
Abstract
Key message
We obtained a complete mutant line of Petunia having mutations in both F3H genes via Cas9-ribonucleoproteins delivery, which exhibited a pale purplish pink flower color.
The CRISPR-Cas system is now revolutionizing agriculture by allowing researchers to generate various desired mutations in plants at will. In particular, DNA-free genome editing via Cas9-ribonucleoproteins (RNPs) delivery has many advantages in plants; it does not require codon optimization or specific promoters for expression in plant cells; furthermore, it can bypass GMO regulations in some countries. Here, we have performed site-specific mutagenesis in Petunia to engineer flower color modifications. We determined that the commercial Petunia cultivar ‘Madness Midnight’ has two F3H coding genes and designed one guide RNA that targets both F3H genes at once. Among 67 T0 plants regenerated from Cas9-RNP transfected protoplasts, we obtained seven mutant lines that contain mutations in either F3HA or F3HB gene and one complete mutant line having mutations in both F3H genes without any selectable markers. It is noteworthy that only the f3ha f3hb exhibited a clearly modified, pale purplish pink flower color (RHS 69D), whereas the others, including the single copy gene knock-out plants, displayed purple violet (RHS 93A) flowers similar to the wild-type Petunia. To the best of our knowledge, we demonstrated a precedent of ornamental crop engineering by DNA-free CRISPR method for the first time, which will greatly accelerate a transition from a laboratory to a farmer’s field.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The CRISPR-Cas system is a versatile and cutting-edge technology that allows researchers to generate various desired mutations in plants at will, enabling the development of plant mutants. In contrast with classical genetic methods that require long-term breeding processes, CRISPR modifies a target trait of interest within a few generations in a site-specific manner (Brooks et al. 2014; Feng et al. 2014; Ma et al. 2016; Pan et al. 2016; Subburaj et al. 2016a; Zhang et al. 2014). The most popular method for CRISPR-mediated genome editing in plants is to use an Agrobacterium tumefaciens-mediated transformation system to deliver DNA encoding the components. However, there are issues with this method; plant transformation by A. tumefaciens is accompanied with T-DNA integration in the host plant genome, frequently inducing unwanted genetic alterations (Clark and Krysan 2010; Jupe et al. 2019; Nacry et al. 1998) and the constitutive expression of CRISPR-Cas9 complexes during the life cycle can increase off-target mutations in the genome (Zhang et al. 2018). Alternatively, DNA-free genome editing via the delivery of preassembled Cas9 protein and single guide RNA (sgRNA) ribonucleoproteins (RNPs) has many advantages in plants. This method does not require codon optimization or specific promoters for expression in host cells, so it can be used directly in various crop species (Malnoy et al. 2016). Furthermore, Cas9 RNPs cleave target DNAs immediately after transfection and are rapidly degraded in cells, which drastically reduces the number of unwanted mutations at off-target sites (Kim et al. 2014). In this regard, use of the Cas9 RNP system can bypass genetically modified organism (GMO) regulations in some countries (Subburaj et al. 2016a). To date, we and other research groups have demonstrated genome editing via Cas9-RNPs in Arabidopsis, rice, lettuce, tobacco, microalgae, grape, apple, and potato (Andersson et al. 2018; Baek et al. 2016; Lin et al. 2018; Malnoy et al. 2016; Subburaj et al. 2016b; Woo et al. 2015). In a continuation of our previous study in which we established a protocol for gene knock-out in Petunia protoplasts via Cas9-RNP delivery (Subburaj et al. 2016b), we have now performed site-specific mutagenesis in Petunia to engineer flower color modifications.
Modern Solanaceae Petunia hybrids (Petunia x hybrida, 2n = 14) are self-incompatible and taxonomically originated from two parental species of P. axillaria (two subspecies: axillaris, parodii) and P. integrifolia (three subspecies: integrifolia, inflata, oxxidentalis) (Bombarely et al. 2016; Stehmann et al. 2009; Vandenbussche et al. 2016). Flower color is an important trait for ornamental plants like Petunia because it has a strong influence on their market value. Petunia flower colors are determined by genes that are involved in the flavonoid/anthocyanin biosynthesis pathway, in which flavanone 3′-hydroxylase (F3H) is one of the key enzymes. F3H catalyzes the 3-hydroxylation of (2S)-flavanones such as naringenin to dihydroflavonols, which are important for the synthesis of anthocyanidin and flavonol pigments. Thus, the F3H gene is thought to be a potential target in flower color engineering. Previously, many studies have aimed to modify flower color by inhibiting or over-expressing the F3H gene through stable genetic transformations in ornamentals of the genera of Torenia, Dianthus and Nierembergia as well as Petunia (Suzuki et al. 2000; Tsuda et al. 2004; Ueyama et al. 2006; Zuker et al. 2002). Therefore, in this study, we selected F3H as a target for editing by the CRISPR-Cas9 system to change Petunia color.
