order to translate the molecular response into the desired macroscopic effects [13–23]. Under these conditions, the particular molecular environment and specific multi-chromophore interactions may affect the photoswitching properties to unknown extent. To obtain detailed mechanistic insight into the fundamental processes on the femtosecond to picosecond time scale [24–28] that govern important photochemical properties such as product quantum yields, photostability and fluorescence, time-resolved studies of the photo-induced dynamics and chemical transformations of photo-excited molecular switches embedded in the complex environments used for applications are mandatory. Beyond fundamental interest, the results provide a basis for a rational design of functional photosensitive systems and devices with new properties and improved performance.
The required femtosecond time-resolved experiments rely on the pump–probe scheme, where an ultrashort pump pulse is used for photoexcitation and is followed by a time-delayed probe pulse that monitors the molecular response. This is repeated for many different delay times to obtain a complete set of time-dependent data set [5, 26, 29]. To acquire the desired information over an extended spectral and temporal range with adequate sensitivity, typical experimental implementations accumulate the signals over multiple detection cycles, which requires refreshing the sample between successive laser shots to avoid photodegradation or buildup of photoproducts. In case of gas- or liquid-phase samples, this is conveniently achieved by flow techniques. However, this approach cannot easily be extended to photoreactive molecules in solid phases because sample refreshing is too slow or cannot be repeated often enough.
Several techniques capable of simultaneous detection at multiple delay times have been developed to reduce the number of pump–probe cycles for femtosecond measurements of crystalline or amorphous solids. In the dual-echelon method, a two-dimensional spatial delay pattern is imposed on the probe pulse by inserting two orthogonally oriented step prisms (echelons) into the beam path, and the resulting array of probe pulses with predetermined time delays is imaged onto a two-dimensional detector [30–32]. The method is intrinsically restricted to single-wavelength detection and requires replacement of the echelons for a different set of delay steps. For pump–probe imaging spectroscopy [33–36], the pump and probe pulses incident on the sample are line-shaped and intersect at a fairly large angle, so that the time delay varies across the illuminated sample area. The time resolution (typically \(\ge\)100 fs) and the accessible temporal range depend on the particular experimental geometry. Reported largest delay times are \(\approx\)20 ps using a large lateral spot size, and sensitivities for the detection of the transient absorption changes are of the order of \(\Delta {\mathrm{OD}} \approx 0.01\). An alternative approach that works without the need for multiple-delay detection has gained popularity recently. Optimization of the single-shot sensitivity in combination with precise single-axis transversal sample translation allowed for signal detection at 50–100 different delay times with 10–20 accumulations resulting in a typical noise of \(\Delta {\mathrm{OD}} \approx 3 \times 10^{-3}\) [37]. This technique has been applied for monitoring the shape transformation of spherical Ag nanoparticles in a glass matrix upon photoexcitation [37].
In the present publication, we report femtosecond transient absorption spectroscopy on solid polymer films doped with photoreactive molecules by making use of a high-sensitivity setup for parallel \(\Delta {\mathrm{OD}}\) measurements via broadband supercontinuum probing in the VIS/UV range and single-color probing at a selected wavelength from deep-UV to IR. Combined with bi-directional fast sample translation and data collection with real-time data discrimination, we achieve a sensitivity of \(\Delta {\mathrm{OD}} \approx 10^{-3}\) (r.m.s. standard deviation) for single-shot detection and a time resolution below 50 fs. Experimental results after \(\pi \pi ^*\) photoexcitation of Disperse Red 1 (DR1, Scheme 1) doped in films of polymethylmethacrylate (PMMA) at \(\lambda =473\) nm are reported as a test case.

Disperse Red 1 (\(\hbox {DR}1\))
2 Experimental section
2.1 Transient absorption experiment
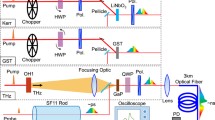
The experimental setup is based on our previously reported time-resolved broadband absorption spectrometer [38]. Two probe pulses are applied, one spectrally broad pulse covering the wavelength range of \(310\,\hbox {nm}<\lambda _{{\mathrm{probe}}}<750\,\hbox {nm}\) and a second, spectrally narrower pulse that can be set to any desired wavelength in the UV to the near-IR. Usually, we select a deep-UV wavelength \(\le\)300 nm to follow the ground state recovery and vibrational cooling processes when exciting in the deep UV. Most of the general description refers to both liquid and solid samples. For solids, translation stages allow for a precise and fast transversal (x–y) sample motion. With this setup, solid samples can be moved to a fresh spot between successive laser shots to ensure their integrity even in case of samples prone to photodegradation. In the following, the experimental details with regard to beam pathways and the timing of the translational stages will be given. A sketch of the setup is given in Fig. 1.

