
Abstract
Key message
An RNAseq-based analysis of the cassava plants inoculated with Xam allowed the identification of transcriptional upregulation of genes involved in jasmonate metabolism, phenylpropanoid biosynthesis and putative targets for a TALE.
Abstract
Cassava bacterial blight, a disease caused by the gram-negative bacterium Xanthomonas axonopodis pv. manihotis (Xam), is a major limitation to cassava production worldwide and especially in developing countries. The molecular mechanisms underlying cassava susceptibility to Xam are currently unknown. To identify host genes and pathways leading to plant susceptibility, we analyzed the transcriptomic responses occurring in cassava plants challenged with either the non-pathogenic Xam strain ORST4, or strain ORST4(TALE1 Xam ) which is pathogenic due to the major virulence transcription activator like effector TALE1 Xam . Both strains triggered similar responses, i.e., induction of genes related to photosynthesis and phenylpropanoid biosynthesis, and repression of genes related to jasmonic acid signaling. Finally, to search for TALE1 Xam virulence targets, we scanned the list of cassava genes induced upon inoculation of ORST4(TALE1 Xam ) for candidates harboring a predicted TALE1 Xam effector binding element in their promoter. Among the six genes identified as potential candidate targets of TALE1 Xam a gene coding for a heat shock transcription factor stands out as the best candidate based on their induction in presence of TALE1 Xam and contain a sequence putatively recognized by TALE1 Xam .
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Gram-negative pathogenic bacteria inject effector proteins into the host plant cell via the type-3 secretion system (T3SS) to overcome or suppress plant defense responses (Chisholm et al. 2006), and to promote the establishment of a favorable environment during bacterial colonization (Deslandes and Rivas 2012). Transcription activator-like effectors (TALEs) form a particular family of effectors, which promote pathogen virulence by inducing the expression of host susceptibility genes upon binding to target boxes or effector binding elements (EBE) at defined promoter sites (Bogdanove et al. 2010). The protein structure of TALEs is well-conserved and includes a C-terminal region with nuclear localization signals (NLS) and an acidic activation domain (AAD), allowing them to act as bona fide eukaryotic transcription factors. TALE DNA-binding specificity is mediated by the central region of the protein (Boch et al. 2009; Moscou and Bogdanove 2009; Bogdanove et al. 2010; review by Muñoz-Bodnar et al. 2013), which is formed by a series of quasi-identical repeats of 33–34 amino acids. Amino acids 12th and 13th are highly variable among repeats and are referred to as repeat variable di-residues (RVDs). Different RVDs bind to different nucleotides in a one-to-one relationship, with amino acid 13th controlling recognition specificity (Boch et al. 2009; Moscou and Bogdanove 2009; Mak et al. 2012; Doyle et al. 2013). The RVD arrangement therefore determines the genes a TALE will be able to induce in a host genome.
A few targets of TALEs have been characterized so far, some of which act as susceptibility genes since their activation is necessary for bacterial growth and symptom development. The SWEET/MtN3 family of sugar transporters represents the best-studied example of this type (Chen et al. 2010). Transcriptional activation of SWEET genes by the vascular pathogen Xanthomonas oryzae pv. oryzae is hypothesized to cause the plant to export sugars into the xylem (Yang et al. 2006; Sugio et al. 2007; Antony et al. 2010; Yu et al. 2011; Streubel et al. 2013), thereby increasing sugar apoplastic concentrations and potentially favoring bacterial growth in planta (Chen et al. 2010). Another set of genes turned on by TALE are transcription factors, including a helix-loop-helix transcriptional activator causing hypertrophy in pepper cells (Kay et al. 2007), a bZIP transcription factor and a homolog of the general transcription factor TFIIA in rice (Sugio et al. 2007).
