
Abstract
Laser-induced breakdown spectroscopy (LIBS) has proven to be extremely versatile, providing multielement analysis in real time without sample preparation. The principle is based on the ablation of a small amount of target material by interaction of a strong laser beam with a solid target. The laser must have sufficient energy to excite atoms and to ionize them to produce plasma. We aimed to improve the LIBS limit of detection (LOD) and the precision of spectral lines emitted from the produced plasma by optimizing the parameters affecting the LIBS technique. LIBS LOD is affected by many experimental parameters such as interferences, self-absorption, spectral overlap, signal-to-noise ratio, and matrix effects. The plasma in the present study is generated by focusing a 6-ns pulsed Nd–YAG laser at the fundamental wavelength of 1,064 nm onto the Al target in air at atmospheric pressure. The emission spectra are recorded using an SE 200 Echelle spectrometer manufactured by the Catalina Corporation; it is equipped with an ICCD camera type Andor model iStar DH734-18. This spectrometer allows time-resolved spectral acquisition over the whole UV-NIR (200–1,000 nm) spectral range. Calibration curves for Cu, Mg, Mn, Si, Cr, and Fe were obtained with linear regression coefficients around 99 % on the average in aluminum standard alloy samples. The determined LOD has very useful improvements for Cu I at 521.85 nm, Si I at 288.15 nm, Mn I at 482.34 nm, and Cr I at 520.84 nm spectral lines. LOD is improved by 83.8 % for Cu, 49 % for Si, 84.3 % for Mn, and 45 % for Cr lower with respect to the previous works.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Laser-induced breakdown spectroscopy (LIBS) is becoming a dominant technology for direct solid sampling in analytical chemistry. LIBS refers to the process in which an intense burst of energy delivered by a short laser pulse is used to sample (remove a portion of) a material. The advantages of laser-induced breakdown spectroscopy in elemental analysis include direct characterization of solids, no chemical procedures for dissolution, reduced risk of contamination or sample loss, analysis of very small samples not separable for solution analysis, and determination of spatial distributions of elemental composition [1–12]. LIBS can be regarded as a universal sampling, atomization, excitation, and ionization source since laser-induced plasmas can be produced in gases [1] or aerosols [1–3] or liquids [4–8], as well as from conducting or non-conducting solid samples [9–12]. Moreover, LIBS offers online measurements in industries with the compact new technologies of spectrometers with ICCD cameras [1, 8, 13–17]. In the strictest sense, LIBS cannot be considered a non-destructive technique since part of the target to be analyzed is vaporized and lost. However, the volumes sampled in this manner are very small. It ranges from 10−8 to 10−5 cm3 depending on the material’s base, the laser wavelength, and the fluence that corresponds to the masses in the ng–μg range. Moreover, sample vaporization and excitation is possible in a single step, i.e., there is no need for particles to be transferred to an external source [18].
LIBS measurements consist of spectral and time-resolved analysis of the atomic and ionic emission lines generated at the surface of a sample by a focused and intense laser pulse [19–21]. Since the early application of LIBS for diagnostic purposes, several systems have been developed in the laboratory and for portable units [22–24]. In the LIBS technique, the very high field intensity instantaneously evaporates a thin surface layer and initiates an avalanche ionization of the sample elements, giving rise to the so-called breakdown effect. LIBS spectra can be detected once the plasma continuum emission is almost extinguished. Time-resolved capability is necessary to discriminate the late atomic line emission from the early plasma continuum. High-resolution spectral analysis is required to detect single-emission lines, i.e., the spectral signatures of each element. The atomic and in some cases ionic lines, once assigned to specific transitions, allow for a qualitative identification of the species present in plasma. Their relative intensities can be used for the quantitative determination of the corresponding elements. The method can be certified for analytical applications of material analysis in industry, assuming that the surface composition is maintained in the plasma and that the ablation process can be modeled in an appropriate temporal window with quasi-equilibrium conditions. Other authors [23, 25] have demonstrated that several different experimental parameters (e.g., laser power and repetition rate, interaction geometry, surface conditions) may affect the effective analytical possibilities of the method, especially if a field application is foreseen that could make a profitable use of its main advantage of no sample pretreatment needed.