Schematic overview of the transient absorption experiment. Two probe pulses are available to allow for simultaneous detection of transient absorption changes at \(310\,\hbox {nm}\) \(\le \lambda _{{\mathrm{probe}}}\le 750\,\hbox {nm}\) (shown in blue) and at a freely selected single wavelength (shown in purple)
The experiment was driven by a Ti:Sa laser (Clark-MXR CPA 2001) delivering pulses with pulse lengths of 150 fs (FWHM) at 775 nm. Half of the overall output of 1,000 \(\upmu \hbox {J}\) was used for the transient absorption experiment, the other half for a fluorescence up-conversion experiment and laser pulse diagnostics. The excitation and single-color probe pulses were each generated in a non-collinear optical parametric amplifier (NOPA) with subsequent temporal compression and, if necessary, frequency doubling. The broadband probe pulses were generated using supercontinuum generation (SCG) in CaF2. All pulses were recollimated using reflective optics to reduce the chirp and obtain an optimal time resolution of the experiment [39]. To characterize both, the cross-phase modulation (XPM) and stimulated Raman scattering (SRS) contributions for the pure solvent (in the case of liquid samples) or non-doped polymer films (for solid samples) were measured independently. The center positions of the XPM at many wavelengths were then fitted with a fifth-order polynomial to obtain the time-zero function, which was subsequently employed to correct the raw transient absorption data matrices for the white light continuum chirp. For the displayed two-dimensional transient absorption maps and transient spectra, the respective time-corrected solvent or non-doped polymer film contributions were subtracted from the sample data with a suitable scaling factor accounting for the pump pulse absorption [40, 41].

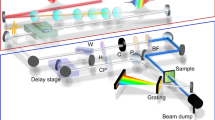
Sketch of the pulse propagation at the sample and the detection units. The broadband probe pulses are traveling perpendicular to the sample and are spatially and temporally overlapped with the pump and the single-color probe pulses. The two reference pulses are located on the right and left of the pump and probe pulses
A detailed view of the optical pathways near the sample and detection units is given in Fig. 2. The broadband and single-color probe pulses were split into probe and reference using the front and back reflection of planar quartz glass plates (\(d=5\,\hbox {mm}\)). All pulses were focused into the sample cell using reflective optics. In the sample, the probe pulses (broadband and single-color) were spatially overlapped with the pump pulse. The angles between the incident pulses were kept as small as possible to reduce the chirp of the pump and probe pulses and consequently the intensity of coherent signals [42]. Since the spectrally broad pulses are especially prone to chirping, tilting of this beam path was avoided as much as possible. A reproducible and straightforward adjustment of the broadband probe beam diameter was ensured using two focusing mirrors after the CaF2 plate. The first mirror was placed so that a slight pre-focussing was achieved, and the second mirror focused the pulses into the sample cell. This resulted in smaller focus diameters than with only one focussing mirror after the CaF2 plate. Generally, the probe foci were of the order of \({\approx } 100\,\upmu \hbox {m}\) and the pump pulse focus of the order of \({\approx } 200\,\upmu \hbox {m}\). Overlapping of the two reference pulses was avoided by placing the two glass plates at an angle of \({\approx } 90^{\circ }\) (see Fig. 2) in case of liquid samples. In case of solid samples, the reference pulses should hit a fresh spot on the sample as is shown in Fig. 3 for the broadband detection pulses.
After passing the sample, the relative displacement of the broadband probe and reference pulses was rotated from horizontal to vertical using a periscope. The pulses were then recollimated and focused onto the entrance slit of a prism spectrometer using either one or a combination of two quartz lenses. An optical filter (BG18, Schott) was used to lower the intensity of residual 775 nm light. In the spectrometer box, the pulses passed a quartz lens, an equilateral quartz prism and a second quartz lens. The vertically displaced probe and reference pulses were separated with a set of two square aluminum mirrors and aligned toward the CCD cameras for detection. The optical pathlength after passing the prism had to be equal for probe and reference pulses to ensure equal horizontal widths of the dispersed pulses on the cameras. The positions were adjusted so that the same lateral pixels of both cameras are illuminated by the same wavelengths using a set of interference filters. For this, a LabView routine allowing for a live scan of the camera signals was employed. The CCD cameras (Series 2000, Entwicklungsbüro Stresing, Berlin) were equipped with back-thinned full frame transfer (FFT) CCD image sensors (S7030-0906, Hamamatsu) with quantum efficiencies of \({\approx } 40\) % in the 300–380 nm range and up to 90 % in the visible range of the spectrum. The sensor area was \(512 (H) \times 58 (V)\) pixels (\(12.3\times 1.4\,\hbox {mm}\)). Both FFT-CCD cameras were read out with the laser repetition rate of 1,010 Hz.
