
Abstract
Adsorption of dynamically diluted ammonia at part-per-billion to low part-per-million concentrations in dry nitrogen was studied with treated and non-treated stainless steel and polymer test tubes. The treatments included electropolishing and two types of coatings based on amorphous silicon. Cavity ring-down spectroscopy with an external cavity diode laser operating in the near-infrared wavelength range was used to monitor the adsorption process in real time in continuous-flow conditions to obtain quantitative assessment of the adsorptive properties of the studied surfaces. The investigated polymers were all less adsorptive than any of the treated or non-treated stainless steel surfaces. Some of the commercial coatings reduced the adsorption loss of stainless steel by a factor of ten or more. Polyvinylidene fluoride was found to be superior (less adsorption) to the four other studied polymer coatings. The number of adsorbed ammonia molecules per surface area obtained at different ammonia gas phase concentrations was modeled with Langmuir and Freundlich isotherms. The time behavior of the adsorption–desorption process occurring in the time scale of seconds and minutes was simulated with a simple kinetic model.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Ammonia (NH3) is an abundant gaseous pollutant in the atmosphere, where it contributes to acid deposition, eutrophication and secondary particulate aerosol formation [1, 2]. Its normal lower atmospheric mixing ratio varies from below one part-per-billion-by-volume (ppbv) to more than 10 ppbv [3], but the concentration may rise to part-per-million-by-volume (ppmv) levels in heavily contaminated areas [4]. Ammonia is mainly produced by agricultural activities linked to animal manure and fertilizers [5] but also by traffic and industrial processes [6]. Ammonia is also an important indoor air contaminant [7] and a potential biomarker in human breath [8].
The National Emission Ceilings (NEC) directive (2001/81/EC) covering four air pollutants including NH3 has set binding emission ceilings to be achieved by each member state by 2010 and not to be exceeded thereafter [9]. This necessitates traceable measurement methods to reliably quantify the prevailing NH3 levels. The gas phase NH3 concentration can be measured with, e.g., acid scrubbers, filter packs, denuders and optical methods [4]. Metal-oxide, catalytic and conducting polymer NH3 sensors used in ammonia analysis are reviewed in [10], and selected inter-comparison studies are presented in Table 3 of [4]. A field inter-comparison of eleven atmospheric ammonia measurement techniques can be found in [11]. Furthermore, real-time gas phase NH3 measurement methods are compared in [12]. The overall correlation between different methods is generally satisfactory, but their inter-correlation tends to worsen at low concentrations. Inlet tubing length and filters, for example, affect the accuracy and time-response of the NH3 measurements. Because the variation in the measured concentrations is large, no single apparatus is suitable for all applications, and all methods are not appropriate for extremely low concentrations. Ammonia is a challenging reactive gaseous compound due to its high water (H2O) solubility and polarity. The quantification is strongly affected by water condensation and adsorption/desorption effects occurring on the surfaces of the sampling system and detection instrumentation. Accurate and reliable measurement data cannot be obtained if such effects are not investigated and understood at the concentration levels appropriate to atmospheric air quality monitoring.
Some studies on the adsorption of NH3 on various metal, glass and polymer surfaces have been performed. Photoacoustic spectroscopy (PAS) was used for testing several materials in [13] and [14]. The authors compared the ammonia concentrations entering and leaving two- to three-meter-long tubes with inner diameters of 4 mm under low-flow (30 standard cubic centimeters per minute, sccm) conditions. Polished and unpolished stainless steel (SS), quartz, Pyrex, polytetrafluoroethylene (PTFE) and perfluoroalkoxy (PFA) surfaces were investigated, and the studied gas phase NH3 concentration varied between 0.5 and 50 ppmv. The researchers used photoacoustic (PA) cells at both ends of the test tubes to measure the gas phase concentration and showed that the adsorption of ammonia leads to a considerable time delay of the signal in the second cell. They found that the surface coverage tends to zero with decreasing concentration for Pyrex, quartz and the polymer surfaces whereas for stainless steel, a sizeable adsorption persists even at the lowest studied concentrations. The adsorption on electropolished SS was further observed to be larger than on non-treated SS. Schmohl et al. [15] used PAS to investigate the effects of adsorption processes on the response time and accuracy of NH3 detection under stopped and continuous-flow conditions. The adsorption was investigated within PA cells made of brass and polypropylene (PP). Their observations established that stopped flow measurements of NH3 are never reproducible, and reliable measurements require continuous-flow conditions. Furthermore, the authors provide a short list of recommendations to substantially reduce the adsorption effect in trace NH3 quantification.
Ellis et al. [16] studied the effect of humidity and heat on the adsorption of NH3 on the PFA sampling line using a quantum cascade tunable infrared laser differential absorption spectrometer (QC-TILDAS) at concentrations of 30–1,000 ppbv in humid and dry conditions. They modeled the decrease in the gas phase NH3 concentration (due to adsorption) by a bi-exponential decay, where, according to the authors, the first, fast decay corresponds to the gas exchange time of the system and the second, slow decay corresponds to adsorption in the inlet, sampling line and sample cell. On the basis of the model, the authors deduced that the relative contribution of adsorption is higher at lower concentrations, under humid conditions and without heating. A similar bi-exponential decay model was employed in [17] to compare 5-m-long inlet tubes (the inner diameter of 4.32 mm) made of plain SS, Silcosteel (current trade name SilcoNert™ 1000, SilcoTek Corporation) coated SS tubes, PTFE and polyethylene (PE). The NH3 concentration in the experiments was 100 ppbv. The authors utilized the value of the first, short (corresponding to fast processes) time constant to assess the strength of adsorption and desorption. They found that the polymer tubes, which were both similar in their adsorptive qualities, outperformed (less adsorption) both Silcosteel-coated and plain SS tubes although the coating improved the adsorptive quality of SS.