Materials and methods
Plant material
Petunia ‘Madness Midnight’ seeds were purchased from the PanAmerican seed Company (West Chicago, IL, USA). The surface sterilized seeds treated with 2% NaClO solution for 20 min were germinated and sub-cultured every 4 weeks on Murashige and Skoog (MS) basic medium. In vitro plants were grown in culture environments of 16 h/8 h light and dark photoperiod at 2000 lx and 25 ºC. Young, fully expanded leaves of the in vitro-grown plants were selected and cut to 1 ~ 2 mm strips for protoplast isolation.
Preparation and isolation of Petunia protoplasts
Young leaves (about 12–16) from 3-week-old in vitro grown P. hybrida cv. ‘Madness Midnight’ seedlings were pre-plasmolysed with cell protoplast washing (CPW) solution containing 13% mannitol (w/v), followed by cell wall digestion with 1.2% Viscozyme, 0.6% PectinEX, and 0.6% Celluclast enzyme combination (Novozymes, Bagsvaerd, Denmark), according to our previously published methods (Subburaj et al. 2016b) with minor modifications. After protoplasts were collected by centrifugation at 600 rpm for 5 min, cells were further purified by flotation on 25% sucrose in CPW (w/v) with subsequent centrifugation at 600 rpm for 5 min. The middle layer containing the isolated cells were collected, diluted in suitable CPW 9% Mannitol (W/V) solution. Protoplasts were then counted to determine the yield and their viabilities were determined by staining 50 µl of cells with 1 µl of fluorescein diacetate solution (5 mg/ml in acetone). The cells reacted with FDA solution then observed with green fluorescence microscope to count green cell number for protoplast activity. To optimize the protoplast enzyme treatments and yield, triplicates of both technical and biological samples were carried out.
Validation of protoplast viability and transfection efficiency using a transient GFP expression
To determine the transfection efficiency, a transient GFP expression system was carried to optimize the concentration of PEG and incubation period as described elsewhere in our previous study with minor modifications (Subburaj et al. 2016b). Purified protoplasts were allowed to stabilize at 4 ℃ for 1 h followed by the resuspension in CPW 9% mannitol (w/v) solution. Then the cells were pelletized (600 rpm for 5 min) and further resuspended in a MMg solution (0.4 M mannitol, 15 mM MgCl2, 5 mM MES) to adjust the cell density into 106 cells/ml according to previous methods. A 250 μl of protoplast (2.5 × 105 cells/ml) cells were mixed with 50 μg of plasmid vector pBI221 carrying green fluorescence protein (GFP) gene (pBI221::GFP). Then a 270 µl of freshly prepared PEG solution [40% (w/v) PEG4000 (Sigma Aldrich), 0.4 M mannitol, 0.1 M Ca(NO3)2] was added and were gently mixed until homogenous appearance. The samples were then incubated at room temperature for 20 min. PEG reaction was stopped by adding 250 µl of CPW 9% Mannitol (w/v) washing solution with an incubation for 10 min. Further, a subsequent addition of CPW washing solution (500 µl, 1 ml, 2 ml, and 3 ml) followed by 10 min incubation, the PEG was completely diluted and finally removed by a final centrifugation at 600 rpm for 5 min. To the protoplast pellets, 1 ml culture media was added and kept for incubation in dark at 25 ºC. After 24 h, the transformed cells were observed under the green fluorescence microscope. Based on the number of GFP transformed cells, transfection efficiency was estimated.