The single-color probe and reference pulses were detected using a set of slow matched Si photodiodes (S1227-66BQ, Hamamatsu) that were read out using differential signaling to reduce noise due to electromagnetic interference. Additionally, the pump pulse intensity was monitored with a third photodiode to monitor the chopper status for every laser shot (see below). The photodiode signals were streamed into the data flow of the CCD cameras. The detection unit consisting of prism spectrometer, CCD cameras, photodiodes and controller was developed in close collaboration with G. Stresing (Entwicklungsbüro Stresing, Berlin).
Measurements were carried out in the following sequence: An optical chopper equipped with a 2-shot blade (MC1F2, Thorlabs) was used to block all pump pulses for a time window that allowed for the translation of the film to a fresh sample spot. For the given pump focus diameter of \(200\,\upmu \hbox {m}\), the pump pulses were blocked for \(380\,\hbox {ms}\) and the film was moved by \(250\,\upmu \hbox {m}\) in the x or y direction (cf. Figs. 3, 4) using computer-controlled translation stages mounted perpendicular to each other (Physik Instrumente, M-404.1PD and M-403.1PD). A second chopper (MC2000, Thorlabs) equipped with a 10-shot blade (MC1F10, Thorlabs) was used to block every second pump pulse, thus enabling the measurement of the sample (pump pulse unblocked) and background (pump pulse blocked) signals. The change of transient absorption was calculated from the respective probe and reference intensities [39].

Detailed scheme of the optical pathways at the solid sample. The sample can be moved vertically and horizontally so that the pump always hits a fresh, unexcited spot (cf. Fig. 4). The reference pulse has to hit a fresh sample spot as well. The single-color pulses have been omitted for clarity

a Photographs of three polymer film samples with optical densities of 0.2, 0.5 and 1.0 (from left to right). b Scheme of the \(7\times 7\) test areas on the film that can be used for time-resolved measurements. c Schematic drawing of one test area with \(10\times 10\) spots. The horizontal and vertical movement is depicted by arrows. Excited spots are depicted in orange, fresh ones in dark red
For a typical polymer film of \(20 \times 20\,\hbox {mm}\), taking into account the area that is needed for the reference pulses, \(7\times 7\) test areas of \(2.5 \times 2.5\,\hbox {mm}^2\) each are accessible. With a typical focus diameter of \(250\,\upmu \hbox {m}\), transient absorption changes at \(10\times 10\) spots per test area can be measured (cf. Fig. 4).
To obtain reliable results, two parameters need to be carefully checked: First, the film needs to be placed exactly perpendicular to the plane of incidence so that moving of the sample does not change the spatial overlap of the probe and pump pulses in the sample (Fig. 3). This can easily be achieved by adjusting the tilt angles of the film so that the back-reflected broadband probe pulse is traveling exactly opposite to the incoming pulses. Second, the homogeneity of the sample needs to be checked in advance. In case of sensitive samples, the use of the pump or probe pulses is not advised because of a potential damage of the sample and the inherent pulse-to-pulse fluctuations. A noninvasive alternative is to use a weak, stable laser diode at a suitable wavelength that can be coupled into the pump pulse path by a flipping mirror (cf. Fig. 2) and focused on the same spot as the pump pulses. The transmitted intensity can be monitored with one of the photodiodes and the change in optical density at each sample spot relative to the mean intensity can be calculated. This procedure is routinely used to check the homogeneity of the polymer films after preparation.
2.2 Single-shot analysis and sensitivity of the setup
The single-shot readout enables a shot-to-shot analysis of the transient absorption signals. This increases the sensitivity since the pulse-to-pulse noise of the laser fundamental is only of the order of \(\le\)0.5 % (r.m.s.), but on the time scale of a few seconds, laser fluctuations are more pronounced [43]. Consequently, if sample and background transmissions [(\(T^{*}(\lambda , \Delta t)\) and \(T^0(\lambda )\)] are averaged over long times before calculating \(\Delta \text {OD}(\lambda ,\Delta t)\), the correlation of both is at least partially lost after several hundred laser shots, which results in higher noise [39]. A detailed analysis of the signal-to-noise ratio has shown that laser fluctuations and correlations of pump and probe pulse intensities are indeed a dominant factor regarding the sensitivity of the transient absorption signal, whereas shot noise and electronic noise contribute less [43]. Exploiting these correlations by shot-to-shot analysis thus yields an improved S/N ratio.