The main parameters affecting the performance of LIBS results are laser intensity, excitation wavelength, laser pulse duration, physical and chemical characteristics of the target material, and its surface conditions, especially its surrounding atmosphere [8]. Although many studies have been done investigating the parameters that influence the plasma characteristics, there is relatively less knowledge about the influence of laser pulse duration on LIBS calibration curves [26, 27]. On the other hand, for accurate quantitative analysis, self-absorption, spectral overlap (spectral line interference and band interference), and matrix effect (chemical interferences) have to be avoided in LIBS [28]. The physical and chemical properties of the sample can affect the plasma composition, a phenomenon known as the matrix effect. The matrix effect can result in the sample being ablated differently from the target sample. Several researches studied the matrix effect under different experimental conditions to specify causes and to find various methods for correction [9, 10, 27–30].
We aimed to improve LIBS LOD and the precision of spectral lines emitted from the produced plasma by optimizing the mentioned parameters that affect LIBS technique. In the present work, spectra from several laser shots are averaged in order to reduce the statistical error due to laser shot-to-shot fluctuation, and measurements were taken at three locations on the sample surface in order to avoid problems linked to sample heterogeneity; this improved the LIBS precision.
2 Experimental setup
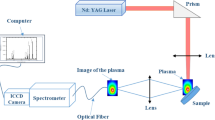
Figure 1 presents the schematic diagram of the experimental setup. A Q-switched Nd–YAG laser type Brilliant B from Quantel was used at the fundamental wavelength of 1,064 nm. The plasma is generated by focusing the laser pulse of 6-ns duration onto the target in air at atmospheric pressure. Three laser energies are used, 440 mJ, 220 mJ, and 23 mJ, measured at the target surface. These correspond to power densities (irradiances) of 4.58 × 1011, 2.29 × 1011, and 2.35 × 1010 W/cm2, respectively, for a laser focal spot diameter of about 144 μm. A time-resolved diagnostic technique was used to investigate the emission spectra from the produced plasma.

Schematic diagram of experimental setup
The plasma was imaged 1:1 by a quartz lens (f = 20 cm) onto the entrance port of an SE 200 Echelle spectrograph produced by Catalina corporation equipped with on ICCD type Andor model iStar DH734-18F. The gain of the camera was fixed at a value of 250 with a binning mode at 1 × 1. This spectrometer allows for a time-resolved spectral acquisition over the whole UV-NIR range (200–1,000 nm). The whole operation of the system and data acquisition was fully controlled by a PC program (Kestrel Spec. Version 3.96).
Six certified reference aluminum-based alloy samples of known elemental composition were used. The surface of the Al samples was polished to a mirror-like state and mounted on a rotating sample stage inside the interaction chamber, which was rotated to provide a fresh surface. However, the experimental data were collected from three different locations on the sample surface; this allows us to present an average and a standard deviation of the results (Table 1 ).
For enhancements and to improve LIBS signal, an accumulation of consecutive measured spectra (10, 20, 50, and 100) will be investigated. In the present work, one of Al alloy references (Al316) is chosen for optimizing the above-mentioned parameters such as self-absorption and signal-to-noise ratio (SNR). Figure 2 presents samples of spectral lines for different numbers of accumulated spectra as consecutively recorded on the ICCD.

Samples of emission lines of Cu I, Mg I, Mn I, and Si I obtained for various accumulated spectra on the ICCD
3 Results and discussions
In the case of high plasma density, the plasma itself absorbs its own emission. This is mainly true for resonance lines connected to the ground state, but other lines may also be affected. The absorption causes a distortion in the spectral line profile, resulting in a broadened line. This effect is known as self-absorption [31]. During the LIBS experiments, the plasma temperature tends to drop toward the outer parts of the plasma plume, and when light passes through these colder parts, an effect known as self-reversal can occur. Such lines show a (severe) dip at the center of the ordinarily well-behaved line-shape function faking two lines.
For extracting quantitative data and evaluating the plasma parameters from the line intensities, it is important to ensure that the plasma is not optically thick for the lines being used. The ratio of emission intensities of resonant and non-resonant lines should be verified according to a procedure for the “optically thin” limit described by Cremers and Radziemski [32], Simeonsson and Miziolek [33], and Sabsabi and Cielo [18].
