
Abstract
CO2 stable carbon isotopes are very attractive in environmental research to investigate both natural and anthropogenic carbon sources. Laser-based CO2 carbon isotope analysis provides continuous measurement at high temporal resolution and is a promising alternative to isotope ratio mass spectrometry (IRMS). We performed a thorough assessment of a commercially available CO2 Carbon Isotope Analyser (CCIA DLT-100, Los Gatos Research) that allows in situ measurement of δ 13C in CO2. Using a set of reference gases of known CO2 concentration and carbon isotopic composition, we evaluated the precision, long-term stability, temperature sensitivity and concentration dependence of the analyser. Despite good precision calculated from Allan variance (5.0 ppm for CO2 concentration, and 0.05 ‰ for δ 13C at 60 s averaging), real performances are altered by two main sources of error: temperature sensitivity and dependence of δ 13C on CO2 concentration. Data processing is required to correct for these errors. Following application of these corrections, we achieve an accuracy of 8.7 ppm for CO2 concentration and 1.3 ‰ for δ 13C, which is worse compared to mass spectrometry performance, but still allowing field applications. With this portable analyser we measured CO2 flux degassed from rock in an underground tunnel. The obtained carbon isotopic composition agrees with IRMS measurement, and can be used to identify the carbon source.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Stable carbon isotopes are a key to study gas exchanges and the carbon cycle in the atmosphere as well as in the geosphere [1, 2]. CO2 carbon isotope analysis is usually performed by Dual-Inlet Isotope Ratio Mass Spectrometry on pure CO2, with a high accuracy of typically 0.01 ‰ (1σ) [3]. However, this technique necessitates time-consuming CO2 purification. For more than ten years, He-continuous-flow isotope ratio mass spectrometry (CF-IRMS) has allowed measurement of δ 13C in small amounts of CO2 in an air matrix, with higher sample throughput and a good precision of typically 0.08 ‰ (1σ) [4]. Although both these techniques are very precise, they are based on ion mass spectrometry which necessitates heavy and delicate hardware and operation in highly controlled conditions (very small ion currents <10-10 A and very high vacuum conditions <10-8 mbar). These conditions are very difficult to achieve outside of a well-conditioned laboratory.
Concurrently, laser-based CO2 Carbon Isotope Analysers (CCIAs) have been proposed for several years. These instruments measure the isotope content of CO2 based on infrared absorption spectroscopy. One main advantage of CCIAs is that there is no need to purify CO2 from other air components. Moreover, the air is analysed in continuous flow, not in batch samples, with a high measurement rate up to 1 Hz. These simplified operating conditions led to the development of research instruments for volcanic [5] or atmospheric monitoring [6, 7]. Field-deployable CCIAs have subsequently become commercially available for continuous measurements of CO2 concentration and isotopic composition. These analysers, primarily devoted to atmospheric CO2, have already found interesting applications, for example in soil sciences [1], ecosystem research [8] and carbon sequestration monitoring [9, 10]. In this study, we focus on measurements in geological media where conditions are specific: large variations in CO2 concentrations (from 0 ppm at the beginning of a flux measurement, up to 30000 ppm in soil gas), large range of δ 13C values (from -27 ‰ for plant respiration [11] to -1.5 ‰ for magmatic CO2 [12]), water vapor content up to 100 % RH, and possibly limited sample volume.
As is always the case for very young technologies, only a reduced set of working conditions have been tested until now. CCIAs have been designed to be applied to atmospheric conditions with around 400 ppm of CO2. Little is known about the analytical sensitivity of such instruments in response to non-standard conditions. A detailed review of the system is necessary before it may be used as a reliable and accurate research instrument. Here we assess the performance of one commercially available analyser, the CO2 Carbon Isotope Analyser CCIA DLT-100 (Los Gatos Research) under geological conditions, and present a field application. McAlexander et al. [10] presented impressive performances for this instrument, demonstrating an accuracy of 0.05 ppm for CO2 concentration and 0.2 ‰ for δ 13C (1σ for 60 s), but only under a limited range of CO2 concentrations.