The detection sensitivities with broadband probe pulses achieved in the single-shot readout mode are depicted in Fig. 5 as function of the number of measured shots. The respective measurements have been performed with a liquid sample in a flow cell and thus are not affected by sample inhomogeneities. The correlation is linear with a slope of \({\approx }0.5\), and transient absorption measurements with sensitivities ranging from \(\Delta {\mathrm{OD}}\approx 1\times 10^{-3}\) for a single shot to \({\approx }2\times 10^{-5}\) after averaging over several thousand single measurements are feasible. The single-shot readout also allows for a pulse discrimination to eliminate poor single measurements resulting from, e.g., dust particles in the optical pathway. This is performed on the fly with a LabVIEW routine. Toward these ends, the mean value \(\left\langle \Delta {\mathrm{OD}}(\lambda )\right\rangle\) and the deviations \(\left| \left\langle \Delta {\mathrm{OD}}(\lambda )\right\rangle -\Delta {\mathrm{OD}}(\lambda )\right|\) of each single measurement is calculated. If the deviation from the mean exceeds a preset limit, the respective single measurement is discarded. Typically, this applies to a fraction of \(<\)1 % of single measurements. In Fig. 5, the sensitivities after applying this correction are shown in blue. The sensitivity increase by this single-shot discrimination analysis for \(\ge\) 500 single measurements per delay step is evident. For a smaller number of shots, the applied simple discrimination method is not feasible. A more sophisticated approach would require more computing capacity or computing time (leading to longer overall accumulation times) than available for our experiment.

Standard deviations of \(\Delta {\mathrm{OD}}\) with broadband probe at \(\lambda _{{\mathrm{probe}}}=350\,\hbox {nm}\) as function of number of single measurements before (gray) and after (blue) single-shot analysis. The sensitivity of the single-color probe measurements was about three times better
3 Measurement of DR1-doped PMMA Films
3.1 Sample preparation
DR1 was dissolved together with polymethylmethacrylate (PMMA, 100 kg/mol, \(M_W=99,400\,\hbox {g mol}^{-1}\), polydispersity index 1.08) powder in toluene (Uvasol, \(\ge\)99 %) or chloroform (\(\ge\)99 %), and mixtures with dye content of 6, 10, and 18.5 % in the solid film were prepared. PMMA was purchased from PSS GmbH, and all other components were purchased from Sigma-Aldrich and used without further purification. Solutions of DR1 in chloroform and toluene were purified by filter devices (Puradisc 25 TF-25 mm) prior to preparing films. Borosilicate glass (Schott, D263M, \(20\times 20\,\hbox {mm}, d=0.5\,\hbox {mm}\)) was used as a substrate. The thin polymer films were prepared by spin-coating [23, 44] on the glass plate for 63 s at a speed of 2,400 rpm and dried for 63 s at a speed of 3,600 rpm [45].
3.2 Static absorption spectra
The static UV/VIS absorption spectra of DR1 in solution in toluene and in the PMMA matrix are displayed in Fig. 6. The absorption band of DR1 in solution features a maximum at \(469\) nm attributed to a \(\pi \pi ^*\) transition. The weak \(\hbox {n}\pi ^*\) transition that is present in azobenzene is hidden underneath [46, 47]. The static absorption spectrum of DR1 embedded in the PMMA film features a red shift of the maximum to 489 nm and a slight broadening of the band. This behavior of push–pull azobenzenes is well known and attributed to the different polarity in the PMMA film compared to the solvent and possible formation of DR1 aggregates in polymeric environments [40, 47–49].

Normalized absorbance spectra of DR1 in toluene (dashed line) and in PMMA (solid line)
3.3 Time-resolved measurement of PMMA films doped with DR1
The excitation wavelength for the time-resolved measurement of DR1 in the PMMA matrix was \(\lambda _{{\mathrm{pump}}}=473\,\hbox {nm}\). The single-color detection was not used for these measurements. To avoid high energy densities at the test spot and potential thermal excitation of neighboring spots, the excitation energy was reduced to \(\le\)200 nJ.
Measurements of the transient absorption at different spots (spot-by-spot stability) on the film were done to check the film homogeneity and orientation relative to the pump and probe pulses (cf. Sec. 2.1). The delay time was fixed to \(\Delta t = 1\,\hbox {ps}\) for these measurements. The results are displayed in Fig. 7a and show a standard deviation of \(\Delta {\mathrm{OD}} = 1\times 10^{-3}\). This sensitivity is of the same order as for liquid samples (see above). It shows the correct positioning of the film as well as the high homogeneity of the film. The shot-by-shot stability is shown by 190 single measurements at the delay time \(\Delta t = 1\,\hbox {ps}\) on one test spot of the film in Fig. 7b. The standard deviation is \(\Delta {\mathrm{OD}}\approx 1\times 10^{-3}\). It can be seen that the signal height drops from \({\approx }10\times 10^{-3}\) to \({\approx }9\times 10^{-3}\) after \(190\) single measurements, indicating a slow photodegradation of the DR1 molecules. Averaging over the first \(50\) single measurements can thus safely be done in case of DR1. The analysis of these plots could be used for the analysis of thermal back-reactions as it was done by Warth et al. [37].