Cassava, Manihot esculenta Crantz, is a starchy root crop essential for food security in developing countries. Its importance is increasing due to its use for industrial starch production. Cassava production has increased by more than 40 % in the last 10 years and it is estimated that the land surface cultivated with cassava will increase up to 300 % over the next 15 years (Rosenthal and Ort 2012). The crop is threatened by several viral, bacterial and fungal diseases. Cassava bacterial blight (CBB) is a vascular disease of global distribution caused by Xanthomonas axonopodis pv. manihotis (Xam). It is the most important bacterial disease of cassava, causing losses of up to 90 % of production (Lozano 1986; López and Bernal 2012).
The most effective available strategy to control CBB is the deployment of resistant cassava cultivars (López and Bernal 2012). However, current resistant cultivars are not well-adapted to the different cassava production zones and often show undesirable agronomic traits (López and Bernal 2012). No major resistance gene has been identified so far for CBB, making the development of new resistant cultivars challenging. CBB resistance is quantitative and several QTL (quantitative trait loci) associated with resistance to different Xam strains have previously been reported (Jorge et al. 2000, 2001; Wydra et al. 2004).
Previous efforts to identify resistance and defense genes used resistance gene analogs (López et al. 2003) and transcriptomic strategies (Santaella et al. 2004; López et al. 2005). The use of microarrays showed that, upon stem inoculation, most of the global gene expression changes occur 7 or 15 days post-inoculation, suggesting a relatively late response as compared to other pathosystems (López et al. 2005). In addition, these studies showed that resistant cassava plants respond to Xam by the activation of genes associated with the oxidative burst, those encoding pathogenesis related (PR) proteins and protein degradation (López et al. 2005). The expression of some of these induced genes was validated by qRT-PCR. These experiments showed that the induction also occurred in a susceptible cultivar, but at a lower level or at later times post-infection. On the other hand, genes involved in photosynthesis and metabolism were down-regulated (López et al. 2005). However, so far no genome-wide transcriptomic studies have been conducted to gain insights into the molecular reprogramming of gene expression involved in early responses. Furthermore, little is known about the mechanisms underlying susceptibility of cassava to Xam.
Recently, we reported the characterization of a major virulence TALE in Xam, which plays an important role during bacterial growth and symptom development in planta (Castiblanco et al. 2013). TALE1 Xam (previously known as pthB) derives from strain CFBP1851 and was discovered upon the analysis of the non-pathogenic Xam strain ORST4 (CFBP1851∆p44 in Castiblanco et al. 2013), which fails to cause symptoms in plants despite of a functional T3SS and many conserved type three effectors, as well as another TALE of yet-unknown sequence (Bart et al. 2012). ORST4 contains an 8 kb deletion within plasmid p44, which includes TALE1 Xam . As shown by Castiblanco et al. (2013), introduction of a plasmid carrying TALE1 Xam back into ORST4 restores its ability to cause disease in susceptible cassava plants. TALEs with identical or similar RVD-sequences were also reported in other virulent Xam strains originating from Colombia and Brazil (Bart et al. 2012).
In this study, we took advantage of next-generation sequencing technologies to obtain a genome-wide picture of the pathogen-induced transcriptomic reprogramming upon infection with contrasting Xam strains ORST4 and ORST4(TALE1 Xam ). This study represents the first global transcriptomic analysis of cassava during a compatible interaction, notably highlighting the suppression of genes involved in jasmonic acid signaling.