Purification of recombinant SpCas9 protein
Plasmid pET28a-SpCas9 was provided by Prof. Jin-Soo Kim. BL21-Pro cells (Ezynomics, CP111) were transformed with pET28a-SpCas9 and grown to mid-log phase (OD600 nm of 0.4–0.5) in LB medium containing kanamycin at 37 ℃ with shaking at 200 rpm. Protein expression was induced with the addition of 0.8 mM IPTG, after which cells were incubated overnight at 18 ℃ with shaking at 200 rpm. Cell pellets were obtained by centrifugation at 4 ℃ (3100×g, 20 min). After cells were resuspended in lysis buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole), 2 mg/ml lysozyme and 1 mM PMSF were added. The lysate was incubated for 1 h on ice and sonicated (amplitude 25%, 1 s ON/1 s OFF, pulse 30 times) eight times. The lysate was then cleared by centrifugation at 4 ℃ (18,000×g, 30 min). The supernatant was filtered through a 0.45-μm syringe filter and then incubated with Ni–NTA agarose for 1 h with rotation at 4 ℃. The resin was then washed twice with wash buffer (50 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole). The bound proteins were eluted with elution buffer (50 mM NaH2PO4, 300 mM NaCl, 250 mM imidazole). The eluted proteins were diluted with storage buffer (150 mM NaCl, 20 mM HEPES, 0.1 mM EDTA, 1 mM DTT, 2% sucrose, 20% glycerol). The concentration of the purified protein was determined on an SDS-PAGE gel.
sgRNA preparation
For sgRNA preparation, a partial DNA duplex was used as the template for in vitro RNA transcription. DNA oligonucleotides are listed in Supplementary Table 2. The partial duplex was annealed and then extended with Phusion polymerase (Thermo Scientific, #F530L) and purified using a PCR cleanup kit (GeneAll, 103-102). sgRNAs were then generated by in vitro transcription at 37 ℃ overnight, after which the template DNA was cleaved with DNA nuclease I (New England Biolabs, M303L) at 37 ℃ for 20 min. The RNA product was further purified using an RNA cleanup kit (Qiagen, 74204). The final RNA concentration was measured with a NanoDrop device.
Cas9-RNP transfection
Ribonucleoprotein (RNP) complexes were made by combining Cas9 and sgRNA at a molar ratio of 1:3 during Petunia protoplast transfection according to the protocol described previously (Subburaj et al. 2016b; Woo et al. 2015). Briefly, 20 µl of transfection reaction mixture, containing 2 µl of Cas9 protein buffer (50 mM Tris–HCl, 10 mM MgCl2, 100 mM NaCl, 1 mM DTT, pH 7.5), 10 µg of Cas9 protein, 30 µg of sgRNA, and an adequate volume of deionized water, was prepared. This mixture was transfected with resuspended protoplasts along with PEG as described in the above validation of protoplast viability and transfection efficiency via the transient GFP expression assay.
NGS-based targeted deep sequencing and mutation analysis
Genomic DNA was prepared from protoplasts and regenerated plants. DNA segments that encompass Cas9 target sites were amplified with Phusion polymerase using pairs of primers that are listed in Supplementary Table 2. PCR amplicons were subjected to paired-end read sequencing using Illumina MiSeq. Obtained next generation sequencing data were analyzed using Cas-Analyzer (https://www.rgenome.net/cas-analyzer). Reads that occurred only once were excluded to remove errors associated with amplification and sequencing. Indels located around the Cas9 cleavage site (3 bp upstream of the protospacer–adjacent motif sequence) were considered to be mutations induced by Cas9.
Petunia protoplast regeneration
RNP-transfected protoplasts were cultured in liquid MS medium supplemented with 6% myo-inositol, 2% sucrose, 2 mg/L 2,4-dichlorophenoxyacetic acid (2,4-D), and 0.5 mg/L 6-benzylaminopurine at 25 ℃ in darkness with shaking (10 rpm). After 1 day, cell division was initiated, multicellular structure was formed at 3 days after Cas9-RNP transfection. Then colonies were formed after 3–4 weeks. Colonies were transferred to callus-induction MS medium supplemented with 3% sucrose, 0.5 mg/L α-naphthaleneacetic acid (NAA) and 2 mg/L BAP. After 3–4 weeks of culture, calli were transferred to shoot induction MS medium supplemented with 3% sucrose and 1 mg/L zeatin. The regenerated plantlets with health roots were transplanted to soil for hardening those whole plants.