Shah et al. [18] evaluated five different polymer tubing materials [PTFE, PFA, fluorinated ethylene propylene (FEP), high-density polyethylene (HDPE) and polyvinylchloride (PVC)] for ammonia adsorption at two nominal concentrations (1 and 10 ppmv) at the room temperature (297 K). Teflon is often used to refer to the three firstly mentioned fluorinated polymers although it is also used exclusively as a trade name for PTFE. The acronym TFE is also occasionally used to designate PTFE Teflon. The authors used 2.5-m-long tubes with an inner diameter of 4.32 mm and a gas scrubber as the detection method. They did not find statistically significant differences in NH3 adsorption between different tubes. They further concluded that the cheapest PVC tube might be the best (least adsorption) option under the conditions of their study. Mukhtar et al. [19] measured the gas phase NH3 concentration at the inlet and outlet of PFA (referred to as Teflon in the paper) and low-density polyethylene (shortened as LDPE or PELD) tubes using a chemiluminescence analyzer. The tubes were 15 or 46 m long, had an inner diameter of 3.2 mm and the measurements were performed at six nominal concentrations between 2 and 35 ppmv at two different temperatures (298 and 310 K). Ammonia adsorption on LDPE was found to be significantly higher than the adsorption on PFA. Adsorption (the difference between the inlet and outlet mixing ratios) on PFA was independent of the gas phase concentration or temperature. The adsorption on LDPE, on the contrary, increased with the gas phase concentration and temperature.
In the current study, we have systematically assessed the adsorption of gas phase NH3 on various surface materials. The studied stainless steel-based materials were non-treated SS316L, electropolished SS316L and three commercial (SilcoTek Corporation) coating materials deposited on an SS 316L substrate: SilcoNert™ 1000, SilcoNert™ 2000 and Dursan™. The investigated polymers were PTFE, PFA, FEP, PELD (low-density polyethylene) and PVDF (polyvinylidene fluoride). The work was performed using a laser spectrometer based on cavity ring-down spectroscopy (CRDS) operating in the 1.5-μm spectral region. The time-resolved adsorption of ammonia was studied in controlled conditions using test tube (TT) samples of different surface materials. The effects of the gas phase NH3 concentration, temperature and H2O content of the experiment were investigated. The observed results were fitted to Langmuir and Freundlich adsorption isotherms, and the real-time adsorption data were modeled using a Langmuir-based rate equation model. The gathered information facilitates the choice of materials and procedures to reduce the measurement time and uncertainty owing to adsorption effects in atmospheric trace gas NH3 measurements.
2 Experimental setup and measurement protocol
2.1 Cavity ring-down setup
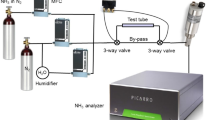
Cavity ring-down spectroscopy is a highly sensitive absorption technique where a laser beam interacts with an optical cavity usually consisting of two highly reflective mirrors [20, 21]. We used a home-built continuous-wave CRD spectrometer based on a tunable external cavity diode laser (ECDL). The details of the spectrometer are given in [22], and a simplified schematic diagram of the setup is presented in Fig. 1. A ring-down cell (RDC) made of stainless steel and with a length of 50 cm and a volume of 40 cm3 was used in the measurements. The RDC is coated with the SilcoNert™ 2000 coating. At the 1,000 sccm gas flow rate and 100 mbar pressure, the theoretical gas exchange time of the cavity is about 240 ms. During the measurements, the empty cavity ring-down (RD) time constant was between 140 and 160 μs.

The measurement setup. ECDL external cavity diode laser, OI optical isolator, AOM acousto-optic modulator, DAQ data acquisition card, V valve
To measure the mixing ratio of gas phase NH3 prior to each adsorption measurement (see Sect. 2.3 for the measurement protocol), the laser wavelength was scanned over the region 6,548.45–6,549.05 cm−1. A typical spectrum for a low (90 ppbv) NH3 concentration is shown in Fig. 2. The spectrum shows three strong lines which all belong to NH3 [23]. Fifty RD events were averaged for each spectral point, and altogether 200 spectral points were recorded using a pressure of 100 mbar. Three Voigt profiles were fitted to the spectrum using the peak fitting software Fityk [24]. The estimated detection limit (3 times the standard deviation, 3σ) for NH3 was ≤1 ppbv for these measurements.

The spectrum of ammonia (91 ppbv) around 6,548 cm−1. All the observed lines belong to NH3
High time resolution is needed to follow the adsorption in real time. For this reason, the laser wavelength was kept fixed (without active stabilization) at the top of the NH3 absorption line at 6,548.79 cm−1. The line strength of this transition is 1.67 × 10−21 cm molecule−1 at 296 K [23]. The specified wavenumber was continuously monitored using a wavemeter (EXFO, WA-1500), which has a frequency uncertainty of 40 MHz (0.0013 cm−1). The full-width at half maximum (FWHM) line width of the NH3 transition is about 0.032 cm−1 at the measurement pressure of 100 mbar. Altogether 25 RD events were averaged for each point, and the average repetition rate for the RD events was about 50 Hz. Thus, the time resolution of the experiment was about 0.5 s. The detection limit for these measurements was calculated (from >20 successive measurement points) to be ~1.5 ppbv (3σ) for low concentrations (≤500 ppbv). In the highest measured concentration (~9 ppmv), the noise in the ring-down signal corresponded to ~15 ppbv (3σ).