2 Materials and methods
2.1 Commercial CO2 carbon isotope analyser
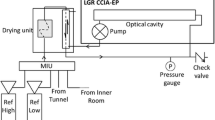
The CCIA is based on Off-Axis Integrated Cavity Output Spectroscopy, a technique described in detail in Baer et al. [13]. The sample gas at atmospheric pressure is drawn through the 0.9 L optical cell [23] of the analyser by an external diaphragm vacuum pump (KNF). This absorption cell is maintained at low pressure (38 torr) and constant temperature (45 ∘C). Sample flow rate can be varied by adjusting the speed of the pump. All measurements were made at maximum speed, corresponding to a sample input flow rate of 0.6 L/min. The wavelength of a diode laser operating in the near infrared region is tuned to scan the absorption features of 12CO2 and 13CO2. Light transmitted through the cell is detected to obtain the absorption spectrum, which is automatically fitted by the analyser and used to determine the mixing ratios of 12CO2 and 13CO2. CO2 concentration and carbon isotope composition, expressed as δ 13C value in ‰ relative to the V-PDB standard, are deduced from both mixing ratios. All measurements were made at 1 Hz, the maximum measurement rate. We used the Multi Inlet Unit (MIU), an optional device available from Los Gatos Research, which allows automatic switching between 16 different sampling ports.
Gas is dried before entering the analyser to prevent water absorption interference [14]. According to the manufacturer, the sensitivity to water vapour is -0.4 ‰ per 1000 ppm H2O for δ 13C value, and we found that it amounts to -0.3 ppm CO2 per 1000 ppm H2O for concentration, and -0.2 ‰ per 1000 ppm H2O for δ 13C value. Although the instrument is now supplied with a sample drying system, this was not the case at the time of purchase; therefore, we used an alternative drying system composed of two Nafion membranes (Permapure ME and Permapure MD). Using this system, water vapour was reduced to around 2000 ppm. Reference gases (see below) were also measured using this drying system. As they were initially dry, they got humidified, and finally also contained around 2000 ppm water vapour. All measured gases, references and samples, thus retained the same concentration of water vapour; therefore, we didn’t take into account its effect in the following analyses.
2.2 Reference gases
Seven commercial reference gas cylinders, lettered A to G, of CO2 mixture in synthetic air (N2+20 % O2) were used in this study. They span a large range of CO2 concentrations (392 to 17800 ppm) and δ 13C values (-35 to -44 ‰). CO2 concentrations were determined by the manufacturer using gravimetric method, with an uncertainty of 2 %. For cylinders A, B and C, CO2 concentrations were also measured at LSCE (France) by gas chromatography and were calibrated against the international WMO mole fraction scale for carbon dioxide in air [15]. The obtained values are not significantly different from those certified by the manufacturer, albeit far more precise. We measured the δ 13C values of the seven cylinder gases by CF-IRMS (Gas Bench II and DeltaPlusXP, Thermo Finnigan). Table 1 shows the values which are taken as references for calibration and estimation of accuracy.
2.3 Internal and external calibration
Because of instrumental drift and sensibility to changing environmental parameters, frequent calibration is necessary to ensure accurate measurements. We distinguish between internal and external calibration. Internal calibration is performed directly within the analyser. A single reference gas cylinder is sampled and the reference CO2 concentration and δ 13C value are entered in the software. Before each measurement sequence, we performed internal calibration with cylinder B (Table 1). As a day-by-day quality control of the analyser, this same cylinder was measured just after calibration.
As shown hereafter, measurements have to be corrected for concentration dependence and instrumental drift. This external calibration can be applied from regular measurements of reference gases. The MIU allows alternating samples and reference gases, and thus long-term monitoring without any permanent operator assistance.
2.4 Performance assessment
In the following, we test four properties of the analytical system: precision, concentration dependence, accuracy and long-term stability.
The precision and noise of optical analysers can be determined by estimating the Allan variances of the CO2 concentration and δ 13C value [6, 16, 17]. Allan deviation (square root of the Allan variance) expresses the measurement precision as a function of the averaging time. Reference cylinders D and G were analysed with the CCIA over a 3 hours period and the Allan deviation was calculated for both CO2 concentration and δ 13C value.
The instrumental response to changing CO2 concentration at constant isotopic composition was determined by progressively diluting reference gases D and G with CO2-free air, using two mass flow controllers (SLA-7850, Brooks). The resulting mixtures were analysed in the CCIA.
In order to test the analyser accuracy, all reference gases were analysed for 5 min, switching automatically from one gas to another with the MIU. The first three minutes of each measurement were discarded because of response and stabilization time; the last two minutes were averaged.