Estimation of anthocyanin content
Petunia flower petals were collected at 0 and 1 day post anthesis. The total anthocyanin content in petals was estimated according to methods in a previous report (Tan et al. 2013). In brief, total anthocyanins were extracted from flower petals by incubation in acidic (1% HCl [w/v]) methanol for 48 h and quantified by measuring the absorbance at 530 nm. Triplicates of three biological replicates were carried out during this estimation.
Data availability
DNA and amino acid sequence information of F3HA and F3HB have been deposited in NCBI GenBank under accession number MN727296 (F3HA) and MN727297 (F3HB). Targeted deep sequencing data used in this study have been deposited in the NCBI Sequence Read Archive database (SRA; https://www.ncbi.nlm.nih.gov/sra) under accession number PRJNA591067.
Results
Design of guide RNAs that target two F3H genes at once in Petunia
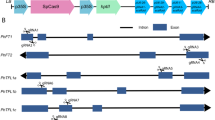
We used the commercial Petunia cultivar ‘Madness Midnight’ as a model system for examining Cas9-RNP-driven Petunia color modification. Whole genome annotation has not been completed for this variety, so we first used Sanger sequencing to examine the F3H gene. This analysis revealed that ‘Madness Midnight’ has two F3H coding genes; one gene (F3HA) originates from P. axillaris and the other (F3HB) from P. inflata (Fig. 1a). The two genes have a high degree of similarity at both the nucleotide (93.3% identity) and amino acid (98.9% identity) levels (Supplementary Figs. 1 and 2). We then designed three sgRNAs that target exon 1 or 2 of both F3H genes at once (Fig. 1b) (Bae et al. 2014; Park et al. 2015). To estimate the activity of each sgRNA, we transfected in vitro transcribed sgRNAs with purified SpCas9 proteins, derived from Streptococcus pyogenes, into ‘Madness Midnight’ protoplasts via polyethylene glycol (PEG)-mediated transformation and performed high-throughput sequencing (or targeted deep sequencing) of bulk genomic DNA isolated from the protoplasts via a next-generation sequencing (NGS) technology, after 24 h (Fig. 1c). As a result, we found that the frequency of insertions and deletions (indels) at the targets ranged from 9.99 to 26.27% (Fig. 1d), which were calculated by Cas-Analyzer (Park et al. 2016). The represent mutation types from NGS outcomes are listed in the Supplementary Fig. 3. For further experiments, we chose the target closest to the beginning of F3H, F3H-g1, to increase the chances of gene disruption after Cas9-mediated indel mutations.

Simultaneous targeting of duplicated F3H genes in P.hybrida cv. ‘Madness Midnight’. a Phylogeny analysis of F3H genes from ‘Madness Midnight’ and the parental wild-type strains. Full nucleotide sequences were aligned with MUSCLE3.8.31 and the phylogeny was analyzed with PhyML3.1/3.0. The tree was prepared using TreeDyn198.3. All of the analysis was accomplished using Phylogeny.fr (https://www.phylogeny.fr/index.cgi). b Design of sgRNAs simultaneously targeting both F3HA and F3HB. The black bars on the gene diagrams indicate the relative positions of each guide RNA. c An experimental scheme of determining indel frequencies after Cas9-RNP transfection in protoplasts. d The frequencies of indels near each target region after Cas9-RNP transfection, which were determined by NGS-based targeted deep sequencing
Successful isolation and regeneration of protoplasts in Petunia
To prepare viable and high-quality protoplasts, we employed a modified enzyme combination comprising cellucast (0.6%), PectinEX (0.6%) and Viscozyme (1.2%), and obtained highest yield of 6.90 ± 0.18 × 106 cells with an incubation period of 3 h, in which 94.3% of the cells were found to be alive by green colored fluorescence from the FDA test. And plasmid vector (pBI221:GFP) containing GFP were transfected to isolated Petunia protoplast cells (2.5 × 105 cells) using a 40% polyethylene glycol (PEG)-mediated transformation, which showed approximately 55% of transfection efficiencies (Supplementary Fig. 4). After Cas9-RNP treatment, we tried to obtain mutant plants through direct protoplast regeneration (Fig. 2a). Briefly, protoplasts transfected with Cas9-RNPs were cultured on colony inducing liquid MS medium. It was found that protoplast cells displayed to divide at or after day 1 of inoculation, and by day 3 the cells appeared to form a mass of multicellular structure. After 3–4 weeks of culture, protoplast derived micro-calli were noticed. When the micro-calli were spread on to the solid callus-inducing medium, friable organogenic calli were appeared after 2–3 weeks. The separated green calli were placed on shooting medium and small protuberances was initially emerged, followed by shoots buds were observed on the surface of the calli after 2–3 weeks. Whole plants with well-developed roots were successfully regenerated in the root-inducing MS medium with 3% sucrose supplementation.

