
Abstract
The structural features of polyacrylonitrile and pitch-based carbon fibers were analyzed from a comprehensive point of view by X-ray measurements and related techniques. The results indicated that the undulating graphite ribbon with embedded microvoid was the main structural unit for graphitic fibers. The void’s parameters for these fibers could be obtained directly by small-angle X-ray scattering following the classic method deduced based on the typical two-phase system (i.e., Porod’s law, Guinier’s law and Debye’s law). The non-graphitic fibers, however, were composed of two-dimensional turbostratic crystallites in the aggregation of microfibril and thus had a quasi two-phase structure (microfibril, interfibrillar amorphous structure and microvoid embedded within the microfibril). The extended Debye or Beaucage model in this case should be applied in order to obtain the structural parameters. It also revealed that the quasi two-phase system would complete its transformation to two-phase system during high-temperature graphitization. Therefore, the degree of graphitization was speculated to be the essential indicator distinguishing graphitic fibers from non-graphitic ones and would be helpful in understanding the transformation of structural features during the graphitization of carbon fibers.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Carbon fiber is an important reinforcing material applied in advanced composites (e.g., metal, ceramic, polymer, carbon-based) [1–5]. The majority of commercial carbon fibers presently produced are based on polyacrylonitrile (PAN) fibers [6]. The production of carbon fibers from mesophase pitch is also expected to gain importance, especially if their strength was greatly improved [7]. PAN-based carbon fibers are characteristically of high strength but low modulus, while pitch-based carbon fibers tend to have higher modulus but lower strength, which is ascribed to the difference in their structures [7, 8]. The designation “graphite fiber” as suggested by Ruland [8] is used for carbon fibers which have been graphitized at temperatures above 2000 °C. By means of improving the dimension and alignment of the crystallites, this graphitization stage realizes the increase in tensile modulus [9]. The resulting specific mechanical properties further confer upon these fibers an unquestionable role in the aerospace industry [8–13].
As for the basic structural feature, the crumpled sheet model proposed by Guigon et al. gives a deep impression: (1) The high-strength carbon fibers are composed of two-dimensional turbostratic crystallites roughly oriented along the fiber axis and have a two-phase structure (crystalline and amorphous phases) [14]; (2) the high modulus fibers are made up of slowly undulating graphite ribbons highly oriented along the fiber axis [13]. These above structural features are confirmed by transmission electron microscopy (TEM) and X-ray diffraction (XRD) and now generally accepted by researchers [8, 15–18]. Besides these features, the microvoids embedded in carbon structures also attract wide attention from researchers [19–23]. Small-angle X-ray scattering (SAXS) has been widely used in previous works and successfully made a lot of beneficial explorations in the study of this microporosity. However, some SAXS reports about the microvoid’s dimension are not always consistent. For example, the longitudinal lengths of microvoids in the works of Sugimoto et al. were all above 14.0 nm, while this parameter from other related papers was as low as merely 1.0–2.6 nm for PAN-based fibers [20–26]. Given the basic structure and mechanical properties of the fibers used in these papers were quite close (bulk density at about 1.8 g cm−3, tensile strength at about 3.5 GPa and tensile modulus at about 242 GPa), why the calculated results were so much different in the order of magnitude?
According to our observation, the existence of large dimensional scatterer is true, but may not really due to the void. In this study, the Debye–Bueche plots of Torayca T300B and its graphitized samples were entirely different, especially in the low angle area. The typical PAN- and pitch-based fibers commercially obtained from companies confirmed this point as well. In combination with these observations and various pieces of evidences gathered from literature, we finally determined an intensity component for amorphous structure in the scattering data of non-graphitic carbon fibers [14, 27–31]. A systematic analysis based on SAXS was thus carried out aiming to gain a comprehensive understanding toward the structural features of carbon fibers. The result of this paper will offer a new idea to the characterization of carbon materials and provide some valuable data feedback to the carbon fiber production.