The long-term stability of the analyser was estimated by measuring cylinder F for 5 min, every hour, for a period of 72 hours. Ambient air from the lab was sampled the rest of the time.
2.5 Field application in the Roselend Natural Laboratory
Our application of CO2 concentration and isotope ratio monitoring in geological media consists of CO2 flux measurement in an underground research laboratory located in the French Alps [18]. Pili et al. [19] demonstrated that the rock surrounding the tunnel is a net source of CO2 to the atmosphere, due to many geochemical processes linked to weathering. CO2 degassing was therefore expected. The carbon isotopic composition of degassing CO2 was needed to better understand its source and the geochemical processes that could convert it into carbonates and dissolved gases.
The closed accumulation chamber method has classically been used to measure soil CO2 flux and the isotopic composition of the source [12]. We adapted this method to a sub horizontal borehole (2.2 m long, 86 mm diameter) drilled into the tunnel wall. Using an inflatable packer, an accumulation volume was isolated inside the rock. The borehole was first flushed with CO2-free air for 30 to 45 min, in an open-system, until CO2 concentration decreased to less than 50 ppm. CO2 accumulation and δ 13C were then measured in a closed system for 2 hours using the CCIA, whose exhaust was looped back into the borehole to minimize depressurization of the borehole that would modify CO2 transfer from the rock. A rubber septum sampler port was installed on a tee. Using an air-tight syringe, two discrete vials were sampled at the end of the experiment and subsequently analysed by CF-IRMS.
When working in a closed-system, leakage and contamination have to be taken into account, especially when working in a high CO2 environment, such as poorly ventilated underground cavities. In such cavities, CO2 concentration can easily reach 5000 ppm due solely to the presence of operators. To quantify ambient CO2 contamination by leakage in the measurement line, we disconnected it from the borehole, which produced CO2. The line was first flushed with CO2-free air, and then ran in closed-loop. The increase in CO2 concentration was measured with the CCIA.
3 Results
3.1 Performance in the laboratory
Figure 1 shows the Allan deviation as a function of averaging time, for both CO2 concentration and δ 13C. For each averaging time, Allan deviation measures the analysers intrinsic precision. For cylinder G, the Allan plots exhibit the typical V shape obtained for spectroscopic analysers [16, 17]. Precision first increases with the averaging time, down to 2 ppm and 0.04 ‰, the best precision being obtained with 200 s averaging. For longer averaging times, precision decreases because of instrumental drift. For cylinder D with lower CO2 concentration, precision for δ 13C value decreases to only 0.50 ‰ and cannot be improved with longer averaging time. This observation has not been explained, yet. We used an averaging time of 60 s which results in a precision of 5 ppm for CO2 concentration and 0.05 ‰ for δ 13C value (1σ for cylinder G).

Allan plots for CO2 concentration (a) and δ 13C value (b). Black: cylinder D (1920 ppm). Grey: Cylinder G (17800 ppm)
Figure 2 displays the measurements of cylinder B after each internal calibration. Without correction for temperature or concentration dependence, the standard deviation (1σ) of each measurement and the shift between the measured and the reference values directly serve as a check of the analyser state. The standard deviations (1σ) of all measurements are 0.37 ppm for CO2 concentration and 0.19 ‰ for δ 13C value, which is an estimation of the long-term reproducibility of the analyser.

CO2 concentration (a) and δ 13C value (b) measured for cylinder B immediately after internal calibration with this same gas. Each point represents the mean and standard deviation of 120 s of measurement. Measurements obtained in the field are indicated by grey circles. Grey lines are the mean (thick) and standard deviation (dotted) of all measurements. Horizontal black lines are reference values. For δ 13C, the mean and reference values are superimposed
The dependence of δ 13C measurement on CO2 concentration is given as the difference between the CCIA measurement and the reference δ 13C value (Fig. 3). A pronounced non-linearity is observed especially in the range 300–2000 ppm. The curve which we obtained has a shape similar to that obtained by Sturm et al. [20] for δ 2H measured using the Los Gatos Research Water Vapour Isotope Analyser, based on the same physical principle. The two curves obtained from cylinders D (δ 13C=-43.99 ‰) and G (δ 13C=-39.37 ‰) overlap well within their uncertainties in the concentration range 1000–2000 ppm. This shows that this concentration dependence of the measured δ 13C values does not depend on the isotopic composition of the CO2 in the range -44 to -39 ‰. Several determinations of this curve over a one month period reveal that its global shape is retained, but it shifts by up to 1 ‰. For our applications, we worked exclusively in the range 300–2000 ppm. Lacking any physical model for this non-linearity, we chose to fit the experimental curve in this range with a polynomial. According to a χ 2 test, a fifth order polynomial is sufficient. This fit provides a calibration curve (Fig. 3(b)) which is used to correct the sample δ 13C values for non-linearity.