Process of Cas9-RNP mediated mutant generation in Petunia. a Schematic procedures showing entire Petunia regeneration from protoplasts for targeted mutagenesis of F3H via Cas9-RNP. b Genotypes of both F3H genes in regenerated T0 plants. Among 67 T0 plants, eight plants showed mutations in either the F3HA or the F3HB gene, and the P4C4 line contained a frameshift mutation in both genes. Red colored sequences indicate nucleotide insertion, blue bars indicate deletion compared to wild-type sequence, and underbars indicate target seqeunces of Cas9 (color figure online)
Construction of complete F3H knockout mutant line in Petunia
Through the regeneration process, we finally obtained 67 regenerated T0 plants from the F3H-g1 transfected protoplasts, which were derived from 67 independent calli. To investigate the gene mutations in both F3H genes, we performed targeted deep sequencing for all regenerated plants. As a result, eight plants (11.9%) showed indel mutations near DNA cleavage sites in F3H genes (Fig. 2b); four plants contained homozygous mutations only in the F3HA gene, three plants contained homozygous mutations only in the F3HB gene, and one plant, named P4C4, contained homozygous mutations in both F3H genes. The other plants did not contain significant mutation frequencies in either gene (Supplementary Table 1). We confirmed the two gene disruptions in P4C4 by Sanger sequencing (Fig. 3a). The 1 bp insertion in the two F3H genes in P4C4 would lead to the production of truncated proteins during translation. We checked that the mutations can induce premature termination codons in 78th and 74th positions of F3HA and F3HB genes, respectively (Supplementary Fig. 5), which eliminate the conserved enzymatic domain found in members of the 2-oxoglutarate and Fe(II)-dependent oxygenase superfamily (pfam03171: https://pfam.xfam.org/family/PF03171) (Fig. 3b).

Isolation of f3ha f3hb mutant and its flower traits. a F3HA and F3HB mutations in the f3ha f3hb mutant (P4C4). F3H sequences of WT and f3ha f3hb mutant were confirmed by Sanger sequencing. b Domain organization in the F3H protein. The rounded green rectangle indicates the conserved enzymatic domain (2OG-Fe(II)_Oxygenase superfamily: pfam03171) in the WT F3H protein (upper). A predicted mutant protein is also represented (lower; 77 aa for F3HA and 73 aa for F3HB). A premature termination codon would lead to a truncated protein that would lack the functional domain. c Flower color phenotypes of WT (left) and the f3ha f3hb (P4C4, right) mutant. d Flower pictures of WT, single gene mutants (P1C7 and P3C5), and the f3ha f3hb (P4C4) at 0 and 1 day post anthesis (DPA). e Estimation of the anthocyanin content in WT and f3ha f3hb mutant flower petals at 0 and 1 DPA. Error bars represent the standard deviation of the mean, and three repeats of the experiment were performed
To allow evaluation of the visible flower color phenotype, the regenerated plants were transplanted to soil. Transplants began to bloom approximately 4 weeks later. Using RHS color chart (Royal Horticultural Society, 5th edition), the WT ‘Madness Midnight’ was found to display a purple violet color (RHS 93A). To our surprise, only the P4C4 plant (f3ha f3hb) exhibited a clearly modified, pale purplish pink flower color (RHS 69D), whereas the others, including the single gene knock-out plants, displayed purple flowers similar to the wild-type (WT) Petunia (Fig. 3c, d). In addition, we quantified the relative total anthocyanin content in flower petals from the WT, single gene mutant, and f3ha f3hb mutant plants at 0 and 1 days post anthesis (Fig. 3e). The absorbance values of the f3ha f3hb mutant flower petal extracts at 530 nm were significantly lower on both days than those of the WT and single gene mutant Petunia petal extracts. This result indicates that F3H function was completely knocked out in the mutant Petunia, thereby reducing flower petal anthocyanin levels.