Normally, during the measurement of major elements in composite solid samples, or in pure samples, self-absorption is observed for many lines during few microseconds after the plasma formation. This phenomenon has less effect in the case of liquid and gaseous samples in comparison with solid, but it is observable at higher analytic concentration or at higher laser intensity. A multiplet is very useful in the assessment of self-absorption [34], since the intensity ratio of its components is well known for the free atoms and ions: Any deviation will indicate the magnitude of self-absorption. A convenient possibility to analyze quantitatively the total self-absorption of a line is offered by the measurement of a second line from the same upper level. If, in addition, the absorption oscillator strength is also much weaker, this transition will hardly be influenced by absorption. A comparison of the measured intensity ratio of both lines with their optically thin limit is required [35].
Doublet lines of Cu I (324.72/327.39), Mn I (478.34/475.4), and Mg I (518.36/517.26) emitted from the same higher-excitation state had been tested at different numbers of accumulated consecutively measured spectra on ICCD (10, 20, 50, 100) corresponding to a number of fired laser shots. The measured intensity ratio was found within the optical thin limit with acceptable error bars for different delay times. The error bars of the intensity ratio are reduced by increasing the number of laser shots, and the best is achieved at accumulation of 50 laser pulses as seen in Fig. 3.

Measured intensity ratio of Cu I, Mn I, and Mg I spectral lines versus the delay time for 50 laser pulses accumulated on the ICCD as an example; solid line is the calculated optically thin limit
However, testing the self-absorption of the resonance transition (2p63 s2 S-3s3p Po) of Mg I at 285.21 nm and the resonance transitions (2p63 s 2S-2p63p 2Po) of Mg II at 279.55 and 280.27 nm in the UV region of the emission spectrum reveals that these lines are strongly influenced by self-absorption for the present concentration (1.16 %) of Mg in the present test sample.
3.1 Study of signal-to-noise ratio (SNR)
For analytical applications, the transient nature of the laser-induced plasmas requires the observation of a plasma in which the continuum is negligible with respect to the line emission (high signal-to-noise ratio), but the line intensity is still strong enough. Consequently, the signal-to-noise ratio (SNR) at different delay times and at a constant ICCD gating at of 2 μs is used.
The SNR for Cu I (324.72, 327.39, 510.55, 515.23, and 521.82 nm), Mg I (516.73, 517.26, and 518.36 nm), Mn I (475.40, 478.35, and 482.34 nm), and Si I (288.15 nm) was studied at different delay times and at different numbers (10, 20, 50, and 100) of accumulated consecutively measured spectra on ICCD. The maximum SNR was found at t d = 4 μs and for 50 accumulated spectra as shown in Figs. 4, 5, 6, and 7.

SNR of Cu I spectral lines (324.72 and 327.39 nm) versus the delay times for a 10, b 20, c 50, and d 100 of accumulated consecutively measured spectrum on ICCD

SNR of Mg I spectral lines (516.73, 517.26, and 518.36 nm) versus the delay times for a 10, b 20, c 50, and d 100 of accumulated consecutively measured spectra on ICCD

SNR of Mn I spectral lines (482.34, 478.35, and 475.40 nm) versus the delay time for a 10, b 20, c 50, and d 100 of accumulated consecutively measured spectra on ICCD

SNR of Si I spectral line (288.15 nm) versus the delay times for a 10, b 20, c 50, and d 100 of accumulated consecutively measured spectrum on ICCD
Compromising the above results, we found that 50 accumulated consecutively measured spectra on ICCD reveal a high SNR and play a role in reducing the effectiveness of self-absorption of different spectral lines tested in the previous sections for the elemental composition of the test sample. Table 2 summarizes these optimized parameters; it also presents the operating parameters at different incident laser energy on the target.
3.2 Analytical calibration function and figures of merit
The quantitative spectral analysis relates the spectral line intensity of an element in the plasma with the concentration of that element in the sample. The emitted spectrum, at early times following plasma formation, is dominated by an intense radiation continuum and strongly broadened ionic emissions. At subsequent times, emissions from neutral atoms become dominant. However, the optimum time delay depends on the selected atomic species (i.e., transition probability) and the energy of the upper level of the analytical line of the element of interest used.
Based on the optimized operating condition, the calibration curves of copper, magnesium, manganese, silicon chromium, and iron have been obtained using six standard reference aluminum alloy samples of known element concentration. The spectral lines that have been used to construct the calibration curves are listed in Table 3.