2.2 Gas generation and test tube setup
The required NH3 gas phase concentration was generated by dynamic dilution of a bottled NH3 standard in nitrogen (AGA). Calibrated mass flow controllers (MKS Inc.) and instrument nitrogen (specified H2O content of <3 ppmv and NH3 content of <1 ppbv) (AGA) were used for dilution. The concentration of the standard was determined to be 8,750 ppbv by the CRDS setup, and it was within the specifications provided by the manufacturer.
A schematic diagram of the gas generation and TT setup is presented in Fig. 3. The gas-generating line and its connection to the RDC were made of PTFE tubing, except for the connectors, valves, mass flow controllers and the input appendix (a short and narrow tube made of SS) of the RDC. The TT was connected to a 3-way valve with a 4 cm long and ~1 mm inner diameter adapter made of PTFE. The contribution of the adapters at the both ends of the test tube was considered negligible to the total adsorption. The 3-way valve attached to the TT was directly connected to the appendix of the RDC (which was coated with SilcoNert™ 2000) with a short (~70 cm) PTFE tube.

The gas generation and test tube setup. The additional by-pass was used in test measurements. Indoor air contains ~1 % of water
2.3 Measurement protocol
The measurement protocol consisted of three successive stages: (1) a cleaning of the test tube (2) an exposure of the rest of the vacuum system (except the test tube) to a known concentration of gas phase NH3 and (3) a time-resolved adsorption measurement by exposure of the cleaned test tube. The stages are explained in detail below.
-
1.
The test tube was flushed with indoor air for at least an hour. Indoor air (with H2O content of ≤1 % by volume and NH3 content of about 5 ppbv) instead of pure nitrogen gas was used because the H2O molecules in indoor air aid to remove NH3 from the TT and the rest of measurement setup surfaces. Without the water “treatment,” it is impossible to remove the adsorbed NH3 in a reasonable time and obtain reproducible results in two successive adsorption measurements. Then, the TT was flushed with pure nitrogen gas for at least 30 min to remove H2O and the remaining adsorbed NH3 molecules in the system. The NH3 concentration was ~1 ppbv and the H2O content 20–30 ppmv after the flushing procedure. The H2O vapor mixing ratio was measured with the current CRDS setup using a line at 6,549.763 cm−1 with a line strength of 3.31 × 10−24 cm molecule−1 at 296 K [25]. Care was taken to ensure that the flushing time for all test tubes with different surface materials was the same and always exceeded the minimum time specified above. Removing the adsorbed NH3 and H2O completely, i.e., achieving the specified NH3 and H2O concentrations of the “dry and pure” nitrogen gas would require longer flushing times, up to days. Furthermore, even a minor leak somewhere in the vacuum system leads to a ppmv level of H2O contamination from the indoor air. This possibility cannot always be ruled out. The flushing time was therefore a compromise between a reasonable total measurement time (at least one measurement per day) and minimal NH3 and H2O contamination of the TT. Heating of the test tubes in an oven was also tested as an alternative method to the flushing procedure but because it did not produce superior results and was found to be impractical, this was not continued. At the end of the first phase, the nitrogen gas was switched to flow through the by-pass line depicted in Fig. 3 using electronic 3-way valves (Bio-Chem Valve 105T212-125). Thus, after this stage, a clean TT contained pure nitrogen gas at a pressure of about 150 mbar (the pressure behind the RDC and further away from the vacuum pump is always higher than the pressure of 100 mbar in the RDC).
-
2.
The RDC and the rest of the vacuum line excluding the TT were exposed to a desired dynamically diluted NH3 concentration utilizing the by-pass line. Depending on the NH3 concentration, time varying from 1–2 h was usually required for the RD signal to adequately stabilize to the studied concentration. This time is necessary for all the surfaces of the vacuum line to be saturated with NH3. Absolute signal stabilization would require longer times and even if these longer stabilization times were used, other long-term effects, such as drift of the mass flow controllers and gas bottle regulator pressures, would step in. After the stabilization, the gas phase concentration of NH3 was recorded by scanning the laser frequency over the spectral region mentioned in Sect. 2.1.
-
3.
After the system had been cleaned and stabilized, and the initial gas phase concentration determined, the actual time-resolved adsorption measurement took place. This stage consisted of three phases (a–c, see Fig. 4). (a) The NH3 gas flow by-passed the (pure N2 containing) TT. This phase took about 3 min. (b) The NH3 flow was switched to pass through the TT with the help of the 3-way valves. This step was instantaneous and resulted in a fast decrease in the measured NH3 concentration in the RDC. The sudden concentration drop is due to the rapid gas exchange in the unexposed TT filled with pure N2 and fast NH3 adsorption on the surface of the TT. (c) The NH3 concentration signal slowly recovered and finally reached the same level as in the phase a. This phase took up to about 15 min depending on the type of the TT and NH3 concentration employed.