To confirm inheritability of the mutations at f3ha and f3hb genes, those in vitro propagated P4C4 cuttings (T0) were self-pollinated to obtain T1 seeds using a bud pollination method to overcome their self-incompatibility. As appeared in Supplementary Fig. 6, all the germinated T1 plants displayed the same flower colors as pale purplish pink flower in P4C4 line, indicating the stable inheritance of the mutant trait.
Discussion
Following to our previous study reporting a protocol for gene knock-out in Petunia protoplasts via Cas9-RNP delivery (Subburaj et al. 2016b), we here performed site-directed mutagenesis to alter two F3H genes in Petunia using direct delivery of DNA-free Cas9-RNPs. Recent studies showed Agrobacterium tumefaciens-mediated plant transformation methods in Petunia (Xu et al. 2020; Zhang et al. 2016) but, to the best of our knowledge, DNA-free mutagenesis of Petunia in whole procedures including gene knock-out and regeneration is demonstrated in this study for the first time. By targeting the two F3H genes with one guide RNA, in one step we obtained total eight mutant plants (11.9%) in 67 regenerated T0 plants without any selection processes such as resistance genes. Among the eight mutants, one mutant line (f3ha f3hb) ultimately contained homozygous mutations in both F3H genes, whereas the others contained mutations in either F3HA or F3HB gene. Notably, only the f3ha f3hb exhibited a clearly modified, pale purplish pink flower color (RHS 69D), whereas the others displayed purple violet (RHS 93A) flowers similar to the wild-type Petunia, indicating that complete knock-out of F3H genes is necessary for flower color modifications. It is well known that F3H is a key enzyme of anthocyanin biosynthesis pathway and that the suppression of F3H gene could induce white and pale shades colors of Petunia flower (Tsuda et al. 2004).
Previously, A. tumefaciens-mediated F3H gene mutations in the model plant torenia (Torenia fournieri L.) generated various flower color phenotypes (Nishihara et al. 2018), which were mainly caused by the chimeric (mosaic) genotypes. It is known that the Agrobacterium tumefaciens-mediated delivery of CRISPR-Cas9 nucleases enable continuous expression of CRISPR complexes, resulting in various chimeric genotypes in tissues or organisms. In this study, however, we adopted a Cas9 RNP-mediated transformation strategy in Petunia and did not any observe chimeric genotypes in each regenerated T0 plant from NGS outcomes, similar to the previous studies (Woo et al. 2015) using the Cas9 RNP-mediated transformation strategy. As described above, it is supposed that Cas9 RNPs cleave target DNAs immediately after transfection and are rapidly degraded in protoplasts, dominantly generating homozygous or heterozygous genotypes, rather than the chimeric genotypes in each plant.
As seen in this study, orthologous or paralogous genes, which occur widely in plant speciation, should be investigated when the CRISPR-Cas system is applied to target-specific gene editing. Furthermore, our method is a feasible means of creating novel traits in Petunia, which can potentially be exempted from GMO regulations because of the absence of any foreign DNA (Waltz 2016); thus, we expect that more extensive applications in ornamental plant breeding will be forthcoming.
Author contribution statement
S.B. and G.J.L. conceived this project; T.L., S.S., and J.Y. performed the experiments. J.Y. performed bioinformatics analyses. J.Y., S.S., G.J.L., and S.B. wrote the manuscript with comments of all other authors. S.B. and G.J.L. supervised the research.