Transient absorption changes of DR1 embedded in a PMMA matrix after excitation at \(\lambda _{{\mathrm{pump}}}=473\,\hbox {nm}\). The delay time was fixed to \(\Delta t = 1\) ps. a Spot-by-spot stability of \(\Delta {\mathrm{OD}}\) measured at 50 neighboring spots each with a diameter of \(250 \upmu \hbox {m}\) on the film. b Shot-by-shot stability of \(\Delta {\mathrm{OD}}\) for 190 consecutive single measurements performed at one selected test spot. The dashed lines in both cases indicate the standard deviation of \({\approx }1\times 10^{-3}\)
3.4 Results of time-resolved transient absorption measurements
For the time-resolved measurements, data were taken on one \(2.5 \times 2.5\,\hbox {mm}^2\) test area at 100 delay times (each at a fresh spot). The measurements were repeated at several other test areas on the sample to ensure the reproducibility of the results. The probe wavelength range was limited to \(360\,\hbox {nm} \le \lambda _{{\mathrm{probe}}}\le 750\,\hbox {nm}\) due to the strong absorption of DR1 at \(\lambda _{{\mathrm{probe}}}\le 360\,\hbox {nm}\). Three films with dye concentrations corresponding to optical densities between \(0.2\) and \(1.0\) were measured. The time-resolved results were virtually identical, and in the following only the results for the PMMA film doped with \(6\) % DR1 (\({\mathrm{OD}}=0.2\)) are shown. Figure 8 depicts the two-dimensional (2D) spectro-temporal transient absorption map after excitation at \(\lambda _{{\mathrm{pump}}}=473\,\hbox {nm}\).

2D spectro-temporal absorption maps of the change in optical density \(\Delta {\mathrm{OD}}\) (transient absorption) following excitation of PMMA film doped with DR1 at \(\lambda _{{\mathrm{pump}}}=473\) nm for delay times up to \(\Delta t= 18\,\hbox {ps}\). a Result for the single-shot mode, b transient signals averaged over 50 single shots. The range at \(\lambda = 420\)–530 nm is cut out because of a strong contribution of scattered pump light
The map features four overlapping bands: three positive absorption signals at \(\lambda \approx 400, 590\) and \(750\,\hbox {nm}\) and a negative signal around \(\lambda \approx 470\,\hbox {nm}\) that is partially obscured by scattered pump light. The negative band can be attributed to the bleaching of the ground state absorption and to stimulated emission (SE). Its decay beyond a few hundred fs reflects the ground state recovery (GSR). The transient spectra in Fig. 9 provide a closer look at the spectro-temporal behavior. The longer-lived positive band centered at 590 nm features a pronounced red shift during the first 400 fs, which could be due to the rapid decay of the stimulated emission contribution overlapping with the positive ESA band. The subsequent blue shift is typical for the absorption of vibrationally excited molecules in the electronic ground state. The band is therefore assigned to hot ground state absorption (HGSA). The bands at 400 and 750 nm feature no pronounced shifts and are attributed to excited-state absorption (ESA). The signals at probe wavelengths \({\le } 700\,\hbox {nm}\) show a residual absorption after the maximum delay time of 18 ps which is attributed to the absorption of the \(Z\) isomer of DR1 and vibrationally excited \(E\) and \(Z\) isomers.

Transient spectra of DR1 at different delay times between a \(0\,\hbox {ps}\le \Delta t \le 0.06\,\hbox {ps}\) and b \(0.06\,\hbox {ps}\le \Delta t \le 18\,\hbox {ps}\). The range at \(\lambda = 420\)–\(530\,\hbox {nm}\) is cut out because of a strong contribution of scattered pump light
A more detailed look into the dynamics was provided by a nonlinear least-squares fitting analysis of four selected time profiles at probe wavelengths \(\lambda _{{\mathrm{probe}}}=400, 531, 590\) and \(700\, \hbox {nm}\). In these fits, the XPM was used as parametrized input by fitting the corresponding time profiles of the background measurement with a sum of Gaussians, treating \(t_0\) as a free fit parameter. The resulting values of \(t_0\) were virtually identical to those determined from the XPM (cf. Sec. 2.1) with deviations of only a few femtoseconds and confirm the validity of the chirp correction procedure. Likewise, the time resolution of the experiment was determined to be \(\Delta r =35 \pm 5\) fs by taking the width of a Gaussian-shaped instrument response function (IRF) as a fit parameter. This value is corroborated by the IRF width of about 40 fs estimated from the SRS contribution of independent solvent measurements. In accordance with our earlier study of DR1 in solution [40], the following relaxation scheme was employed:
Here, \({E}^{*}\) and \({E}^{**}\) denote excited molecules in two different electronically excited “states” that give rise to prompt and delayed ESA, respectively, and \({E}^{\#}\) and \({Z}^{\#}\) denote vibrationally hot molecules in the electronic ground state. \({E}\) and \(Z\) are the final products. The data analysis resulted in decay times of
Additionally, a constant contribution was necessary for the description of the product absorption.