2 Experimental
2.1 Materials
Carbon fiber used in this study included PAN-based non-graphitic fibers, PAN-based graphitic fibers and pitch-based graphitic fibers, as shown in Table 1. The graphitization of these fibers was carried out in helium in a graphite element resistance furnace (Tongxin Electric Heating Apparatus Co., Ltd, Xi’an, China) at the set temperature [32].
2.2 Characterization
The XRD experiment was performed on PANalytical X-ray diffractometer (X’Pert PRO, CuKa, λ = 0.1541 nm, 40 kV, 40 mA) with a fiber specimen attachment. Measurements were taken by performing equatorial and meridional scan. The step size was about 0.05°, and the scan time was set as 30 s per step. The diffraction curves were fitted by MDI Jade 5.0, and the structural parameters were obtained according to Bragg’s equation and Scherrer formula [9]. The Raman experiment was performed on Raman spectrometer (Jobin Yvon, LabRAM HR Evolution). The wavelength of the exciting laser beam was 532 nm, the laser spot diameter was about 1 μm, and the spectra were recorded at 0.5 cm−1 resolution. The structural parameters were obtained through a Lorentz fitting to the data [33]. The SAXS experiment was performed for all the samples on a long-slit collimating SAXSess mc2 instrument (λ CuKα = 0.15418 nm, Anton Paar GmbH) operated at 40 kV, 50 mA in vacuum. Approximately 1-mm-thick bundles of fibers were arranged parallel on the sample holder to conduct the experiment. The scattered X-ray intensity was recorded using an image plate detector, and each measurement was recorded for an hour. The desmearing of collimating error and the background correction of SAXS data were done by SAXS-quant 1.01 software included with the instrument. TEM analysis was carried out to obtain the high-resolution crystalline morphology of carbon fibers on FEI Tecnai G2 F20 transmission electron microscope (accelerating voltage 200 kV).
2.3 Data analysis
For the determination of size parameters by SAXS several options are available, namely Porod, Guinier and Debye–Bueche method. They are generally well established and applicable for most of the complex structures, especially for the quasi two-phase system of carbon fibers as suggested by many related articles [8, 17]. Based on Porod’s law the influence of the local density fluctuation on the structural analysis can be examined [34]:
where I obs(q) is the observed SAXS intensity using a pinhole collimating system, I(q) is the intensity component followed Porod’s law, I fl(q) is a constant modeling the scattered intensity from electron density fluctuation in the phases, b 1 and b 2 are the constants, the scattering vector q = 4πsinθ/λ, and λ is the wavelength of the X-ray. From Guinier’s law and other related methods the size parameters of the scatterers were estimated [35, 36]:
where I(0) represents the incident X-ray intensity, and R g is the radius of gyration of scatterers. The distance of heterogeneity is given by [22, 23, 35]:
Beaucage developed a general equation that we find capable of describing scattering functions containing multiple length scales. The unified fit model for a two-phase system is expressed as [37, 38]:
where G is the exponential prefactor, B is a power law prefactor, and p is the power law exponent. For surface fractals 4 > p > 3 and for mass fractals p < 3 [39, 40]. In the cases of a complex system, the model realizes the modeling of the scattering data by adding another Guinier exponential forms and the corresponding structural limited power laws [37, 38, 41]:
Among the classic methods Debye–Bueche correlation length analysis is of unique importance to understand the intensity distribution of carbon fiber in this study. The Debye–Bueche scattering function for a two-phase system is expressed as [42]:
where A is an intensity scaling factor and a is the correlation length [34]. From the above equation, a linear relationship between I(q)−1/2 and q 2 can be directly obtained for an ordinary two-phase system structure. It was further established that if there are two distinct distributions of interfaces originating from objects of different sizes, then the two can be regarded as having separate contributions to the scattering intensity [42, 43]. A deviation from the linear relationship at the low q 2 area of the I(q)−1/2 ~ q 2 plot may thus be observed. In this case a “double Debye” function can be construed [43]:
3 Results and discussion
3.1 Degree of graphitization
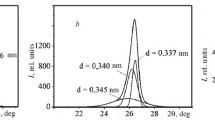
The XRD profiles of three kinds of typical carbon fibers are shown in Fig. 1a. The result showed that the crystalline structure differed greatly among various kinds of fibers. The PAN-based non-graphitic fibers showed a very broad “hump” for 002 reflection, while the graphitic fibers showed a relatively sharp peak. The crystalline parameters (the crystalline size L a, interlayer spacing d 002 and stack thickness L c in Table 2) further established that the graphitic fibers were generally better crystallized than non-graphitic ones. The Raman spectra also indicated a similar structural difference among these samples as shown in Fig. 1b. All of the samples showed the main E2g2 line near 1580 cm−1 (G band) and additional lines at about 1360 cm−1 (D band) and 1620 cm−1 (D’ band). The G band intensities of graphitic fibers were obviously higher than that of the non-graphitic ones, and the graphitization degree A D/A G (the integral area ratio of D to G band) of graphitic samples was likewise lower than that of the non-graphitic ones [32, 33, 44]. It should be pointed out, whether the broad “hump” for 002 reflection or the high D band at about 1360 cm−1, the current experimental phenomena of non-graphitic fibers indicated that these fibers contain large amount of amorphous structures. Based on the Lorentz fitting results, an order of the degree of graphitization for various kinds of fibers can be concluded as: pitch-based graphitic fibers >PAN-based graphitic fibers >PAN-based non-graphitic carbon fibers.

The a XRD profiles and b Raman spectra of T300B, TGGF and PBGF
3.2 Structural features revealed by SAXS
The intensity distribution of non-graphitic carbon fibers is dramatically different from that of graphitic ones. As can be seen from their lnI(q) ~ q 2 plots (Fig. 2a–c), lnI(q) values of graphitic fibers continuously decreased with increasing q 2. The intensity distribution of T300B, however, showed both a rapid decrease in the lower q 2 range and an obvious slowing down in the higher q 2 range. Furthermore, the radius of gyration calculated based on Guinier approximation (about 9.10 nm for T300B) was found much larger than 1.39 nm reported by Shioya et al. and 0.7 nm by Gupta et al. [20, 22, 23]. No direct evidence from TEM, etc., actually supported the existence of such a large pore in high-strength fibers until now. Therefore, it was suspected that the radius given by Guinier method was not absolutely due to the microvoids within the carbon structures. A Debye–Bueche correlation length analysis was carried out in this case to investigate the scatterers within carbon fibers. As shown in Fig. 2d, a small drop-off from the straight line at the lower angle area of I(q)−1/2 ~ q 2 plots was observed for both directions of the non-graphitic samples. This meant that these samples actually did not belong to an ordinary two-phase system. According to Debye theory, it was speculated that there were at least two distinct distributions of interfaces originating from objects of different sizes within these samples [42, 43]. For graphitic samples (Fig. 2e, f), however, a straight line without deviation fitted reasonably well to the data, indicating that the sample had developed into a two-phase system after graphitization process. The similar phenomenon could be examined in the reports by Shioya and Takaku [22], Gupta [23] and Johnson and Tyson [45].

The Guinier approximation to the lnI(q) ~ q 2 plot of a T300B, b TGGF and c PBGF; the Debye plot (I(q)−1/2 ~ q 2) of d T300B, e TGGF and f PBGF
The unified fit model in this case was applied to the fitting for non-graphitic fibers [37, 38, 41]. The results indicated that a one-level model could only describe either the lower (q < 0.4 nm−1) or higher angle area (q > 0.8 nm−1) of the observed intensity for non-graphitic fibers, because the scattering originated essentially from two distinct distributions of interfaces as depicted in Fig. 3a. The two-level model, nevertheless, fitted reasonably well to the data as indicated by the fitting parameters and statistic results in Table S1. On the other hand, the structure of graphitic fibers did not seem as complex as that of non-graphitic ones. As shown in Fig. 3b, c, these fibers whether they were PAN or pitch based could be described simply by a one-level unified fit model.