Fig. 4 
The total loss of NH3 molecules due to gas exchange and adsorption. a stable NH3 concentration before actual adsorption experiment, b sharp decrease in NH3 concentration due to gas exchange and adsorption, c slow recovery of NH3 concentration due to adsorption

The measurement protocol was repeated using different surface materials. The same TT was used for three consecutive measurements with a given surface material, but tests with tubes belonging to other manufacturing batches were also performed. For comparing the total adsorption of different SS and coated SS surfaces, an NH3 gas phase concentration of about 425 ppbv and a total flow rate of 1,000 sccm were employed. Three consecutive measurements with the same experimental conditions were performed without touching the setup except actions required for flushing it. In another experiment, the SS316L TT temperature was raised stepwise from 295 to 333 K to test the temperature effect on adsorption at an NH3 gas phase concentration of about 425 ppbv. These measurements were performed only once in the same conditions. The TT length was 30 cm and inner diameter 4 mm in all the above-mentioned measurements. For the comparison of the total adsorption on various polymer surfaces, a non-diluted bottled NH3 standard with a concentration of 8,750 ppbv was used. Additionally, the TT length was increased to 200 cm, and a flow rate of 200 sccm was used. The increase in the NH3 concentration and TT length were necessary to observe the adsorption phenomenon with an adequate signal-to-noise ratio for all of the studied polymers in similar conditions. For the PFA isotherm studies, the gas phase NH3 concentration was varied between 150 and 8,750 ppbv using flow rates of 200 and 1,000 sccm.
The measurement temperature in the RDC was 295 K. This was also the estimated temperature of the NH3 gas in the experiments when the TT itself was heated. Small changes in the room temperature (±1 K) occurred during the measurement campaign, but this has only a minor (~1 %) effect on the retrieved concentration. It has not been taken into account because it is well within the experimental uncertainty.
3 Results
3.1 General remarks
In Fig. 5, the time evolution of the recorded gas phase NH3 concentration is shown in an experiment where the concentration was changed in a stepwise fashion (the step size ~180 ppb and 20 min) by diluting a bottled NH3 standard with indoor air (H2O content ~0.6 %). A careful inspection of the data presented in the figure reveals several phenomena, which are ubiquitous in the context of adsorptive molecular species such as NH3. First, if no adsorption were present, the concentration steps would be of a square shape, evenly spaced and the corresponding observed concentrations should be the same on the both sides of the concentration “ladder” which is not the case. Second, the shape of the “ladder” form is skewed: The decrease in the concentration on the right-hand side of the figure is always steeper than the respective increase in the concentration. Third, the calculated (on the basis of the dilution ratio) NH3 concentration value is closer to the decreasing concentration than the increasing concentration at the end of each 20-min step. This means that it is easier to remove than to enrich NH3 molecules on a surface, and the rise time from 0 to 90 % of an actual concentration in an experiment is longer than the corresponding decay time from 100 to 10 %.

Stepwise increase and decrease in dynamically diluted NH3 gas phase (diluted in indoor air with a H2O content ~0.6 %) concentration
3.2 Calculation of total adsorption
Figure 4 shows the time evolution of the adsorption signal during the measurement protocol. There are two processes which contribute to the observed loss of gas phase NH3 molecules: gas exchange within the TT and adsorption on the surface of the TT. Therefore, in the figure, the fast decrease in the signal can be associated with gas exchange and the “fast adsorption component” of NH3 and the slow re-increase with the “slow adsorption component.” The number of missing molecules in the TT due to the gas exchange, N g, can be calculated from the ideal gas law. The number of adsorbed molecules N ads is achieved by subtracting N g from the total number of the lost molecules N loss obtained by integration (see Fig. 4):
where t is time, t 0 is the time point when the adsorption process starts and N is the number of the NH3 molecules. The total number of adsorbed molecules per unit surface area, N s, is then obtained by a division of the TT surface area.
3.3 Treated and non-treated stainless steel surfaces
Table 1 presents the results obtained for the SS and treated SS surfaces. The NH3 concentration of the measurements was about 425 ppbv, and the gas flow rate was 1,000 sccm. The TT pressure and measurement temperature were 176 mbar and 295 K, respectively. The variation in three consecutive measurements performed with an identical setup and in the same experimental conditions using the same TT was always within 20 %. In Fig. 6, a typical example of the reproducibility of two consecutive measurements is presented. The calculated standard deviation is in the case of the lowest adsorbing coating, SilcoNert™ 2000, considerably smaller than the stated 20 %. However, the actual uncertainty in the adsorption value is estimated to be about 3 × 1012 molecule cm−2, which is about 50 % of the observed total adsorption. As the amount of adsorption decreases, the signal-to-noise ratio of the measurement also decreases which increases the uncertainty in the integration. This introduces a possibility for a systematic error larger than 20 %.

The reproducibility of the measurement in two successive recordings. The studied surface material was stainless steel 316L
Conspicuous adsorptivity inconsistencies of about a factor of two were also occasionally observed for the same individual SS316L tube with a reassembled (but schematically identical) setup in measurements performed a few months apart, despite the fact that no obvious problems in the setup or in the measurement protocol (e.g., obvious leakages or sudden pressure changes due to irregular closure or opening of the 3-way valves) were observed. The reasons for such inconsistencies are currently unknown. Possible explanations include a leakage of indoor air (leading to a H2O contamination) to the TT during the saturating phase of the protocol or, more likely, variable H2O content of the trace gas used in the measurements. Water plays an important role through competitive adsorption with ammonia, and the adsorption of NH3 was observed to be strongly dependent on the H2O concentration of the measurement (Fig. 7). Because it was impossible to monitor the H2O concentration in real time with the current measurement setup, the actual H2O mixing ratio during the individual adsorption measurements is not exactly known. According to the data in Fig. 7, a change by a factor of two in H2O concentration, which is conceivable, might lead up to a factor of two change in the recorded NH3 adsorption. In order to obtain identical conditions, the whole measurement setup was leak-tested before a series of the three consecutive measurements. However, because our simple leak-testing procedure is not impeccable and it is impossible to monitor the pressure change in the TT during the saturation phase, minor leaks may have existed during the protocol. For these reasons, care should be taken in interpreting the results presented in Table 1 and in estimating their uncertainty. The adsorption values should be considered as minimum values at an H2O concentration of about 20 ppmv which was the smallest concentration achieved.