The graph between the concentrations of elements and the intensity of the spectral line of a certain wavelength is known as the analytical calibration curve. Both the terms “intensity” and “concentration” can be defined in different ways, which often reflect the actual experimental conditions. Several authors define the line intensity as the area of the analytic function used to fit the line profile (usually a Lorentzian or Voigt function). In this case, the background is automatically subtracted. Other researchers take the line intensity to be the peak height, i.e., the maximum of the line profile minus the baseline, which is evaluated in a spectral region free from significant line emission. Peak height of the spectral lines is used to present the intensity of spectral lines, and concentrations are usually expressed as the percent of the mass of the analyte per unit mass of the sample, i.e., (w/w).
The error bars shown in Figs. 4, 5, 6, and 7 represent the standard deviation of three measurements taken for each sample. The typical relative standard deviations obtained vary from 2 to 20 % of the line peak. These values are comparable to those usually reported in LIBS analysis using ns laser pulses [36] while the relative standard deviation of the background varies from 1 to 5 %.
Figure 8 presents the analytical calibration curves for Cu I, Mg I, Mn I, and Si I, for the fundamental laser at 1,064 nm for different laser energies.

Examples of calibration curves of Cu I, Mg I, Mn I, and Si I for different laser energies
3.3 Precision and detection limit
The ultimate goal of an analytical technique is to determine the concentration of a certain substance or element in the sample under consideration. The relation between the signal “x” and the concentration “c” is expressed by the analytical calibration function c = f(x). In optical emission spectroscopy, the measured signal is the intensity of a spectral line I. The available analytical calibration function originates from a well-known empirical equation after Scheibe and Lomakin, that is, I = S Cm, where S is the sensitivity and m is a parameter characterizing the linearity of the calibration curve. In an ideal case, m = 1 and the concentration over which the relation is linear is called the dynamic range. In this case, S will be the slope of linear plot between the intensity and concentration.
3.3.1 The dynamic range
This denotes the concentration interval over which a calibration plot is linear. A large dynamic range is convenient because it permits simultaneous multielement determination with one spectral line per element and with a single sample solution without requiring different dilution for different elements. Possible reasons for departure from linearity of the calibration function are self-absorption, erroneous background correction, and nonlinear response of the detector. As it is well known, the limit of detection (C L) is the concentration corresponding to the smallest line intensity that can be distinguished with a given level of confidence from the random fluctuation of the background. In practice, the magnitude of the fluctuation can be found numerically by taking a sufficiently large number of measurements of the background IB. In order to determine the limit of detection c L, the smallest detectable signal I L is equalized to z times the standard deviation of the background intensity σ (I B), where z is a number.
Accepted values of z are 3 or 3√2.
From the analytical calibration curve, when the relation between the concentration and the net line intensity can be taken to be a straight line that holds especially at low concentrations, we obtain the limit of detection c L as follows:
3.3.2 Sensitivity
It is often confused with the detection limit. The detection limit is the smallest amount of analyte that can be determined with confidence. The detection limit, therefore, is a statistical parameter, and sensitivity is the change in signal per unit change in the amount of analyte. The sensitivity S is defined as the slope of the calibration curve, i.e.,
Thus, the limit of detection will be given by:
The limit of detection thus defined is useful for comparison between different instruments or between differing operating conditions on a single instrument. However, these limits of detection are unrealistically low to most practical analyses and should be qualified as instrumental detection limits. For a given spectral line and under optimized conditions, the detectability can be increased by the increase in the sensitivity.
3.3.3 The precision term
Precision represents the random uncertainty in the value of the net line intensity or the corresponding uncertainty in the estimate of the concentration. It is defined as the relative standard deviation (RSD) of the signal counts.
Table 4 shows the detection limits and the precision obtained in the present study at different laser pulse energy compared with the other results by different authors [10, 18, 36–43]. Improvements of the determined LOD for the fundamental wavelength in the present work are obtained for Cu I at 521.85 nm, Si I at 288.15 nm, Mn I at 482.34 nm, and Cr I at 520.84 nm spectral lines and are shown in Table 5 in comparison with other works [37, 40, 42]. The LOD is 83.8 % for Cu I line at 521.85 nm, 49 % for Si I at line 288.15 nm, 84.3 % for Mn I line at 482.34 nm, and 45 % for Cr I at line 520.84 nm lower than the previously reported works.