Results
Pathogenic and non-pathogenic strains of Xam cause similar transcriptomic responses in cassava
RNA-seq libraries were generated from cassava tissues challenged with either Xam strain ORST4, which is unable to cause disease in susceptible plants, or ORST4 transformed with TALE1 Xam which recovers wild-type growth in planta and the ability to cause disease (Castiblanco et al. 2013). Eight-week-old cassava plants of the cultivar MCOL 1522 were stem-inoculated with each of the two strains. RNA was extracted from tissues collected at 0 (mock), 5 and 7 days post-inoculation (dpi). As already shown in previous studies (Restrepo et al. 2000; Jorge et al. 2000), stem inoculation has proven to best discern between resistant and susceptible genotypes of cassava, leading to visible disease symptom development as early as 5–7 dpi. At these time-points plants are mostly asymptomatic but there is extensive transcriptional reprogramming occurring (López et al. 2005). Two technical replicates were performed for each treatment and each library comprised a minimum of 2.7 million paired-end reads. These reads were mapped to the publicly available cassava genome (v. cassava4 from http://www.phytozome.net) and gene expression was calculated for the reference transcriptome using Tophat and Cufflinks from the Tuxedo suite (Trapnell et al. 2012). 64 % of the total reads were mapped to 27,937 reference cassava genes, which corresponds to 91 % of the annotated transcriptome. The data have been deposited in NCBI’s gene expression omnibus (Edgar et al. 2002) and are accessible through GEO series accession number GSE53369 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=mrkbmwsqxpejdajandacc=GSE53369).
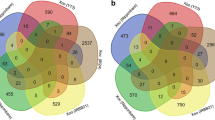
Gene expression values (FPKMs = Fragments Per Kilobase Of Exon Per Million Fragments Mapped) were then used to evaluate similarities among libraries using hierarchical clustering and principal component analysis (PCA). Interestingly, our analyses demonstrated a greater difference in gene expression across time points than between treatments at each time point (Fig. 1a). As shown in the PCA, only 0.9 % of the variation could be explained by component 4, which was the component that allowed association of genes to either one of the two treatments (Fig. 1b, c). As expected, virtually no variation was detected between the two control libraries (cassava stems, 0 days post-inoculation) (Fig. 1). Differential expression for cassava genes was then computed using NOISeqBIO (Tarazona et al. 2011), by comparing the libraries made from RNA extracted at 5 and 7 dpi, to the control libraries. This generated a set of 1,348 and 1,288 genes that were up- or down-regulated, respectively, in response to Xam strains ORST4 or ORST4(TALE1 Xam ) (supplementary Table 1). As shown in Fig. 2a, b, we observed some overlap between the cassava genes found to be induced or repressed upon inoculation with either strain. In total, 338 genes (54 %) that were up-regulated by Xam strain ORST4(TALE1 Xam ) were also induced by ORST4. Likewise, 338 (76 %) of the genes that were down-regulated by ORST4(TALE1 Xam ) were also down-regulated by ORST4.

Gene expression values (FPKMs) show similarities in transcriptomic responses of cassava to non pathogenic ORST4 and pathogenic ORST4(TALE1 Xam ) Xam strains. a Heat map showing Euclidean distances (the darker, the closer) between RNA-seq libraries. The dendrogram shows hierarchical clustering of the treatments; numbers to the left and right of each branch indicate, respectively, Bootstrap probabilities and approximately unbiased p values, obtained upon multi-scale bootstrap resampling using pvclust. b Scatter plot showing genes and treatments in the two main components of a PCA based on FPKM values. Differentially expressed (DE light dots) and non-differentially expressed (Not_DE dark dots) genes are highlighted as identified by NOISeqBIO analysis. c Scatter plot showing genes and treatments in the third and fourth components in a PCA based on FPKM values. Differentially expressed (DE light dots) and non-differentially expressed (Not_DE dark dots) genes are highlighted as identified by NOISeqBIO analysis

Similar processes are altered upon infection of Xam strains ORST4 and ORST4(TALE1 Xam ). Venn diagrams illustrating the overlap among differentially up-regulated (a) or down-regulated (b) genes in response to ORST4 and ORST4(TALE1 Xam ), when comparing each time-point to the control treatments (0 dpi). Heatmaps representing adjusted p-values for over-representation of functional terms among either up-regulated (c) or down-regulated (d) genes in response to Xam as calculated by hypergeometric tests with Benjamini–Hochberg–Yekutieli FDR correction