Ammonia adsorption as a function of water concentration. The studied surface material was stainless steel 316L
Despite the insufficiencies discussed above, our results compare well with the literature values obtained for similar coatings [13, 14] and match qualitatively with the data provided by the manufacturer of the silicon-based coatings [26]. In [13] and [14], the H2O concentration of the trace gas was not quantified, and the phrasing “dry” was used for apparently water-free conditions. However, the authors in [14] have observed that ammonia can almost completely be degassed at room temperature by using H2O-containing gas flow. This notion corroborates our observations and the data presented in Fig. 7. The finding is also the foundation of our TT cleaning procedure.
In [14], at a concentration of 1 ppmv, a total adsorption of 2 × 1014 molecule cm−2 for SS316 was observed. This matches well with our result of 1.4 × 1014 molecule cm−2 for SS316L because in their measurement, the mass concentration was four times higher due to an increased NH3 concentration and pressure. Electropolishing does not improve much, if at all, the adsorptive properties of steel. Electrolysis used in the electropolishing process removes atoms from the SS surface but does not change the surface qualitatively. Melander et al. [14] observed, analogously to our results, more adsorption on electropolished SS316 than on non-polished SS304. They, however, compared two different SS types which likely have an effect on adsorptivity (there is 2 % of molybdenum on SS316 which improves its corrosion resistivity; otherwise, these two SS types are similar). On the other hand, in the current study, a direct comparison is also compromised because, firstly, the non-polished and polished SS test tubes were from different batches (and therefore have possibly somewhat different composition) and, secondly, because of the reproducibility problems addressed earlier.
The SilcoNert™ 2000 coated surface was the least adsorptive of the studied metal-based materials with an improvement by a factor of 25 over non-treated SS316L. SilcoNert™ 2000 has two layers: a hydrogenated amorphous silicon (a-Si) layer on a plain SS substrate and an extra alkyl layer on the top of the a-Si layer for capping the remaining active adsorption sites [27]. This additional layer may explain the threefold superiority of SilcoNert™ 2000 over SilcoNert™ 1000 which has only a single a-Si layer. For a quartz surface, Melander et al. [14] measured a total adsorption of about 1 × 1013 molecule cm−2. This is approximately the same value as we have observed using a similar NH3 concentration for our “quartz” surfaces, SilcoNert™ 1000 and SilcoNert™ 2000 (both of these coating are often referred as quartz). There is not enough information available from the manufacturer regarding the possible reasons for the third coating produced by SilcoTek Corporation, Dursan™, being more adsorptive than the other studied SilcoTek coatings. In any case, this observation has also been confirmed by real-time proton-transfer-reaction mass spectrometry (PTR-MS) studies performed elsewhere (G. Barone, SilcoTek Corporation, personal communication).
When the SS316L TT temperature was increased stepwise from 295 to 333 K, there was a decreasing trend in adsorption, as expected. However, the overall change in the total adsorption was surprisingly small and almost within the experimental uncertainty of 20 %. This is an interesting result because the adsorption effects of NH3 are often claimed to be considerably alleviated even by a relatively small increase in temperature, especially in humid samples. This superficially contradictory (with respect to our study) observation can be explained by the role of H2O (which is almost always present at percent level in, e.g., atmospheric trace gas analysis if no action is taken to remove it), even a minimal H2O condensation results in NH3 dissolving from the gas phase and reducing its apparent concentration. This happens every time the relative humidity reaches 100 % anywhere in the sampling and recording apparatus. An increase in T reduces H2O condensation, and consequently, less NH3 is dissolved in H2O droplets.
In addition to the surface materials listed in Table 1, adsorption on a plain copper surface was measured to be about double (~2.5 × 1014 molecule cm−2) to that of SS under similar conditions. For comparison, Schmohl et al. [15] observed higher but comparable adsorption of ≥4.4 × 1014 molecule cm−2 for a brass surface in a desorption study employing a considerably higher NH3 concentration (61 ppmv) than in our experiment.
3.4 Polymer surfaces
Polymer surface adsorption results are displayed in Table 2. The NH3 concentration of the measurements was about 8,750 ppbv, and the gas flow rate was 200 sccm. The TT pressure was 119 mbar, and the temperature was 295 K. The standard deviation of three consecutive runs was within 25 % in these measurements. However, in the case of the least adsorbing polymer, PVDF, the actual maximum uncertainty in the total number of adsorbed NH3 molecules is estimated to be as much as 50 %, corresponding to 5 × 1011 molecule cm−2. This is due to the low adsorptivity of this material in relation to the sensitivity of the measurement, which causes uncertainty in the integration of N loss. Furthermore, as with the SS coatings, the absolute H2O concentration is not exactly known, and therefore, the uncertainty in the measurements could be larger than the given standard deviation. Similar large variations in the adsorption as in the case of SS surfaces were on the other hand never observed which gives confidence on the validity of the results. In any case, the N s values should be taken as minimum values and not as absolute quantities. In the case of polymer surfaces, where adsorption is, in general, less significant than in the case of SS surfaces, the effect of the gas exchange is considerable. For example, for PVDF, the gas exchange constitutes ~70 % of N loss. Once again, despite these effects, our results for PTFE and PFA match those of [14] taking into account different measurement conditions. They, however, did not observe any statistical difference between those two similar fluoropolymers.