The results are shown in Fig. 10. The fast-decaying component (\(\tau _1\)) is assigned to internal conversion of initially excited molecules \({E}^{*}\) to \({E}^{**}\). The latter species gives rise to the delayed appearance of the ESA contribution decaying with \(\tau _2\). This decay time is in turn the rise time of the HGSA contribution of hot \({E}\) and \({\mathrm{E}}\) isomers (\({E}^{\#}\) and \({Z}^{\#}\)) decaying to the vibrational ground state with \(\tau _3\). The product formation of the \(Z\) isomer and the permanent GSR contribution is accounted for by a step function with an IRF-limited rise as it cannot be separated from the ESA and HGSA at early times.

Absorption-time profiles at four representative probe wavelengths for delay times up to \(\Delta t = 2.0\,\hbox {ps}\) (left column) and \(\Delta t = 18\,\hbox {ps}\) (right column). Triangles represent the data points and black lines the overall fit curves. The individual contributions to the latter are shown in green (\(\tau _1\)), red (\(\tau _2\)) and blue (\(\tau _3\)). The contribution of product absorption is shown in gray
4 Discussion
4.1 Transient absorption experiment
The reported femtosecond time-resolved transient absorption experiment for transmissive solid samples allows for highly sensitive measurements of broadband transient absorption spectra in the wavelength range of 310–750 nm and the simultaneous detection of transient absorption at a single additional wavelength. In a single-shot mode, typical sensitivities of the order of \(\Delta {\mathrm{OD}}\approx 1\times 10^{-3}\) enable the investigation of sensitive samples that are prone to photodegradation. Data accumulation over a larger number of laser shots on the same spot or, if necessary, after x–y translation of the sample to a new spot, enables a corresponding improvement of detection sensitivity.
Furthermore, the setup can easily be transformed to a pump–repump–broadband probe or pump–dump–broadband probe experiment in the solid state. This only requires a third chopper in the single-color probe beam path, which then serves as the second pump or dump pulse. Such experiments could be useful for the investigation of multichromophores [50, 51] or the control of complicated photo-induced reactions such as the ring closure and opening of fulgides as was already shown for indolylfulgides [52–57].
4.2 Time-resolved experiments of DR1 in PMMA matrices
The time-resolved experiments on \(\hbox {DR}1\)-doped PMMA films on glass substrates revealed molecular dynamics that are comparable with those obtained in solution [40, 46]. The initial dynamics and the subsequent isomerization via a conical intersection are virtually unaffected, apart from minor shifts of the ESA bands, by the polymeric environment. The similar relaxation behavior of DR1 in solution and embedded in the PMMA matrix can be rationalized by the fact that the \(\hbox {DR}1\) molecules are not covalently linked to the polymer, and hence, the isomerization is less hindered compared with systems in which the azobenzene unit is sterically unrestrained. In contrast, the dynamics of azobenzene derivatives cross-linked into a polymer matrix via covalent bonds has been found to be drastically slower compared with the free monomeric switches in solution [51]. These effects are currently under investigation in solid films.
The ensuing vibrational cooling dynamics in the electronic ground state is slowed down compared with the vibrational cooling dynamics in the solvent. The differences include spectral broadening of the HGSA contribution and an increase of the vibrational cooling time from \(\tau _3 = 6.0\pm 0.1\,\hbox {ps}\) to \(\tau _3 = 14\pm 1\,\hbox {ps}\). The dependence of the rate of vibrational relaxation on the environment could be due to a viscosity effect. As has been previously discussed, vibrational relaxation is highly dependent on the solvent viscosity and higher viscosity leads to slower vibrational cooling [58, 59]. A detailed discussion of this point is, however, beyond the scope of the present paper and will be subject to future studies with the described setup.