References
Andersson M, Turesson H, Olsson N, Fält A-S, Ohlsson P, Gonzalez MN, Samuelsson M, Hofvander P (2018) Genome editing in potato via CRISPR-Cas9 ribonucleoprotein delivery. Physiol Plant 164:378–384
Bae S, Park J, Kim J-S (2014) Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 30:1473–1475
Baek K, Kim DH, Jeong J, Sim SJ, Melis A, Kim J-S, Jin E, Bae S (2016) DNA-free two-gene knockout in Chlamydomonas reinhardtii via CRISPR-Cas9 ribonucleoproteins. Sci Rep 6:30620
Bombarely A, Moser M, Amrad A, Bao M, Bapaume L, Barry CS, Bliek M, Boersma MR, Borghi L, Bruggmann R, Bucher M, D'Agostino N, Davies K, Druege U, Dudareva N, Egea-Cortines M, Delledonne M, Fernandez-Pozo N, Franken P, Grandont L, Heslop-Harrison JS, Hintzsche J, Johns M, Koes R, Lv X, Lyons E, Malla D, Martinoia E, Mattson NS, Morel P, Mueller LA, Muhlemann J, Nouri E, Passeri V, Pezzotti M, Qi Q, Reinhardt D, Rich M, Richert-Pöggeler KR, Robbins TP, Schatz MC, Schranz ME, Schuurink RC, Schwarzacher T, Spelt K, Tang H, Urbanus SL, Vandenbussche M, Vijverberg K, Villarino GH, Warner RM, Weiss J, Yue Z, Zethof J, Quattrocchio F, Sims TL, Kuhlemeier C (2016) Insight into the evolution of the Solanaceae from the parental genomes of Petunia hybrida. Nature Plants 2:16074
Brooks C, Nekrasov V, Lippman ZB, Van Eck J (2014) Efficient gene editing in tomato in the first generation using the clustered regularly interspaced short palindromic repeats/CRISPR-Associated9 system. Plant Physiol 166:1292–1297
Clark KA, Krysan PJ (2010) Chromosomal translocations are a common phenomenon in Arabidopsis thaliana T-DNA insertion lines. Plant J 64:990–1001
Feng Z, Mao Y, Xu N, Zhang B, Wei P, Yang D-L, Wang Z, Zhang Z, Zheng R, Yang L, Zeng L, Liu X, Zhu J-K (2014) Multigeneration analysis reveals the inheritance, specificity, and patterns of CRISPR/Cas-induced gene modifications in Arabidopsis. Proc Natl Acad Sci 111:4632–4637
Jupe F, Rivkin AC, Michael TP, Zander M, Motley ST, Sandoval JP, Slotkin RK, Chen H, Castanon R, Nery JR, Ecker JR (2019) The complex architecture and epigenomic impact of plant T-DNA insertions. PLoS Genet 15:e1007819
Kim S, Kim D, Cho SW, Kim J, Kim J-S (2014) Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res 24:1012–1019
Lin C-S, Hsu C-T, Yang L-H, Lee L-Y, Fu J-Y, Cheng Q-W, Wu F-H, Hsiao HC-W, Zhang Y, Zhang R, Chang W-J, Yu C-T, Wang W, Liao L-J, Gelvin SB, Shih M-C (2018) Application of protoplast technology to CRISPR/Cas9 mutagenesis: from single-cell mutation detection to mutant plant regeneration. Plant Biotechnol J 16:1295–1310
Ma X, Zhu Q, Chen Y, Liu Y-G (2016) CRISPR/Cas9 platforms for genome editing in plants: developments and applications. Mol Plant 9:961–974
Malnoy M, Viola R, Jung M-H, Koo O-J, Kim S, Kim J-S, Velasco R, Nagamangala Kanchiswamy C (2016) DNA-free genetically edited grapevine and apple protoplast using CRISPR/Cas9 ribonucleoproteins. Front Plant Sci 7:1904
Nacry P, Camilleri C, Courtial B, Caboche M, Bouchez D (1998) Major chromosomal rearrangements induced by T-DNA transformation in Arabidopsis. Genetics 149:641–650
Nishihara M, Higuchi A, Watanabe A, Tasaki K (2018) Application of the CRISPR/Cas9 system for modification of flower color in Torenia fournieri. BMC Plant Biol 18:331
Pan C, Ye L, Qin L, Liu X, He Y, Wang J, Chen L, Lu G (2016) CRISPR/Cas9-mediated efficient and heritable targeted mutagenesis in tomato plants in the first and later generations. Sci Rep 6:24765
Park J, Bae S, Kim J-S (2015) Cas-Designer: a web-based tool for choice of CRISPR-Cas9 target sites. Bioinformatics 31:4014–4016
Park J, Bae S, Lim K, Kim J-S (2016) Cas-analyzer: an online tool for assessing genome editing results using NGS data. Bioinformatics 33:286–288