According to our studies, the least adsorbing polymer is PVDF. We have two potential explanations for this observation. First, PVDF has both hydrogen (H) and fluorine (F) atoms attached to its carbon chain, and therefore, there is a possibility of hydrogen bonds between H and F atoms belonging to different polymer branches. This might make the polymer more inert against adsorption due to decreased surface energy. Second, PVDF is able to uptake (absorb) more water than the other fluoropolymers listed in Table 2 [28]. It is not unconceivable that H2O absorption into the polymer is also associated with a high concentration of H2O on the surface (adsorption). This inhibits the adsorption of NH3 molecules. Intriguingly, Shah et al. [18] discovered that another polymer, PVC, which has a similar, relatively high water absorption propensity as PVDF, had lower adsorptivity than the other studied polymers. It should be noted that the possible adsorptive edge of PVDF over the other studied polymers in dry conditions might be lost when the H2O concentration of the measured trace gas becomes high, as happens in actual atmospheric conditions.
We observed similar adsorptivities for all Teflon polymers (PTFE, PFA, FEP) although PFA reproducibly produced almost a factor of two higher values than PTFE or FEP. This is similar to the results of the studies of Shah et al. [18] who found no statistically significant difference between PTFE, PFA, FEP and HEPD/PEHD (high-density polyethylene). In contrast to our results, Mukhtar et al. [19] observed that PELD is more adsorptive than PFA. The authors also claimed that adsorption on PFA does not depend at all on NH3 gas phase concentration. In the light of the adsorption isotherm presented later in this paper, the latter claim seems untenable.
4 Modeling the adsorption equilibrium and kinetics
Adsorption isotherms are used to model the equilibrium surface adsorption coverage as a function of the gas phase concentration at a fixed temperature. Langmuir [29] proposed one of the most widely used isotherms, which can be derived theoretically based on two assumptions: (1) all adsorption sites are equivalent and can accommodate at most one molecule and (2) there is no interaction between the adsorbed molecules of adjacent sites. The first assumption leads to a maximum adsorption coverage of a single monolayer. The isotherm can be described by
where S ads.eq is the equilibrium surface concentration of adsorbed molecules [molecules cm−2], S mono is the total number of adsorption sites (monolayer) [molecules cm−2], K is the equilibrium constant for adsorption and desorption [cm3 molecules−1] and C gas is the gas phase concentration [molecules cm−3].The empirical Freundlich isotherm [30], like the Langmuir isotherm, includes two independent parameters and has been used successfully to model adsorption in different systems
where A and n are fitting parameters without an explicit physical meaning.
Figure 8 shows the adsorption as a function of gas phase concentration for PFA, in the concentration range of 0.7–43 × 10−12 molecule cm−3. Superimposed are the non-linear least-squares fits to the Langmuir and Freundlich adsorption isotherms. The agreement with the Freundlich isotherm is visibly better but since the Freundlich model is purely empirical, no real physical insight can be gleaned from this comparison. According to the Langmuir model, the adsorption should saturate at high concentrations. Unfortunately, we were limited by the concentration of our NH3 gas standard and could not explore higher concentration ranges.

Ammonia adsorption on PFA Teflon as a function of the gas phase NH3 concentration. The round markers represent measurements with a flow rate of 200 sccm and a pressure of 119 mbar, and the star-shaped marker represents a measurement with a flow rate of 1,000 sccm and a pressure of 176 mbar. See Sect. 4 for details and symbols for the Langmuir and Freundlich adsorption isotherms. The quantity R 2 is the coefficient of determination of the fitted curves
Because our experimental setup allowed an instrumental time resolution of less than one second, we attempted to model the real-time adsorption inside the test tube with a suitable kinetic model. A simple rate equation model can be constructed from the Langmuir adsorption model, assuming homogeneous adsorption energy, monolayer adsorption and non-interacting adsorbent molecules. It is given by
where P is the gas flow rate [cm3/s], V is the test tube volume [cm3], R is the test tube radius [cm], C in is the concentration of the gas supplied to the test tube, C gas(t) is the gas phase concentration inside the test tube at the time t, k a is the rate coefficient for adsorption, k d is the rate coefficient for desorption and S ads(t) is the surface concentration of adsorbed molecules [molecules cm−2] at the time t. The rate coefficients for adsorption and desorption are related to the equilibrium constant of the Langmuir isotherm by K = k a /k d.
The rate equations were solved using the Berkeley Madonna software (version 8.3.18, Macey and Oster). Figure 9 displays the real-time adsorption data for an experiment where a PFA test tube was exposed to an NH3 gas flow of 200 sccm and 8,781 ppbv. Superimposed is a solution to the rate equation where the adsorption rate coefficient was optimized to fit the experimental data by the curve fitting feature of the Berkeley Madonna software. The values for the monolayer coverage (S mono) and desorption rate coefficient (k d = k a /K) were derived from the fit to the Langmuir isotherm for PFA measurements with different concentrations (see Fig. 8). The agreement is not perfect, but the model retains the main features of the adsorption signal, both the rapid changes and the slow recovery to the equilibrium. Unfortunately, the deficiencies of the Langmuir model become apparent as we try to apply the optimized parameters to measurements performed with lower concentrations. A satisfactory agreement with the data can be achieved by re-fitting the adsorption rate coefficient for different concentrations but then the model loses its physical meaningfulness and predictivity.

Real-time ammonia adsorption and gas exchange using a PFA Teflon test tube. See Eq. (4) in Sect. 4 for details and symbols for the real-time model
The kinetic model can be improved by introducing heterogeneity into the adsorption energy. This can be done by including an additional sorption term in the rate equation, with three new parameters for monolayer coverage and adsorption/desorption rate coefficients. The fit to the individual measurements improves, as can be expected by adding more fitting parameters, but the predictivity of the model does not improve significantly. We also tried to separate the test tube into five successive compartments and solve these differential equations simultaneously using the same Langmuir parameters, but this did not improve the agreement between the experimental data and model results in any significant fashion.