(a) Non-linearity of δ 13C in the range 200–20000 ppm. Data from reference cylinders D (circles) and G (squares) progressively diluted with CO2-free air. (b) Non-linearity of δ 13C in the range 200–2000 ppm (cylinder D only) with a fifth order polynomial fit of the data (black line). This fitting curve is used as a calibration curve to correct for non-linearity
Table 1 summarizes the results of the reference gas measurements. Figure 4 shows the agreement between measured and reference CO2 concentration values. Accuracy, defined as the standard deviation of the residuals, is 8.7 ppm for CO2 concentration in the range 300–2000 ppm.

Linear relationship between CO2 concentrations measured by the CCIA DLT-100 and reference values for all reference cylinders (squares). Inset: residuals defined as [CO2]CCIA-[CO2]ref (triangles)
Figure 5 shows the agreement between measured and reference δ 13C values. In the range 300–2000 ppm, accuracy for raw measurements is 2.7 ‰. After correction for concentration dependence as described above, the accuracy is improved to 1.3 ‰ for δ 13C (1σ of residuals).

Calibration of the δ 13C values. Grey squares are raw measurements. Black squares are corrected for CO2 concentration dependence using the fitted curve in Fig. 3(b)
We explored the system stability by frequently analysing reference gas F. As illustrated in Fig. 6, instead of a regular drift, a diurnal cycle for both CO2 concentration and δ 13C value is observed. This cycle is strongly correlated with a similar diurnal cycle for cell temperature. Temperature in the laboratory also follows a diurnal cycle with an amplitude of up to 4 ∘C. This is certainly the cause of the cell temperature variation. Both CO2 concentration and δ 13C value vary linearly with the cell temperature. The dependence is respectively 16.7 ppm and -1.3 ‰ per 0.1 ∘C in the cell. For long-term monitoring, we thus correct the measurements for variations in cell temperature. All measurements of CO2 concentration and δ 13C value are normalized to a reference temperature, chosen as the mean temperature of the measurements (in this case 46.0 ∘C). The diurnal cycle is thus suppressed (Fig. 6). The obtained stability over 72 h is found to be ±8.0 ppm for CO2 concentration and ±0.52 ‰ for δ 13C value (1σ).

Long-term measurement of cylinder F. Black squares and error bars are respectively the average and standard deviation of 60 s measurements made every hour, for CO2 concentration (a) and δ 13C value (b). Temperature of the cell is plotted as a dotted line (b). The diurnal cycle for both CO2 concentration and δ 13C value is due to temperature variations in the optical cell
3.2 Field application
Contamination tests show a CO2 increase rate of 100 to 150 ppm/1000 s in the measurement line when it is connected as a closed-loop. During the flux measurement the 7 L packed-off borehole is connected to the line, which increases the volume of the line by around ten times. A precise quantification of the contamination rate is beyond the scope of this paper, we, however, estimated it to be ten times lower, i.e. around 10 ppm/1000 s.
As shown, temperature dependence is a major issue. In the tunnel, temperature is stable and constant at 7 ∘C (within 0.2 ∘C), which considerably reduces the potential instrumental drift. As the flux measurement must be performed in a closed system, it is not possible to measure a reference gas with the CCIA without contaminating the borehole. As the time required for flux measurement is less than 4 hours the instrumental drift is considered to be negligible and internal calibration at the beginning of the measurement is sufficient.
Figure 7 shows the result of the CO2 flux measurement in one borehole. Because of the concentration dependence previously determined, δ 13C values of raw data have to be corrected using the calibration curve. In the first 50 min, CO2 concentration increase can be considered linear. The initial CO2 increase rate is 40 ppm/1000 s, only four times higher than the estimated contamination rate. The isotopic composition of CO2 in the ambient air was measured by CF-IRMS and is around -22.6 ‰, quite similar to the composition of the CO2 in the line. The potential effects of contamination can thus be neglected in this case. The CO2 source is supposed to be of constant isotopic composition, this is supported by an almost constant corrected isotopic composition of the carbon source of -23.7±0.5 ‰ (1σ). The late increase of 3 ‰ in the measured δ 13C value after 190 minutes may indicate a variation in the source. One main advantage of the CCIA is the capability to measure such evolution. Two discrete samples taken at the end of the measurement and analysed by CF-IRMS gave a mean δ 13C value of -21.9±0.2 ‰ (1σ), fully consistent with the CCIA result, as shown in Fig. 7(b).