In order to check the accuracy of the present technique and its suitability, different samples from the Aluminum Company in Egypt of known elemental compositions as a reference sample were tested; the determined concentrations have acceptable values with respect to the certified ones.
4 Conclusion
Successful results are achieved in improving the limit of detection (LOD) and the precision (RSD) of different spectral lines emitted from plasma generated by an incident nanosecond Nd–YAG laser at its fundamental wavelength under optimized operating parameters as discussed above. To reach this important goal, different important parameters were optimized. These are the number of accumulation of consecutively measured spectra on the ICCD and the optimum delay time that corresponds to the high signal-to-noise ratio (SNR), for spectral lines free of self-absorption. These optimized parameters for different laser energy on the target are also obtained. The selected accumulation number of consecutively measured spectra is 50 on the ICCD; it gives a higher SNR, and it plays a role in the reduction in the self-absorption of most of lines.
The calibration curves of the studied elements (Cu, Mg, Si, Cr, Fe, and Mn) were constructed in the aluminum matrix. They pass nearly through the origin and have good linear fitting (R 2 > 0.95) within the experimental uncertainty. In fact, this result gives the expectation that the proposed LIBS technique has the capability for a good linearity of the calibration curves attaining a wide dynamic range of elemental concentrations.
The study on the limit of detection (LOD) of the spectral lines of different elemental composition emitted from the plasma produced in the present study is performed. It can be seen that the detection limits are a function of the element studied. This observation is due to many factors: (1) the intensity of the analytical line, which is related to the transition probability; (2) the upper energy of the emitting analytical line (in fact, with plasma sources in LTE, it is difficult to populate the higher-level energy); and (3) the spectral region of the analytical line, which is related to the detector sensitivity. The present study reveals the following:
-
For the Cu I spectral lines at 324.72 and 327.39 nm, these transitions are resonant with the ground state (4s2S-4p2P), and intense self-absorption is not negligible even at the copper concentration of 0.5 % for Cu I at 324.72 nm; the calibration curve exhibits a curvature while it is still linear up to 1 % for Cu I at 327.39 nm. While the higher transitions (4p2P-4d2D) and (4 s22D-4p2P) of Cu I at 521.81, 515.32, and 510.55, respectively, are a transition of lower transition probabilities and are hence hardly affected by self-absorption, they show a linear calibration curve up to 2 %.
-
Calibration curves for the Mg I at 285.2-nm line are approximately straight lines up to 1 % and exhibit a curvature for higher concentrations. The leveling of the curve indicates a decrease in the sensitivity of this technique at high concentration of magnesium. This observation is due to the self-absorption of Mg I at 285.2-nm resonance line. At higher concentration, self-absorption is no longer negligible for resonance lines. However, we can reduce self-absorption using a transition that terminates on a higher energy level. Higher transitions of Mg I spectral lines (3s3p-3s4s) are in the visible region of the spectrum at 517.26 and 518.36 nm: These lines are suitable for higher concentrations of Mg. Both magnesium lines have relatively high excitation energies (5.12 eV), and while the Mg I at 285.2-nm line is a resonance line, the 518.2 and 517.3-nm lines are due to transitions that end at 2.72 and 2.71 eV, respectively, above the ground state. The calibration curves are linear, and they pass, within the experimental uncertainty, through the origin. In conclusion, resonance lines may be used when the concentration of the analyte is low on the sample.
-
The determined LOD for the fundamental wavelength in the present work has very useful improvements for CuI at 521.85-nm, SiI at 288.15-nm, MnI at 482.34-nm, and CrI at 520.84-nm spectral lines with respect to other works performed in other laboratories. The LOD is 83.8 % for Cu I at 521.85 nm, 49.08 % for Si I at 288.15 nm, 84.3 % for Mn I at 482.34 nm, and 45 % for Cr I at 520.84 nm lower with respect to the previous works.