The unified fitting to the data of a T300B, b TGGF and c PBGF; the Debye fitting to the data of d T300B, e TGGF and f PBGF
The Debye–Bueche analysis as shown in Fig. 3 and Table 3 further indicated that a single Debye model based on Eq. 6 was solely suitable for fitting the data of graphitic samples. Given the intensity distributions generally followed Porod’s law, it could be deduced that a two-phase system with a sharp density transition at the interface, i.e., a distribution of voids in the matrix of rather homogeneous crystallites, was the main structural feature for these samples [27, 46]. For non-graphitic fibers, however, the model was no longer satisfactory. It seemed likewise that a single Debye model could only describe either the lower (q < 0.5 nm−1) or higher angle area (q > 1.0 nm−1) of the observed intensity as depicted in Fig. 3d. In this case, another correlation distance referring to a larger heterogeneous region should be taken into account, i.e., the “double Debye” based on Eq. 7 could be applied in the interpretation of the intensity distribution [42, 43]. The results (Table S2) showed that “double Debye” model fitted reasonably well to the data for both the longitudinal and transverse sections of these samples.
By applying “double Debye” model or two-level unified fit model, the intensity components due to different scatterers were separated (the fitting parameters and statistic results are shown in Table S1, S2 and S3.). The first Debye fitting (or the first-level unified fit model, similarly hereinafter) was the major intensity component at the lower angle area of the scattering, while the second fitting (or the second-level unified fit model, similarly hereinafter) was the dominant one at the higher angle. Guinier approximation was then applied to the fitting results, and the size parameters were determined to each kind of scatterers as listed in Table 4. According to the parameters, the results from the two models are consistent and both results may thus be confirmed. Then what were the two intensity components really due to in the structure of carbon fibers? The second Debye fitting should be ascribed to the microvoids within the fibrils structure, judging from the q range it appears [20, 22, 23]. The dimensions listed in Table 4 are also apparently very close to the reported values of voids [19, 22, 23]. For the first Debye fitting, we speculated that it was due to the structure of interfibrillar transition regions, i.e., the amorphous structures [14, 27, 47]. An alternative explanation for this was that the scattering signals could originate essentially from any transition interphases of considerable density fluctuation [43, 48]. The scattering power contrast of the first Debye fitting against the second fitting was estimated based on the integral invariant as shown in Table S2, and the results (all contrast values range from 0.190 to 0.405 for non-graphitic fibers) indicated that the density fluctuation for the two interfaces was detectable [35]. It was also established that if there were two distinct, non-interacting distributions of interfaces originating from objects of considerably different sizes, then the two could be considered to have separate, independent contributions to the scattering intensity [43]. With relevance to this study these two objects were no other than the amorphous structure and the much smaller microvoid [14, 27, 47]. They were, respectively, the causes for the first and second Debye fitting, the parameters of which were quite close to the reports [20, 27]. In addition, the power law exponent p for the first-level unified fit model was less than three, corresponding to the mass fractal of low-density amorphous structure. The second-level unified fit model, however, showed an exponent between 3 and 4, which probably indicated a fractal structure for the void’s surface [39]. Basically, the unified fit model returned structural parameters regardless of what the scattering was really due to. The power law exponent, however, indicated that the scatterers for the two intensity components were significantly different in structure.