Both strains induce genes related to photosynthesis and phenylpropanoid biosynthesis and repress genes related to jasmonic acid signaling
We further analyzed the annotations of sets of differentially expressed genes to search for over-represented functional terms (supplementary Tables 2, 3). Interestingly, these analyses revealed that similar processes are induced or repressed in response to both treatments, albeit apparently responses being delayed in ORST4 (Fig. 2c, d). More precisely, the set of induced genes is over-represented with genes related to photosynthesis, gluconeogenesis and phenylpropanoid biosynthesis (Figs. 2c, 3a–c), which may be connected to the production of defense compounds. We also observed the induction of genes involved in the biosynthesis of suberin, a macromolecule composed of derivatives of the phenylpropanoid biosynthesis pathway and which can provide cell wall reinforcement (Fig. 2c; supplementary Tables 2, 3). In contrast, down-regulated genes included those involved in cellular glucan metabolic process (Fig. 3e), such as various xyloglucan endotransglycosylase, which are enzymes involved in cell-wall degradation. Interestingly, among the down-regulated genes, we also observed a pronounced representation of genes related to jasmonic acid biosynthesis (Fig. 3f). Also, since the conjugate JA-Ile has an active role in JA signaling (Katsir et al. 2008), we reasoned that JA down-regulation may also be related to the repression of the isoleucine metabolism observed in response to both strains (Fig. 3g). Furthermore, the down-regulation of genes related to plant-pathogen interactions may also be related to the suppression of JA signaling (Fig. 3h), since six out of the 15 down-regulated genes associated to this term correspond to different Jasmonate ZIM/TIFY domain proteins (JAZ).

Stacked bar plots showing the induction or repression of cassava genes upon inoculation of Xam strains ORST4 and ORST4(TALE1 Xam ), and their association with functional terms. a–d Groups of Xam-induced genes associated to overrepresented terms. E through H, groups of Xam-repressed genes associated to overrepresented functional terms. LogFC values were obtained from FPKM values for each gene at a given time point compared with the controls (0 dpi)
No functional term was found to be specifically enriched in response to only one of the strains at both post-inoculation times. However, some notable differences were observed. First, some processes seem to be induced earlier in response to TALE1 Xam . These include gluconeogenesis, responses to oxidative stress and phenylpropanoid biosynthesis (Figs. 2c, 3b, c). Second, some differences in the magnitude of changes in expression of genes were detected; for example, for genes associated to proteolysis, which seemed to be induced to a higher extent overall in response to TALE1 Xam (Fig. 3d). Whether or not these differences are biologically relevant remain to be investigated.
To further explore the transcriptional regulation of these genes, we grouped them according to their expression profiles and searched for motifs in their promoter regions that may be associated to their regulation. For this, all the differentially expressed genes were clustered according to the fold-change values in all comparisons using k-means (k = 10) (supplementary Fig. 1, supplementary Table 1). Subsequently, known transcription factor binding sites (TFBS, 64 motifs) were predicted in the promoter regions of these genes and hypergeometric tests were used to assess for an over-representation of these TFBS within each cluster when compared to the promoters of all genes in cassava (supplementary Table 4). As a result, among Xam down-regulated clusters (Cluster 1, 2 and 8), we found overrepresentation of light responsive elements (G-box) and elements related to abscisic acid signaling (ABRE). These motifs are in fact very similar and may be related to the same process [ABRE = (C/T)ACGTGGC, G-box = CACGTG] (Yilmaz et al. 2011). This may suggest that many of the down-regulated genes in response to both strains may be regulated by only a few transcription factors. Overrepresentation of TFBS was also assessed for groups associated to specific functional terms (supplementary Table 4). As result of this analysis, an over-representation of MYB binding sites in the promoter region of Xam-induced genes related to phenylpropanoid biosynthesis was observed (supplementary Table 4).