Stehmann JR, Lorenz-Lemke AP, Freitas LB, Semir J (2009) The genus Petunia. In: Gerats T, Strommer J (eds) Petunia: evolutionary, developmental and physiological genetics. Springer, New York, pp 1–28
Subburaj S, Tu L, Jin Y-T, Bae S, Seo PJ, Jung YJ, Lee G-J (2016a) Targeted genome editing, an alternative tool for trait improvement in horticultural crops. Hortic Environ Biotechnol 57:531–543
Subburaj S, Chung SJ, Lee C, Ryu SM, Kim DH, Kim JS, Bae S, Lee GJ (2016b) Site-directed mutagenesis in Petunia x hybrida protoplast system using direct delivery of purified recombinant Cas9 ribonucleoproteins. Plant Cell Rep 35:1535–1544
Suzuki K-i, Xue H-m, Tanaka Y, Fukui Y, Fukuchi-Mizutani M, Murakami Y, Katsumoto Y, Tsuda S, Kusumi T (2000) Flower color modifications of Torenia hybrida by cosuppression of anthocyanin biosynthesis genes. Mol Breeding 6:239–246
Tan J, Wang M, Tu L, Nie Y, Lin Y, Zhang X, Zhang J (2013) The flavonoid pathway regulates the petal colors of cotton flower. PLoS ONE 8(8):e72364
Tsuda S, Fukui Y, Nakamura N, Katsumoto Y, Yonekura-Sakakibara K, Fukuchi-Mizutani M, Ohira K, Ueyama Y, Ohkawa H, Holton TA, Kusumi T, Tanaka Y (2004) Flower color modification of Petunia hybrida commercial varieties by metabolic engineering. Plant Biotechnol 21:377–386
Ueyama Y, Katsumoto Y, Fukui Y, Fukuchi-Mizutani M, Ohkawa H, Kusumi T, Iwashita T, Tanaka Y (2006) Molecular characterization of the flavonoid biosynthetic pathway and flower color modification of Nierembergia sp. Plant Biotechnol 23:19–24
Vandenbussche M, Chambrier P, Rodrigues Bento S, Morel P (2016) Petunia, your next supermodel? Front Plant Sci 7:72
Waltz E (2016) Gene-edited CRISPR mushroom escapes US regulation. Nature 532:293
Woo JW, Kim J, Kwon SI, Corvalán C, Cho SW, Kim H, Kim S-G, Kim S-T, Choe S, Kim J-S (2015) DNA-free genome editing in plants with preassembled CRISPR-Cas9 ribonucleoproteins. Nat Biotechnol 33:1162
Xu J, Kang B-C, Naing AH, Bae S-J, Kim J-S, Kim H, Kim CK (2020) CRISPR/Cas9-mediated editing of 1-aminocyclopropane-1-carboxylate oxidase1 enhances Petunia flower longevity. Plant Biotechnol J 18:287–297
Zhang H, Zhang J, Wei P, Zhang B, Gou F, Feng Z, Mao Y, Yang L, Zhang H, Xu N, Zhu J-K (2014) The CRISPR/Cas9 system produces specific and homozygous targeted gene editing in rice in one generation. Plant Biotechnol J 12:797–807
Zhang B, Yang X, Yang C, Li M, Guo Y (2016) Exploiting the CRISPR/Cas9 system for targeted genome mutagenesis in Petunia. Sci Rep 6:20315
Zhang Q, Xing H-L, Wang Z-P, Zhang H-Y, Yang F, Wang X-C, Chen Q-J (2018) Potential high-frequency off-target mutagenesis induced by CRISPR/Cas9 in Arabidopsis and its prevention. Plant Mol Biol 96:445–456
Zuker A, Tzfira T, Ben-Meir H, Ovadis M, Shklarman E, Itzhaki H, Forkmann G, Martens S, Neta-Sharir I, Weiss D, Vainstein A (2002) Modification of flower color and fragrance by antisense suppression of the flavanone 3-hydroxylase gene. Mol Breed 9:33–41
Acknowledgments
This work was supported by grants from the Next Generation BioGreen 21 Program (No. PJ01319301 to S.B. and No. PJ01319303 to G.J.L). J.Y. was also supported by the Stadelmann-Lee Scholarship Fund, Seoul National University, Seoul, Korea.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
G.J.L., T.L., and S.S. are co-inventors on a patent application related to the methods described in this manuscript.
Additional information
Communicated by Neal Stewart.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Yu, J., Tu, L., Subburaj, S. et al. Simultaneous targeting of duplicated genes in Petunia protoplasts for flower color modification via CRISPR-Cas9 ribonucleoproteins. Plant Cell Rep 40, 1037–1045 (2021). https://doi.org/10.1007/s00299-020-02593-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00299-020-02593-1