As discussed above, a single Debye model (or a one-level unified fit model) was suitable for fitting the data of graphitic samples. However, there was an exception for this expression as shown in Fig. 4. The 2000 °C-graphitized T300B fiber showed a small drop-off from the straight line at lower q range as what the non-graphitic fibers did in the Debye plots. As expected, the “double Debye” model rather than the single Debye fitting was satisfactory in describing the density distribution of this sample. On the other hand, the 2500 °C-graphitized sample behaved like other graphitic fibers and showed no deviation from the Debye’s law for two-phase system. Given their significant structural differences indicated by Raman spectra and XRD (Table 2), great changes should take place during the graphitization of these fibers. Thus, it can conclude that (1) the basic structural feature differentiating the two-phase system of graphitic fibers from the quasi two-phase system of non-graphitic samples was the degree of graphitization; (2) the amorphous structure within these graphitic fibers was generally eliminated due to the recrystallization of the carbon structures during the graphitization process; (3) the 2000 °C-graphitized fiber was just in the intermediate process of the transformation for the structural features. The graphitization results obtained by Raman spectra and XRD in Table 2 could be evidences supporting the above conclusions. The transformation of the scattering system is of interest and will be discussed in detail in a separate paper [46].

The Debye plot (I(q)−1/2 ~ q 2) of a T3-2000 and b T3-2500; the Debye fitting to the scattering data of c T3-2000 and d T3-2500
3.3 Structure morphology
Figure 5 shows the TEM images of various kinds of carbon fibers. It could be drawn from the images that the structural features of these fibers were significantly different in both mesoscopic and microcosmic level. In the low-resolution image of T300B, the profiles of the axially oriented fibrils with a breadth at about 50 nm were observed (the same as the values estimated by Williams et al. [27, 47]). Most areas within the boundaries of these fibrils were interlinked small carbon planes of amorphous structure, which tended to have a lower density than that in the fibril domains [14, 46]. The areas indicated by the red ovals in the high-resolution images might be the representations for amorphous structures. In most cases, these amorphous regions were compressed by the neighboring fibrils and took the shape of discontinuous slender ellipsoid (corresponding to the fusiform patterns in SAXS) [10, 46]. Within the fibrils, the short and twisted carbon layers aligned with a rough preferred orientation in the longitudinal section and stacked disorderly in the transverse section [46]. The crystallites which had a relatively larger interlayer spacing were generally very small in dimension. The orange arrows in Fig. 5 indicated the existence of microvoids in the fibrils. These voids (about 1 nm in dimension) were much smaller than 9.10 nm, and the radius roughly estimated from the Guinier curve of the observed intensity [46]. However, they were dimensionally very close to 1.16 nm, and the radius calculated based on the second-level unified fit model.

TEM images of carbon fibers: low-resolution images of a T300B, b TGGF and c PBGF; high-resolution images of the longitudinal sections of d T300B, e TGGF and f PBGF; high-resolution images of the transverse sections of g T300B and h TGGF [46]
The aggregation structure was observed to decrease in dimension, and carbon ribbon became the main structural unit for graphitic fiber TGGF as shown in Fig. 5b. Evolved from the original fibril structure, the carbon ribbons were well crystallized with better orientation along the fiber axis [32]. Within these ribbons, the crystallite became the absolute main composition of the domains as shown in Fig. 5e, h. The voids indicated by the orange arrows grew a lot beside the crystallites, while the amorphous structure generally disappeared in graphitic fibers. It should be noted that new voids might appear in the original amorphous regions, because the amorphous structure was generally lower in density and thus might result in some new voids after the recrystallization of carbon body. According to the SAXS results of T300B, TGCF and their graphitic fibers, these new voids were dimensionally larger than the previous ones in most cases. In fact, we also believe that some of the previous small voids were decreased in dimension or even eliminated due to the recrystallization, while new voids generated during the graphitization process. Therefore, a final comprehensive effect for all these transformations was the increase of both the fiber’s bulk density (Table 1) and the void’s scales (Tables 3, 4).
These above structural features were even more obvious in pitch-based graphitic fibers. Large crystallites were ubiquitous in PBGF, and the ribbon structures grew a lot in dimension as shown in Fig. 5c. Besides the large long ribbons, the void’s dimensions also increased and were very close to the calculated values by SAXS (Fig. 5f). It can be deduced that a two-phase system with a sharp density transition at the interface, i.e., a distribution of voids in the matrix of rather homogeneous crystallites, became the main structural feature for these samples [7, 27, 46]. Based on the X-ray measurements and TEM images, the structural features of various kinds of carbon fibers are summarized in Table 5.