References
L.J. Radziemski, “Review of selected analytical applications of laser plasmas and laser ablation”, 1987–1994. Microchem. J. 50, 218–234 (1994)
L.J. Radziemski, T.R. Loree, D.A. Cremers, N.M. Hoffman, Anal. Chem. 55, 1246 (1983)
D.W. Hahn, W.L. Flower, K.R. Hencken, Appl. Spectrosc. 51, 1836–1844 (1997)
W.T.Y. Mohamed, Quantitative elemental analysis of seawater by laser induced breakdown spectroscopy. Int. J. Pure Appl. Phys. 2(1), 11–21 (2006)
L.J. Radziemski, D.A. Cremers, Laser-Induced Plasmas and Applications (Marcel Dekker, Inc., New York, 1989)
K. Song, Y.-I. Lee, J. Sneddon, Recent developments in instrumentation for laser-induced breakdown spectroscopy. Appl. Spectrosc. Rev. 37, 89–117 (2002)
E. Tognoni, V. Palleschi, M. Corsi, G. Cristoforetti, Quantitative microanalysis by laser-induced breakdown spectroscopy: a review of the experimental approaches. Spectrochim. Acta Part B 57, 1115–1130 (2002)
D.A. Cremers, The Analysis of Metals at a Distance Using Laser-Induced Breakdown Spectroscopy. Appl. Spectrosc. 41(4), 572 (1987)
M. Sabsabi, V. Detalle, M.A. Harith, W. Tawfik, H. Imam, Comparative study of two new commercial Echelle spectrometers equipped with intensified CCD for analysis of laser-induced breakdown spectroscopy. Appl. Opt. 42(30), 6094–6098 (2003)
M.A. Ismail, H. Imam, A. Elhassan, W.T. Youniss, M.A. Harith, LIBS limit of detection and plasma parameters of some elements in two different metallic matrices. J. Anal. At. Spectrom. 19, 489–494 (2004)
P. Fichet, A. Toussaint, J.F. Wagner, Laser-induced breakdown spectroscopy: a tool for analysis of different types of liquids. Appl. Phys. A 69, 591–592 (1999)
P. Fichet, P. Mauchien, J.F. Wagner, C. Moulin, Quantitative elemental determination in water and oil by laser induced breakdown spectroscopy. Anal. Chim. Acta 42(9), 269–278 (2001)
D.A. Rusak, B.C. Castle, B.W. Smith, J.D. Winefordner, Fundamentals and applications of laser-induced breakdown spectroscopy. Crit. Rev. Anal. Chem. 27, 257–290 (1997)
J. Sneddon, Y.-I. Lee, Novel and recent applications of elemental determination by laser-induced breakdown spectrometry. Anal. Lett. 32, 2143–2162 (1999)
V. Majidi, M.R. Joseph, Spectroscopic applications of laser-induced plasmas. Crit. Rev. Anal. Chem. 23, 143–162 (1992)
A. Ciucci, V. Palleschi, S. Rastelli, R. Barbini, F. Colao, R. Fantoni, Trace pollutants analyses in soil by a time-resolved laser induced breakdown spectroscopy technique. Appl. Phys. B 63, 185–190 (1996)
A. Ciucci, M. Corsi, V. Palleschi, S. Rastelli, A. Salvetti, E. Tognoni, A new procedure for quantitative elemental analyses by laser induced plasma spectroscopy. Appl. Spectrosc. 53, 960–964 (1999)
M. Sabsabi, P. Cielo, Appl. Spectrosc. 49(4), 499–507 (1995)
E.H. Piepmeier, Laser Ablation for Atomic Spectroscopy, Analytical Application of Laser (Wiley, New York, 1986)
H.E. Bauer, F. Leis, K. Niemax, Laser induced breakdown spectrometry with an echelle spectrometer and intensified charge coupled device detection. Spectrochim. Acta Part B 53, 1815–1825 (1998)
R.E. Russo, X.L. Mao, Chemical analysis by laser ablation, in Laser Experimental Ablation and Desorption, ed. by J.C. Miller, R.F. Haglund (Academic Press, San Diego, 1998), p. 375
K.Y. Yamamoto, D.A. Cremers, M.J. Ferris, L.E. Foster, Detection of metals in the environment using a portable laser-induced breakdown spectroscopy instrument. Appl. Spectrosc. 50, 222–233 (1996)
B.C. Castle, K. Talabardon, B.W. Smith, J.D. Winefordner, Variables influencing the precision of laser-induced breakdown spectroscopy measurements. Appl. Spectrosc. 52, 649–657 (1998)