CO2 flux measurement in a packed-off borehole. Evolution of CO2 concentration (a) and δ 13C value (b) measured by the CCIA DLT-100 after initial flush with CO2-free air. Dotted black line (a) is the temperature in the cell, which has here only small amplitude variations. The δ 13C values in grey are raw data and in black are after correction for concentration dependence. The star is the mean δ 13C value of two discrete samples analysed by CF-IRMS
4 Discussion
4.1 Performance and practical use of the analyser
As shown by the computed Allan deviation (Fig. 1), the averaging time must be chosen as a trade-off between precision and temporal drift. An averaging time of 60 s allows a high temporal resolution with a precision of 0.05 ‰ for 17800 ppm, a performance comparable to mass spectrometry. With the same instrument, McAlexander et al. [10] found the same precision of 0.05 ‰ for 60 s averaging time, but in the range 378–944 ppm. We show that precision decreases when CO2 concentration decreases, and we have no explanation for the better performance obtained by McAlexander et al. [10]. All sources of uncertainty and their magnitude are summarized in Table 2. The short-term precision from Allan deviation is often given as the sole performance indication [10], but the main sources of error on the δ 13C value appear to be temperature and concentration dependences. As demonstrated by comparison with CF-IRMS measurements during our field application, and considering these uncertainties, CCIA results can be considered accurate and reliable.
Measurements have to be normalized to international reference scales. These are the WMO scale for CO2 concentration [21], and the VPDB scale for δ 13C value [22]. The δ 13C value can be easily normalized with CF-IRMS using calcite working standards. CO2 concentration is measured by infrared spectroscopy or gas chromatography, and normalized to the WMO scale using standard gases. The WMO scale is not defined for concentrations above 900 ppm [15], decreasing accuracy in this range.
Temperature in the cell appears sensitive to ambient room temperature, and light absorption depends on temperature. The analyser software corrects light absorption for cell temperature fluctuations, but it seems that a complete and accurate correction is not achieved. This temperature sensitivity is of concern for environmental monitoring, where air temperature may easily vary by 20 ∘C between night and daytime. At present, the only method we have to reduce these artefacts is to limit environmental temperature variations as much as possible where the analyser is operating. Los Gatos Research now provides an enhanced version of this analyser, with better thermal insulation and stabilization of the cell temperature [23].
As seen before, it is possible to correct for concentration dependence, by using the fitting curve (Fig. 3(b)). For long-term continuous monitoring, it is necessary to determine this curve frequently. The temporal drift of this curve has not been studied, yet. As we are not currently able to dilute progressively and automatically a reference gas, monitoring cannot run unattended for more than one week.
In order to correct for all of these dependences, the following procedure was adopted:
-
1.
All measurements are normalized to a reference cell temperature using the temperature dependence factors determined above. This reference temperature is chosen as the mean temperature during the measurement of both samples and references.
-
2.
δ 13C values of all measurements are then corrected for concentration dependence with one single fitted curve (determined at the beginning of the measurement campaign).
-
3.
Measured δ 13C values of two reference gases, corrected for temperature and concentration effects, are reported versus the reference δ 13C values in order to define the linear calibration curve (Fig. 5). This isotopic calibration reports δ 13C values in the international VPDB scale.