5 Discussion and conclusions
Our results agree to a large extent with previous published investigations. The adsorption of NH3 on plain and coated SS surfaces falls in the same rank order as the data presented in the literature or provided by the coating manufacturer. This happens although the measurement methods (e.g., PAS, CRDS, chemiluminescence and PTR-MS) and protocols used to estimate the adsorptivity differ. The common observation that polymers generally adsorb less NH3 than stainless steel or even the best-performing coated metal surfaces was also reinstated in this study. Due to the low adsorptivity of polymers in general, a comparison of our results to the literature is more ambiguous.
On the basis of their model, Whitehead et al. [17] ranked their studied polymer and stainless steel surfaces in the order of increasing adsorption: PTFE = PE < SilcoNert™ 1000 < non-treated SS. This is also the rank order of our study with a side note that we found a small difference (the same order of magnitude as the maximum measurement uncertainty) between polyethylene (PELD in our case) and PTFE. The order is also in accordance with other studies where both treated and non-treated stainless steel and polymer surfaces have been investigated [14].
The relative contribution of adsorption to the measurement uncertainty of gas phase NH3 concentration is higher at lower concentrations as corroborated by the observations in [16]. However, on the contrary to their further claim that added humidity would increase the surface interactions (more NH3 adsorption), we have established that NH3 adsorption is decreased as the H2O content is increased. This effect is demonstrated in Fig. 7. The apparent disagreement between the studies might be explained by H2O condensation on the measurement system of [16]. Indeed, the authors of the study observed a significant decrease in “adsorption” when heating the tested PFA line to 313 K, and they further hypothesized that the temperature change might reduce the H2O condensation within the sampling line. Mukhtar et al. [19] have also observed that increasing the temperature of a polymer might actually increase its adsorption.
Although our measurement was performed in real time, the adsorption was not recorded exactly at the point of adsorption (i.e., inside the TT). This induces not only a certain time lag in the observed gas phase NH3 concentration but also creates a potential problem in the quantification of the adsorptivity. During the adsorption measurement, and especially at and immediately after the time point when the 3-way valves are switched from the by-pass to the test tube position, there is a dynamic interaction between the TT and the rest of the vacuum system including the RDC. Immediately after ammonia begins to flow through the TT, there are less NH3 molecules reaching the RDC due to the gas exchange and adsorption. This means that the adsorbed NH3 molecules in the saturated surface of the RDC (and elsewhere in the system) begin to desorb (this is the net effect—both adsorption and desorption processes are always present) to the gas stream. When a new equilibrium (within the whole vacuum system now including also the TT) is again reached within tens of seconds at a lower NH3 gas phase concentration, the molecules begin to re-adsorb to all surfaces. As a consequence, a rounding effect in the “adsorption valley” (see Fig. 4) is observed. Fortunately, this interaction does not affect the total measured number of adsorbed molecules because the same number of molecules is re-adsorbed that has initially been desorbed. The interaction within the vacuum system affects only the time behavior of the observed NH3 concentration and consequently its real-time modeling. To damp these effects, and to observe the actual real-time profile of the adsorption process in the TT, we tested an extra by-passing PTFE tube to create a faster total flow rate in the RDC than in the TT (see Fig. 3). Measurements with this setup confirmed that the effect of adsorption inside the RDC is not negligible but the practical limitations of the setup lie elsewhere.
On a few occasions, an identical and apparently fault-free setup and an unchanged measurement protocol produced inconsistent results exceeding the experimental uncertainty. Explanations include variation in the H2O content of the N2 gas used for dilution and potential contamination of the test tubes by a minor leakage during the saturation phase of the experiment. Fully reproducible (10–20 % standard deviation) adsorption measurements would require real-time monitoring and tight control of these parameters, which is impossible with the current measurement setup and protocol. For example, considerably longer flushing times (days), robust and frequent leak-testing and real-time pressure control would be required. The measurement of both the NH3 and H2O concentrations simultaneously in real time could be a partial solution to this problem.
The results of the current study are qualitatively equivalent to the ones obtained in other meticulously performed investigations. In many of them, considerably less complicated measurement setups, straightforward protocols and shorter flushing times were used as compared to our study. In the light of this, it is, in most cases, sufficient to render to such simple procedures. For example, in choosing the right sampling tube material for NH3 or other adsorptive trace gas analysis, it is often enough to connect a selection of potential long (up to tens of meters) sampling tubes to the analyzer, allow the analyzed trace gas to flow via the tested tubing, follow the time evolution of the recorded signal and rank the tubes on the basis of this information. Rather than accentuating the finesse of the adsorption measurement setup and protocol itself, it is more important to ensure that the tests are performed in the same conditions (especially with the same water content) as what will be encountered in the actual trace gas studies such as atmospheric pollution measurement field campaigns.
Despite the agreement between this study and most of the referenced literature, up to a factor of ten differences in the values of adsorptivity have been presented [18, 19]. The competitive adsorption between ammonia and water might explain many of these inconsistencies. Usually no information beyond a statement “dry” or “zero gas” has been given about the H2O concentration in the adsorption studies even though the actual H2O mixing ratio has, in all likelihood, been in the ppmv range. In our opinion, it is meaningless to compare the results of different studies (or results even within a study) without at least a rough estimate of the H2O concentration of the NH3 trace gas used in the adsorption measurement. The interaction between ammonia and water molecules calls for further and more detailed studies of this phenomenon.