In conclusion, the undulating graphite ribbon with embedded microvoid was the main structural unit for graphitic fibers. The void’s parameters for these fibers could be obtained following the classic method deduced based on the typical two-phase system. The non-graphitic fibers, however, were composed of two-dimensional turbostratic crystallites in the aggregation of microfibril and thus had a quasi two-phase structure. It also revealed that the quasi two-phase system would complete its transformation to two-phase system during high-temperature graphitization, and the final elimination of amorphous structure was a unique key point for the transformation. In this case, the degree of graphitization was speculated to be the essential indicator distinguishing graphitic fibers from non-graphitic ones and would be helpful in understanding the transformation of structural features during the graphitization of carbon fibers.
4 Conclusions
The basic structural features for graphitic fibers (whether they were PAN or pitch based) as detected by X-ray measurements were very similar, in spite of the dimensional differences for their structure units, whereas there was an essential structural distinction between graphitic fibers and non-graphitic ones. According to our observation, undulating graphite ribbons with embedded microvoids were the main structural unit for all kinds of graphitic fibers. The voids’ parameters for these samples could be obtained by SAXS following the classic methods deduced based on the typical two-phase system (i.e., Porod, law, Guinier’s law and Debye’s law). The non-graphitic fibers, however, were composed of two-dimensional turbostratic crystallites in an aggregation of microfibril and thus had a quasi two-phase structure (fibril, interfibrillar amorphous structure and microvoid embedded in fibril). The extended Debye or Beaucage model could be a good analytic method in this case to obtain the structural parameters. The difference in the degree of graphitization was speculated to be the main reason for bringing about the above structural distinctions among these fibers. The amorphous structure was also observed to be generally eliminated from the original quasi two-phase system due to the changes on graphitization degree during the high-temperature graphitization.
References
M.S. Dresselhaus, G. Dresselhaus, K. Sugihara, I.L. Spain, H.A. Goldberg, Graphite Fibers and Filaments (Springer, Berlin, 1988)
E. Frank, F. Hermanutz, M.R. Buchmeiser, Carbon fibers: precursors, manufacturing, and properties. Macromol. Mater. Eng. 297, 493–501 (2012)
L. Li, Cyclic fatigue behavior of carbon fiber-reinforced ceramic–matrix composites at room and elevated temperatures with different fiber preforms. Mater. Sci. Eng. A 654, 368–378 (2016)
L. Feng, K. Li, Z. Si, Q. Song, H. Li, J. Lu, L. Guo, Compressive and interlaminar shear properties of carbon/carbon composite laminates reinforced with carbon nanotube-grafted carbon fibers produced by injection chemical vapor deposition. Mater. Sci. Eng. A 626, 449–457 (2015)
T. Lin, D. Jia, P. He, M. Wang, In situ crack growth observation and fracture behavior of short carbon fiber reinforced geopolymer matrix composites. Mater. Sci. Eng. A 527, 2404–2407 (2010)
D.A. Baker, T.G. Rials, Recent advances in low-cost carbon fiber manufacture from lignin. J. Appl. Polym. Sci. 130, 713–728 (2013)