The corrections needed to improve the analyser accuracy require regular external calibration. Three reference gases must be measured regularly, the first two for the calibration and the third as a quality check to estimate the overall accuracy of the calibration process. We recommend that the concentrations of the chosen reference gases bracket the working range. For our applications, we chose concentrations of 500, 900 and 2000 ppm (cylinders A, C and D). The calibration time interval is chosen as a trade-off between gas consumption and accuracy. A survey of methodologies for two other commercial CCIAs deployed for long-term continuous monitoring reveals contrasting situations: either the CCIA has an intrinsic high stability so that there is almost no need for calibration (Friedrichs et al. [24] for atmospheric and water-equilibrated CO2 monitoring with a Picarro analyser), or a regular calibration is realized with four reference gases analysed for 60 s every 10 min, to ensure high accuracy (Schaeffer et al. [21] with a Campbell Scientific analyser for atmospheric CO2 monitoring). In the latter case, consumption of reference gases is high. In these two cases, no correction for temperature or CO2 concentration dependence is explicitly taken into account. In our set-up, we measure each reference cylinder for 5 min every 3 hours. With a sample flow rate of around 0.6 L/min, the total consumption is 24 L per day. At this consumption rate a typical 200 bar cylinder of 20 L will last 83 days. Larger gas cylinders are too heavy to be handled in our field applications. This means that several reference cylinders must be purchased and fully characterized each year.
4.2 Field application
The CO2 flux \(\varPhi_{\mathrm{CO}_{2}}\) (in g m-2 d-1) is obtained from the initial linear increase of concentration dCO2/dt, expressed as fraction per day, and using (1) from Chiodini et al. [25]:

where P atm is atmospheric pressure (Pa), \(M_{\mathrm{CO}_{2}}\) CO2 molecular mass (44 g mol-1), R the perfect gas constant, T absolute temperature (K), V volume of the packed-off borehole (m3), S area of rock producing CO2 behind the packer (m2). From data presented in Fig. 7 we found a CO2 flux of 0.11 g m-2 d-1 consistent with previous results from Pili et al. [19]. We measured an isotopic composition of -23.7±0.5 ‰ for the CO2 degassed from the rock. At 7 ∘C the fractionation factor for carbon between CO2 and \(\mbox{HCO}_{3}^{-}\) is -9.6 ‰ [26]. Pili et al. [19] measured δ 13C values of -10.9±1.5 ‰ for \(\mbox{HCO}_{3}^{-}\) in drip water. Therefore, the measured CO2 may result from degassing of this reservoir, even if other processes such as fractionation may be involved [27].
5 Conclusion
We assessed the analytical performance of the commercial CO2 Carbon Isotope Analyser DLT-100 from Los Gatos Research. Even if the basic use seems simple, careful review in the laboratory is required to ensure accuracy before any reliable field measurement. Short term Allan precision for 60 s averaging is 5 ppm for CO2 concentration and 0.05 ‰ for δ 13C value (for 17800 ppm CO2 concentration). This parameter largely overestimates the performance of laser-based isotope ratio analysers. Temperature and CO2 concentration dependences are the main sources of error. It is therefore important to carefully determine them, as they may result in shifts of the measured values by several per mill. An appropriate environmental setting for the instrument is required for reliable measurements. In particular we suggest running the analyser in an air-conditioned room, and carefully drying the sample before measurement. Several reference gases must be analysed every few hours. Finally, time consuming data processing must not be underestimated. In the 300–2000 ppm range, the achieved accuracy of 1.3 ‰ for δ 13C is acceptable considering the large variations of carbon isotopic compositions in geological media. The accuracy of this laser-based isotope analyser is still poor compared to mass spectrometry.
Using the CCIA to measure CO2 flux and the isotopic composition of the source gas is very promising, in boreholes, but also for soil respiration. Simple and fast measurements allow studying of the spatial and temporal variability of both fluxes and sources. Until now, monitoring of CO2 isotopic composition in underground cavities required frequent visits for sampling, with poor spatial and temporal resolution [2] and constant risk of human breath contamination. The CCIA DLT-100 and Multi Inlet Unit allow long-term continuous measurement of multiple reservoirs (e.g. atmosphere, tunnel, borehole, etc.), and will certainly help to investigate CO2 sources and migration in geological media, providing the analyser is regularly calibrated and data are carefully corrected for drifts and dependences.
IRMS measurements remain compulsory to determine the δ 13C values of reference gases, as often as new reference cylinders have to be used. They are also recommended during field measurement to punctually verify its accuracy.