Candidate virulence targets of TALE1Xam
We have previously shown that TALE1 Xam is sufficient to cause disease in cassava, probably by inducing a set of so far unknown susceptibility gene(s) (Castiblanco et al. 2013). To search for such TALE1 Xam -responsive genes in our transcriptomic data, we focused on all genes induced by ORST4(TALE1 Xam ), as compared to inoculation with strain ORST4 at any time-point. This resulted in 206 genes, mostly from clusters 5 and 6 (supplementary Fig. 1). We next performed qRT-PCR analysis to evaluate the expression profiles of three of these TALE1 Xam -upregulated gene candidates (supplementary Fig. 2). This enabled to validate the RNA-seq data confirming the TALE1 Xam -associated up-regulation of these genes in cassava. Because TALE binding elements are now relatively easily predictable by computational means, we ran the two complementary programs Talvez (Pérez-Quintero et al. 2013) and Talgetter (Grau et al. 2013), in order to predict potential TALE1 Xam binding sites in each cassava promoter. When cross-referencing the top 200 predicted potential targets with the gene expression profiles obtained from the RNA-seq experiments, one gene (cassava 4.1_014976m.g, cluster 5) was found both to have a strong TALE1 Xam binding box candidate in its promoter (Fig. 4a) and to be up-regulated in a TALE1 Xam -dependent manner (significantly induced when comparing the ORST4(TALE1 Xam ) libraries to either control or ORST4) (Fig. 4b, c). In fact, cassava4.1_014976m.g was, respectively, ranked at position 1 and 9, with Talvez, and Talgetter, and was found among the top 100 genes induced upon inoculation of Xam strain ORST4(TALE1 Xam ) vs. ORST4, thus satisfying previously-established criteria for known TALE targets (Pérez-Quintero et al. 2013). The gene is annotated as encoding a heat shock transcription factor B3 (HsfB3). Other potential targets were also found in this analysis, mainly five genes were found that contained predicted binding sites for TALE1Xam (albeit with lower scores than cassava4.1_014976m.g) and were differentially expressed in at least one relevant comparison, however none of these genes matched the expected profile for a TALE1Xam target, i.e., they were also induced by ORST4 or were only induced by ORST4(TALE1 Xam ) at one time point (supplementary Table 5).

HsfB3 is a possible target for TALE1 Xam . a Representation of the positional weight matrix used by Talvez to screen for TALE1 Xam binding sites, binding probabilities for each RVD-nucleotide pair are shown from lighter (less-preferred) to darker (preferred). Below the matrix, the predicted binding sequences in the promoter regions of HsfB3 are shown, suboptimal matching nucleotides in the promoter regions are underlined b FPKM values for HsfB3 in RNA-seq libraries. c Relative expression of HsfB3 obtained by qRT-PCR, values correspond to (2-ΔCt 0dpi)/(2-ΔCt X dpi), where ΔCt = CtHsfB3a-Ct18S. D) RNA-seq reads mapped to the HsfB3 genomic region, mapped reads from the different timepoints were pooled together, scale represents raw number of reads. Bottom of the graph represents the coding sequence of HsfB3 (2 exons, 1 intron) and the arrow highlights the position of the predicted TALE1 Xam binding site
Discussion
In this work we employed a transcriptomic analysis based on RNAseq with two aims: to gain insights into transcriptomic responses in cassava to both pathogenic and non-pathogenic strains of Xam and to identify potential candidate targets for the virulence factor TALE1 Xam . For our first aim, we compared the transcriptome of cassava plants inoculated with pathogenic vs. non-pathogenic Xam strains at 5 and 7 dpi, obtaining important changes in the expression of genes related to photosynthesis, gluconeogenesis, the biosynthesis of phenylpropanoids and JA biosynthesis and signaling. For our second aim, we detected changes in the transcriptome specifically associated to the effector TALE1 Xam and found potential virulence targets for this effector, the validation of which awaits further functional experimentation.