X.Y. Qin, Y.G. Lu, H. Xiao, Y. Wen, T. Yu, A comparison of the effect of graphitization on microstructures and properties of polyacrylonitrile and mesophase pitch-based carbon fibers. Carbon 50, 4459–4469 (2012)
W. Ruland, Carbon fibers. Adv. Mater. 2, 528–536 (1990)
D.F. Li, H.J. Wang, X.K. Wang, Effect of microstructure on the modulus of pan-based carbon fibers during high temperature treatment and hot stretching graphitization. J. Mater. Sci. 42, 4642–4649 (2007)
R.J. Diefendorf, E. Tokarsky, High-performance carbon fibers. Polym. Eng. Sci. 15, 150–159 (1975)
H. Rennhofer, D. Loidl, S. Puchegger, H. Peterlik, Structural development of pan-based carbon fibers studied by in situ X-ray scattering at high temperatures under load. Carbon 48, 964–971 (2010)
M. Guigon, A. Oberlin, Heat-treatment of high tensile strength pan-based carbon fibres: microtexture, structure and mechanical properties. Fibre Sci. Technol. 27, 1–23 (1986)
M. Guigon, A. Oberlin, G. Desarmot, Microtexture and structure of some high-modulus, pan-base carbon fibres. Fibre Sci. Technol. 20, 177–198 (1984)
M. Guigon, A. Oberlin, G. Desarmot, Microtexture and structure of some high tensile strength, pan-base carbon fibres. Fibre Sci. Technol. 20, 55–72 (1984)
J.B. Donnet, R.C. Bansal, Carbon Fibers (Marcel Dekker, New York, 1984)
C.Z. Zhu, X.F. Liu, X.L. Yu, N. Zhao, J.H. Liu, J. Xu, A small-angle X-ray scattering study and molecular dynamics simulation of microvoid evolution during the tensile deformation of carbon fibers. Carbon 50, 235–243 (2012)
W. Ruland, Apparent fractal dimensions obtained from small-angle scattering of carbon materials. Carbon 39, 323–324 (2001)
O. Paris, D. Loidl, H. Peterlik, M. Müller, H. Lichtenegger, P. Fratzl, The internal structure of single carbon fibers determined by simultaneous small- and wide-angle scattering. J. Appl. Cryst. 33, 695–699 (2000)
R. Perret, W. Ruland, Single and multiple X-ray small-angle scattering of carbon fibres. J. Appl. Cryst. 2, 209–218 (1969)
A. Takaku, M. Shioya, Characterization of microvoids in polyacrylonitrile-based carbon fibres. J. Mater. Sci. 21, 4443–4450 (1986)
Y. Sugimoto, M. Shioya, K. Yamamoto, S. Sakurai, Relationship between axial compression strength and longitudinal microvoid size for pan-based carbon fibers. Carbon 50, 2860–2869 (2012)
M. Shioya, A. Takaku, Characterization of microvoids in carbon fibers by absolute small-angle X-ray measurements on a fiber bundle. J. Appl. Phys. 58, 4074–4082 (1985)
A. Gupta, I.R. Harrison, J. Lahijani, Small-angle X-ray scattering in carbon fibers. J. Appl. Cryst. 27, 627–636 (1994)
Y. Sugimoto, T. Kato, M. Shioya, T. Kobayashi, K. Sumiya, M. Fujie, Structure change of carbon fibers during axial compression. Carbon 57, 416–424 (2013)
C.N. Tyson, Fracture mechanisms in carbon fibres derived from pan in the temperature range 1000–2800 °C. J. Phys. D Appl. Phys. 8, 749–758 (1975)
C.N. Tyson, J.R. Marjoram, Measurement of disorder in turbostratic carbons by small-angle X-ray scattering. J. Appl. Cryst. 4, 488–494 (1971)
W. Li, D.H. Long, J. Miyawaki, W.M. Qiao, L.C. Ling, I. Mochida, S. Yoon, Structural features of polyacrylonitrile-based carbon fibers. J. Mater. Sci. 47, 919–928 (2012)
Y.J. Bai, C.G. Wang, N. Lun, Y.X. Wang, M.J. Yu, B. Zhu, Hrtem microstructures of pan precursor fibers. Carbon 44, 1773–1778 (2006)
N.M.D. Brown, H.X. You, A scanning tunnelling microscopy study of pan-based carbon fibre in air. Surf. Sci. 237, 273–279 (1990)