References
L. Wingate, J. Ogee, R. Burlett, A. Bosc, Glob. Change Biol. 16, 3048 (2010)
S. Frisia, I.J. Fairchild, J. Fohlmeister, R. Miorandi, C. Spotl, A. Borsato, Geochim. Cosmochim. Acta 75, 380 (2011)
S. Assonov, P. Taylor, C.A.M. Brenninkmeijer, Rapid Commun. Mass Spectrom. 23, 1347 (2009)
K.P. Tu, P.D. Brooks, T.E. Dawson, Rapid Commun. Mass Spectrom. 15, 952 (2001)
A. Castrillo, G. Casa, M. van Burgel, D. Tedesco, L. Gianfrani, Opt. Express 12, 6515 (2004)
L. Croize, D. Mondelain, C. Camy-Peyret, M. Delmotte, M. Schmidt, Rev. Sci. Instrum. 79, 043101 (2008)
B. Tuzson, J. Mohn, M.J. Zeeman, R.A. Werner, W. Eugster, M.S. Zahniser, D.D. Nelson, J.B. McManus, L. Emmenegger, Appl. Phys. B, Lasers Opt. 92, 451 (2008)
D.R. Bowling, S.D. Sargent, B.D. Tanner, J.R. Ehleringer, Agric. For. Meteorol. 118, 1 (2003)
S. Krevor, J.C. Perrin, A. Esposito, C. Rella, S. Benson, Int. J. Greenh. Gas Control 4, 811 (2010)
I. McAlexander, G.H. Rau, J. Liem, T. Owano, R. Fellers, D. Baer, M. Gupta, Anal. Chem. 83, 6223 (2011)
R. Amundson, L. Stern, T. Baisden, Y. Wang, Geoderma 82, 83 (1998)
G. Chiodini, S. Caliro, C. Cardellini, R. Avino, D. Granieri, A. Schmidt, Earth Planet. Sci. Lett. 274, 372 (2008)
D.S. Baer, J.B. Paul, M. Gupta, A. O’Keefe, Proc. SPIE Int. Soc. Opt. Eng. 4817, 167 (2002)
L. Gianfrani, M. Gabrysch, C. Corsi, P. DeNatale, Appl. Opt. 36, 9481 (1997)
C.L. Zhao, P.P. Tans, J. Geophys. Res., Atmos. 111, D08S09 (2006)
P. Werle, R. Mucke, F. Slemr, Appl. Phys. B 57, 131 (1993)
P. Werle, Appl. Phys. B, Lasers Opt. 102, 313 (2011)
A.-S. Provost, P. Richon, E. Pili, F. Perrier, S. Bureau, Eos 85, 113 (2004)
E. Pili, M. Dellinger, L. Charlet, P. Agrinier, F. Chabaux, P. Richon, Geochim. Cosmochim. Acta 73, A1029 (2009)
P. Sturm, A. Knohl, Atmos. Meas. Tech. 3, 67 (2010)
S.M. Schaeffer, J.B. Miller, B.H. Vaughn, J.W.C. White, D.R. Bowling, Atmos. Chem. Phys. 8, 5263 (2008)
D. Paul, G. Skrzypek, I. Forizs, Rapid Commun. Mass Spectrom. 21, 3006 (2007)
D.S. Baer, personal communication (2011)
G. Friedrichs, J. Bock, F. Temps, P. Fietzek, A. Kortzinger, D.W.R. Wallace, Limnol. Oceanogr., Methods 8, 539 (2010)
G. Chiodini, R. Cioni, M. Guidi, B. Raco, L. Marini, Appl. Geochem. 13, 543 (1998)
P. Deines, D. Langmuir, R.S. Harmon, Geochim. Cosmochim. Acta 38, 1147 (1974)
D. Scholz, C. Muhlinghaus, A. Mangini, Geochim. Cosmochim. Acta 73, 2592 (2009)
Acknowledgements
The expertise of Carine Chaduteau was invaluable during IRMS measurements. We thank Doug Baer, Feng Dong and other people at Los Gatos Research for their availability, advice and help in using this analyser in specific conditions. Martina Schmidt, Marc Delmotte and Michel Ramonet from LSCE (France) introduced us to the WMO scale and performed the precise CO2 concentration measurements of the reference cylinders. Didier Mondelain introduced us to laser-based isotope measurement and the Allan variance. Comments and language corrections from Chris Parson have been greatly appreciated. We thank the editors and an anonymous reviewer for their suggestions and comments. This is IPGP contribution number 3251.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Guillon, S., Pili, E. & Agrinier, P. Using a laser-based CO2 carbon isotope analyser to investigate gas transfer in geological media. Appl. Phys. B 107, 449–457 (2012). https://doi.org/10.1007/s00340-012-4942-8
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00340-012-4942-8