5 Conclusion
In this paper, we presented a highly sensitive femtosecond time-resolved transient absorption experiment that allows us to observe transient absorption changes in transmissive solid samples with a broadband probe pulse and simultaneously with a single-color probe pulse. With this setup, complex reaction and deactivation dynamics in the solid state can be investigated. In a single-shot mode, the detection sensitivity of \(1\times 10^{-3}\) allows for the investigation of sensitive samples that are easily photodegraded. Using a combination of optical choppers, the sample can be moved between successive laser shots and the transient absorption at each delay time can be monitored at a fresh sample spot. A detailed description of the setup including a convenient, non-invasive method to check the homogeneity of the sample in advance of time-resolved experiments was given. The setup can easily be extended to a pump–repump or pump–dump experiment that could be useful for the investigation of coupled chromophore systems or complex molecular switching mechanisms under solid-state conditions similar as in applications. The setup has been tested with PMMA films doped with the push–pull azobenzene DR1, and the results were discussed in comparison with corresponding solution phase experiments.
References
B.L. Feringa (ed.), Molecular Switches (Wiley-VCH, Weinheim, 2001)
A.A. Beharry, G.A. Woolley, Chem. Soc. Rev. 40, 4422 (2011)
H.M.D. Bandara, S.C. Burdette, Chem. Soc. Rev. 41, 1809 (2012)
Y. Yokoyama, Chem. Rev. 100, 1717 (2000)
F. Renth, R. Siewertsen, F. Temps, Int. Rev. Phys. Chem. 32, 1 (2013)
M. Irie, Chem. Rev. 100, 1685 (2000)
K. Matsuda, M. Irie, J. Photochem. Photobiol. C 5, 169 (2004)
S. Kawata, Y. Kawata, Chem. Rev. 100, 1777 (2000)
D. Gust, J. Andréasson, U. Pischel, T.A. Moore, A.L. Moore, Chem. Commun. 48, 1947 (2012)
F. Ercole, T.P. Davis, R.A. Evans, Polym. Chem. 1, 37 (2010)
S.W. Hell, Science 316, 1153 (2007)
B. Huang, M. Bates, X. Zhuang, Annu. Rev. Biochem. 78, 993 (2009)
G.S. Kumar, D.C. Neckers, Chem. Rev. 89, 1915 (1989)
A. Natansohn, P. Rochon, Chem. Rev. 102, 4139 (2002)
T. Hugel, N.B. Holland, A. Cattani, L. Moroder, M. Seitz, H.E. Gaub, Science 296, 1103 (2002)
N.B. Holland, T. Hugel, G. Neuert, A. Cattani-Scholz, C. Renner, D. Oesterhelt, L. Moroder, M. Seitz, H.E. Gaub, Macromolecules 36, 2015 (2003)
M. Häckel, L. Kador, D. Kropp, H.W. Schmidt, Adv. Mater. 19, 227 (2007)
I. Willerich, F. Gröhn, Macromolecules 44, 4452 (2011)
S. Wiktorowicz, H. Tenhu, V. Aseyev, Macromolecules 46, 6209 (2013)
Z. Sekkat, D. Morichère, M. Dumont, R. Loucif-Saïbi, J.A. Delaire, J. Appl. Phys. 71, 1543 (1992)
C.C. Hsu, Y.T. Wang, A. Yabushita, C.W. Luo, Y.N. Hsiao, S.H. Lin, T. Kobayashi, J. Phys. Chem. A 115, 11508 (2011)
F. Fabbri, D. Garrot, K. Lahlil, J.P. Boilot, Y. Lassailly, J. Peretti, J. Phys. Chem. B 115, 1363 (2011)
C. Pakula, C. Hanisch, V. Zaporojtchenko, T. Strunskus, C. Bornholdt, D. Zargarani, R. Herges, F. Faupel, J. Mater. Sci. 46, 2488 (2011)
M. Klessinger, J. Michl, Excited States and Photochemistry of Organic Molecules (VCH, New York, 1995)
W. Domcke, D.R. Yarkony, H. Köppel (eds.), Conical Intersections: Electronic Structure, Dynamics and Spectroscopy, Advanced Series in Physical Chemistry, vol. 15 (World Scientific, Singapore, 2004)
W. Domcke, D.R. Yarkony, H. Köppel (eds.), Conical Intersections: Theory, Computation and Experiment, Advanced Series in Physical Chemistry, vol. 17 (World Scientific, Singapore, 2011)