References
European Environment Agency: Ammonia (NH 3 ) Emissions (www.eea.europa.eu/data-and-maps/indicators/eea-32-ammonia-nh3-emissions-1/assessment-1)
P. Korhonen, M. Kulmala, A. Laaksonen, Y. Viisanen, R. McGraw, J.H. Seinfeld, J. Geophys. Res. 104, 26349 (1999)
J.D. Whitehead, I.D. Longley, M.W. Gallagher, Water Air Soil Pollut. 183, 317 (2007)
S.B. Shah, P.W. Westerman, J. Arogo, J. Air Waste Manag. Assoc. 56, 945 (2006)
K.W. van der Hoek, Atmos. Environ. 32, 315 (1998)
M.A. Sutton, U. Dragosits, Y.S. Tang, D. Fowler, Atmos. Environ. 34, 855 (2000)
H. Järnstöm, K. Saarela, P. Kalliokoski, A.-L. Pasanen, Indoor Built Environ. 17, 303 (2008)
F.M. Schmidt, O. Vaittinen, M. Metsälä, M. Lehto, C. Forsblom, P.-H. Groop, L. Halonen, J. Breath Res. 7, 017109 (2013)
Directive 2001/81/EC of the European Parliament and of the Council of 23 October 2001 on national atmospheric ceilings for certain atmospheric pollutants (eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri = OJ:L:2001:309:0022:0030:EN:PDF)
B. Timmer, W. Olthuis, A. van der Berg, Sens. Actuators, B 107, 666 (2005)
K. von Bobrutzki, C.F. Braban, D. Famulari, S.K. Jones, T. Blackall, T.E.L. Smith, M. Blom, H. Coe, M. Callagher, M. Ghalaieny, M.R. McGillen, C.J. Percival, J.D. Whitehead, R. Ellis, J. Murphy, A. Mohacsi, A. Pogany, H. Junninen, S. Rantanen, M.A. Sutton, E. Nemitz, Atmos. Meas. Tech. 3, 91 (2010)
J.J. Schwab, Y. Li, M.-S. Bae, K.L. Demerjian, J. Hou, X. Zhou, B. Jensen, S.C. Pryor, Environ. Sci. Technol. 41, 8412 (2007)
J. Henningsen, N. Melander, Appl. Opt. 36, 7037 (1997)
N. Melander, J. Henningsen, in Photoacoustic and Photothermal Phenomena: 10th International Conference CP463, eds. by F. Scudieri and M. Bertolotti (American Institute of Physics, New York, 1999) pp 78–80
A. Schmohl, A. Miklos, P. Hess, Appl. Opt. 40, 2571 (2001)
R.A. Ellis, J.G. Murphy, E. Pattey, R. von Haarlem, J.M. O’Brien, S.C. Herndon, Atmos. Meas. Tech. 3, 397 (2010)
J.D. Whitehead, M. Twigg, D. Famulari, E. Nemitz, M.A. Sutton, M.W. Gallagher, D. Fowler, Environ. Sci. Technol. 42, 2041 (2008)
S.B. Shah, G.L. Grabow, P.W. Westerman, Appl. Eng. Agric. 22, 919 (2006)
S. Mukhtar, A.J. Rose, S.C. Capareda, C.N. Boriack, R.E. Lacey, B.W. Shaw, C.B. Parnell Jr, Agric. Eng. Int. CIGR J. 5 (2003)
D. Romanini, A.A. Kachanov, N. Sadeghi, F. Stoeckel, Chem. Phys. Lett. 264, 316 (1997)
G. Berden, R. Engeln (eds.), Cavity Ring-Down Spectroscopy: Techniques and Applications (Wiley-Blackwell, Chippenham, 2009)
F.M. Schmidt, O. Vaittinen, M. Metsälä, P. Kraus, L. Halonen, Appl. Phys. B 101, 671 (2010)
M.E. Webber, D.S. Baer, R.K. Hanson, Appl. Opt. 40, 2031 (2001)
The Fityk software (www.unipress.waw.pl/fityk)
P. Macko, D. Romanini, S.N. Mikhailenko, O.V. Naumenko, S. Kassi, A. Jenouvrier, V.G. Tyuterev, A. Campargue, J. Mol. Spectrosc. 227, 90 (2004)
SilcoTek Corporation (http://www.silcotek.com/)
U.S. Patent No.: 6,444,326 B1 (2002)
C.M. Hansen, L. Just, Prog. Org. Coat. 42, 167 (2001)
I. Langmuir, J. Am. Chem. Soc. 40, 1361 (1918)
H. Freundlich, Kapillarchemie (Academische Bibliotek, Leipzig, 1909)
Acknowledgments
This work is part of European Metrology Research Programme (EMRP) Joint Research Project (JRP) “Metrology for Chemical Pollutants in Air”—MACPoll (www.macpoll.eu). O. V. operated as a Research Excellency Grant (REG) beneficiary in the Project. The research was carried out with funding by EURAMET and the European Union. The EMRP is jointly funded by the EMRP participating countries within EURAMET and the European Union. Academy of Finland is thanked for financial support. SilcoTek Corporation is thanked for providing detailed information about their coatings and for offering the test tube coatings free of charge.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Vaittinen, O., Metsälä, M., Persijn, S. et al. Adsorption of ammonia on treated stainless steel and polymer surfaces. Appl. Phys. B 115, 185–196 (2014). https://doi.org/10.1007/s00340-013-5590-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00340-013-5590-3