Y.F. Yueh, J.P. Singh, H. Zhang, Laser-induced Breakdown Spectroscopy: Elemental Analysis, in Encyclopedia of Analytical Chemistry, ed. by R.A. Meyers (Wiley, Chichester, 2000), pp. 2066–2087
V. Bulatov, R. Krasniker, I. Schechter, Study of matrix effects in laser plasma spectroscopy by combined multi-fiber spatial and temporal resolutions. Anal. Chem. 70, 5302–5310 (1998)
K.L. Eland, D.N. Stratis, D.M. Gold, S.R. Goode, S. Michael Angel, Energy dependence of emission intensity and temperature in a LIBS plasma using femtosecond excitation. Appl. Spectrosc. 55, 286–291 (2001)
A. Semerok, C. Chale´ard, V. Detalle, J.L. Lacour, P. Mauchien, P. Meynadier, C. Nouvellon, B. Salle´, P. Palianov, M. Perdrix, G. Petite, Experimental investigations of laser ablation efficiency of pure metals with femto, pico and nanosecond pulses. Appl. Surf. Sci. 138, 311–314 (1999)
L. Xu, V. Bulatov, V. Gridin, I. Schechter, Absolute analysis of particulate materials by laser-induced breakdown spectroscopy. Anal. Chem. 69, 2103–2108 (1997)
S.R. Goode, S.L. Morgan, R. Hoskins, A. Oxsher, Identifying alloys by laser-induced breakdown spectroscopy with a time-resolved high resolution echelle spectrometer. J. Anal. At. Spectrom. 15, 1133–1138 (2000)
A.S. Eppler, D.A. Cremers, D.D. Hickmott, M.J. Ferris, A.C. Koskelo, Matrix effects in the detection of Pb and Ba in soils using laser-induced breakdown spectroscopy. Appl. Spectrosc. 50, 1175–1181 (1996)
V.N. Raia, S.N. Thakurb, Physics of plasma in laser-induced breakdown spectroscopy, in Laser-Induced Breakdown Spectroscopy, ed. by J.P. Singh, S.N. Thakur (Elsevier press, Amsterdam, 2007)
D.A. Cremers, L.J. Radziemski, Anal. Chem. 55, 1252 (1983)
J.B. Simeonsson, A.W. Miziolek, Appl. Opt. 32, 939 (1993)
H.J. Kunze, Spectroscopy of Optically Thick Plasmas, 3rd Workshop on Plasma and Laser Technology, Ismailia Oct, 31, 3–7 (1993), ed. by Ph. Mertens (Forschungszentrum Juelich, GmbH, 1994) pp. 31–46
H. Hegazy, Oxygen spectral lines for diagnostics of atmospheric laser induced plasmas. Appl. Phys. B 98, 601–606 (2010)
B. Le Drogoff, J. Margot, T.W. Johnston, M. Chaker, S. Laville, M. Sabsabi, F. Vidal, O. Barthelemy, Y. von Kaenel, Spectrochim. Acta Part B 56, 987–1002 (2001)
W.T.Y. Mohamed, Opt. Laser Technol. 40, 30–38 (2008)
H-K. Li, M. Liu, Z-J. Chen, R-H. Li, Trans. Nonferrous Met. Soc. China 18, 222–226 (2008)
V. Detalle, R. Heon, M. Sabsabi, L. St-Onge, Spectrochim. Acta Part B At. Spectrosc. 56, 1011–1025 (2001)
B. Nemet, L. Kozma, Spectrochim. Acta Part B 50, 1869–1888 (1995)
D. Body, B.L. Chawick, Spectrochim. Acta Part B 56, 725–736 (2006)
M.A. Ismail, S. Legnaioli, V. Palleschi, E. Tognoni, G. Cristofretti, L. Pardini, A. Salvetti, M.A. Harith, J. Anal. Bioanal Chem. 385, 316–325 (2006)
M. Sabsabi, R. Heon, L. St-Onge, Spectrochim. Acta Part B 60, 1211–1216 (2005)
Acknowledgment
Authors deeply thanks Prof. H.-J. Kunze, EPV, Ruhr Universität Bochum, Germany whose advices, revising and discussions was invaluable and sincere thanks to Dr. Sintayehu Woldemariam Physics Department, Faculty of Science, Jazan University, Saudi Arabia for revising this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hegazy, H., Abdel-Wahab, E.A., Abdel-Rahim, F.M. et al. Laser-induced breakdown spectroscopy: technique, new features, and detection limits of trace elements in Al base alloy. Appl. Phys. B 115, 173–183 (2014). https://doi.org/10.1007/s00340-013-5589-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00340-013-5589-9