W.P. Hoffman, Scanning probe microscopy of carbon fiber surfaces. Carbon 30, 315–331 (1992)
M. Kaburagi, Y.Z. Bin, D. Zhu, C.Y. Xu, M. Matsuo, Small angle X-ray scattering from voids within fibers during the stabilization and carbonization stages. Carbon 41, 915–926 (2003)
D.H. Li, C.X. Lu, G.P. Wu, Y. Yang, Z.H. Feng, X.T. Li, F. An, B.P. Zhang, Heat-induced internal strain relaxation and its effect on the microstructure of polyacrylonitrile-based carbon fiber. J. Mater. Sci. Technol. 30, 1051–1058 (2014)
A. Sadezky, H. Muckenhuber, H. Grothe, R. Niessner, U. Poschl, Raman microspectroscopy of soot and related carbonaceous materials: spectral analysis and structural information. Carbon 43, 1731–1742 (2005)
W. Ruland, Small-angle scattering of two-phase systems: determination and significance of systematic deviations from porod’s law. J. Appl. Cryst. 4, 70–73 (1971)
A. Guinier, G. Fournet, Small-Angle Scattering of X-Rays, vol. 14 (Wiley, New York, 1955)
O. Glatter, O. Kratky, Small Angle X-Ray Scattering, vol. 102 (Academic Press, London, 1982)
G. Beaucage, Approximations leading to a unified exponential/power-law approach to small-angle scattering. J. Appl. Cryst. 28, 717–728 (1995)
G. Beaucage, D. Schaefer, T. Ulibarri, E. Black, Multiple Size Scale Structures In Silica/Siloxane Composites Studied by Small-Angle Scattering (Sandia National Labs., Albuquerque, 1994)
G. Beaucage, Small-angle scattering from polymeric mass fractals of arbitrary mass-fractal dimension. J. Appl. Cryst. 29, 134–146 (1996)
P. Schmidt, Small-angle scattering studies of disordered, porous and fractal systems. J. Appl. Cryst. 24, 414–435 (1991)
G. Beaucage, D. Schaefer, Structural studies of complex systems using small-angle scattering: a unified Guinier/power-law approach. J. Non-Cryst. Solids 172, 797–805 (1994)
P. Debye, A. Bueche, Scattering by an inhomogeneous solid. J. Appl. Phys. 20, 518–525 (1949)
B.R. Pauw, M.E. Vigild, K. Mortensen, J.W. Andreasen, E.A. Klop, Analysing the nanoporous structure of aramid fibres. J. Appl. Cryst. 43, 837–849 (2010)
N. Melanitis, P.L. Tetlow, C. Galiotis, Characterization of pan-based carbon fibres with laser Raman spectroscopy. J. Mater. Sci. 31, 851–860 (1996)
D.J. Johnson, C.N. Tyson, Low-angle X-ray diffraction and physical properties of carbon fibres. J. Phys. D Appl. Phys. 3, 526–534 (1970)
D.H. Li, C.X. Lu, G.P. Wu, J.J. Hao, Y. Yang, Z.H. Feng, X.T. Li, F. An, B.P. Zhang, Structural evolution during the graphitization of polyacrylonitrile-based carbon fiber as revealed by small-angle X-ray scattering. J. Appl. Cryst. 47, 1809–1818 (2014)
W.S. Williams, D.A. Steffens, R. Bacon, Bending behavior and tensile strength of carbon fibers. J. Appl. Phys. 41, 4893–4901 (1970)
D.T. Grubb, K. Prasad, W. Adams, Small-angle X-ray diffraction of kevlar using synchrotron radiation. Polymer 32, 1167–1172 (1991)
Acknowledgments
The authors wish to thank Dr. Liyong Wang of Key Laboratory of Carbon Materials for the assistance in sample preparation and measurement. This work was financially supported by Transportation Construction Project of Shanxi Province (Grant No. 15-2-07).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Li, D., Lu, C., Du, S. et al. Structural features of various kinds of carbon fibers as determined by small-angle X-ray scattering. Appl. Phys. A 122, 956 (2016). https://doi.org/10.1007/s00339-016-0487-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00339-016-0487-8