O. Kühn, L. Wöste (eds.), Analysis and Control of Ultrafast Photoinduced Reactions (Springer, New York, 2007)
N. Turro, V. Ramamurthy, J. Scaiano, Modern Molecular Photochemistry of Organic Molecules (University Science Books, Sausalito, California, 2010)
A.H. Zewail, J. Phys. Chem. A 104, 5660 (2000)
G.P. Wakeham, K.A. Nelson, Opt. Lett. 25, 505 (2000)
G.P. Wakeham, D.D. Chung, K.A. Nelson, Thermochim. Acta 384, 7 (2002)
P.R. Poulin, K.A. Nelson, Science 313, 1756 (2006)
N. Furukawa, C.E. Mair, V.D. Kleiman, J. Takeda, Appl. Phys. Lett. 85, 4645 (2004)
Y. Makishima, N. Furukawa, A. Ishida, J. Takeda, Jpn. J. App. Phys. 45, 5986 (2006)
J. Takeda, A. Ishida, Y. Makishima, I. Katayama, Sensors 10, 4253 (2010)
Y. Minami, H. Yamaki, I. Katayama, J. Takeda, Appl. Phys. Express 7, 022402 (2014)
A. Warth, J. Lange, H. Graener, G. Seifert, J. Phys. Chem. C 115, 23329 (2011)
K. Röttger, R. Siewertsen, F. Temps, Chem. Phys. Lett. 536, 140 (2012)
A.L. Dobryakov, S.A. Kovalenko, A. Weigel, J.L. Pérez-Lustres, J. Lange, A. Müller, N.P. Ernsting, Rev. Sci. Instrum. 81, 113106 (2010)
J. Bahrenburg, K. Röttger, R. Siewertsen, F. Renth, F. Temps, Photochem. Photobiol. Sci. 11, 1210 (2012)
F. Renth, R. Siewertsen, F. Strübe, J. Mattay, F. Temps, Phys. Chem. Chem. Phys. 16, 19556 (2014)
M. Lorenc, M. Ziolek, R. Naskrecki, J. Karolczak, J. Kubicki, A. Maciejewski, Appl. Phys. B 74, 19 (2002)
C. Schriever, S. Lochbrunner, E. Riedle, D.J. Nesbitt, Rev. Sci. Instrum. 79, 013107 (2008)
N.G. Semaltianos, Microelectron. J. 38, 754 (2007)
C.B. Walsh, E.I. Franses, Thin Solid Films 429, 71 (2003)
M. Poprawa-Smoluch, J. Baggerman, H. Zhang, H.P.A. Maas, L. De Cola, A.M. Brouwer, J. Phys. Chem. A 110, 11926 (2006)
C. Toro, A. Thibert, L. De Boni, A.E. Masunov, F.E. Hernandez, J. Phys. Chem. B 112, 929 (2008)
D. Brown, A. Natansohn, P. Rochon, Macromolecules 28, 6116 (1995)
T. Buffeteau, F. Lagugné Labarthet, M. Pézolet, C. Sourisseau, Macromolecules 31, 7312 (1998)
J. Bahrenburg, C.M. Sievers, J.B. Schönborn, B. Hartke, F. Renth, F. Temps, C. Näther, F.D. Sönnichsen, Photochem. Photobiol. Sci. 12, 511 (2013)
J. Bahrenburg, F. Renth, F. Plamper, W. Richtering, F. Temps, Phys. Chem. Chem. Phys. 16, 11549 (2014)
A. Rupenyan, I.H.M. van Stokkum, J.C. Arents, R. van Grondelle, K.J. Hellingwerf, M.L. Groot, J. Phys. Chem. B 113, 16251 (2009)
Z. Wei, T. Nakamura, S. Takeuchi, T. Tahara, J. Am. Chem. Soc. 133, 8205 (2011)
S. Draxler, T. Brust, S. Malkmus, J.A. DiGirolamo, W.J. Lees, W. Zinth, M. Braun, Phys. Chem. Chem. Phys. 11, 5019 (2009)
S. Draxler, T. Brust, J. Eicher, W. Zinth, M. Braun, Opt. Commun. 283, 1050 (2010)
T. Brust, S. Malkmus, S. Draxler, S.A. Ahmed, K. Rück-Braun, W. Zinth, M. Braun, J. Photochem. Photobiol. A 207, 209 (2009)
T. Brust, S. Draxler, J. Eicher, W.J. Lees, K. Rück-Braun, W. Zinth, M. Braun, Chem. Phys. Lett. 489, 175 (2010)
R.M. Stratt, M. Maroncelli, J. Phys. Chem. 100, 12981 (1996)
I. Martini, G.V. Hartland, J. Phys. Chem. 100, 19764 (1996)
Acknowledgments
Financial support by the DFG through the CRC 677 “Function by Switching” is gratefully acknowledged. We cordially thank Prof. Dr. F. Faupel, Dr. T. Strunskus and V. Schneider from the University of Kiel for valuable information on the sample preparation and characterization.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Röttger, K., Wang, S., Renth, F. et al. A femtosecond pump–probe spectrometer for dynamics in transmissive polymer films. Appl. Phys. B 118, 185–193 (2015). https://doi.org/10.1007/s00340-014-5967-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00340-014-5967-y