We observed changes in the transcriptome at 5 and 7 days post-inoculation, with more than 1,000 genes identified as induced or repressed at either time point. We expect these changes to be accentuated at later times post-inoculation, as has been previously reported (López et al. 2005). There was some overlap in cassava responses after inoculation with pathogenic and non-pathogenic Xam strains (approximately 54 and 76 % induced and suppressed genes were shared, respectively). This suggests that early responses (triggered by unknown recognition signals) are activated under both conditions. This result is surprising given how different the symptoms are in response to these strains (Castiblanco et al. 2013). This may indicate that the differences in disease development are related to the activation/repression of only a few genes, for example HsFB3 and its targets. Alternatively, differences in disease development between the two strains can be explained by the subtle attenuation or strengthening of the early responses. Other studies have reported large overlaps between compatible, incompatible and non-host responses (Tao et al. 2003), as well as between effector triggered-immunity and PAMP triggered-immunity (Navarro et al. 2004), which has prompted suggestions of conceiving plant responses to bacteria as a continuum with differences in amplitude (robust and weak responses) that determine the outcome of the interaction (susceptibility or resistance) (Abramovitch et al. 2006; Thomma et al. 2011).
The responses observed in cassava under the two conditions included various processes possibly related to known defense responses, particularly responses against vascular pathogens (Yadeta and Thomma 2013). There was a marked induction in biosynthesis of aromatic amino acid derivatives such as flavonoids and phenylpropanoids as well as genes involved in suberin biosynthesis (Fig. 2c). The induction of these genes can be correlated with previous immuno-histological studies showing that phenolic compound production and suberin deposition increase in cassava plants inoculated with Xam (Kpémoua et al. 1996). These responses were observed in resistant and susceptible cassava cultivars indicating that they are common early responses but that they were faster and more intense in the resistant cultivars (Kpémoua et al. 1996). This induction of phenolic compounds has been observed in many other pathosystems and may encompass the production of metabolites with direct antibacterial activity (Maddox et al. 2010), or the reinforcement of cell walls to prevent the spread of bacteria (Yadeta and Thomma 2013). Other changes observed in our results and associated to cell wall reinforcement included the induction of peroxidases (GO: response to oxidative stress) (Hilaire et al. 2001) and the down-regulation of xyloglucan endotransglycosylases (GO: glucan cellular metabolic proccess) (Hématy et al. 2009).
Another major transcriptomic change observed in response to both strains was the general repression of genes related to jasmonic acid biosynthesis and signaling. JA is known to be involved in several physiological responses in plants including biotic and abiotic stress (Wasternack and Hause 2013). It is traditionally believed that JA is the main signaling molecule in defense against necrotrophic pathogens, while SA signaling is involved in defense against biotrophic pathogens (Glazebrook 2005) and that these two hormones are mutually antagonistic to each other (Glazebrook 2005; Pieterse et al. 2009). Although this has not been proven in cassava, it could indicate that JA-signaling repression in response to Xam could lead to an SA-mediated defense even though a clear effect on transcriptomic regulation of the SA pathway was not detected in our data. However, the great extent of cross-talk between hormone signaling pathways (Kunkel and Brooks 2002; Robert-Seilaniantz et al. 2011), as well as the intricate feedback regulation mechanism of the jasmonic acid pathway (Katsir et al. 2008; Staswick 2008) makes it difficult to accurately predict the effect that JA down-regulation would have on the Xam-cassava interaction. A notable observation from our transcriptome analysis was the over-representation of binding sites for the MYB transcription factor in the promoter regions of Xam-induced genes related to phenylpropanoid biosynthesis (supplementary Table 4). This may be a link between the down-regulation of JAZ1 genes and the phenylpropanoid pathway, given that it is hypothesized that the degradation of JAZ proteins abolishes the interactions of JAZ proteins with bHLH and MYB factors, allowing the transcriptional function of WD-repeat/bHLH/MYB complexes (Qi et al. 2011).
Some differences were observed when comparing our findings with previous transcriptomic analysis of the cassava-Xam interaction (López et al. 2005). For example, in the present work we found an overall induction of photosynthesis-related genes in response to both strains, in contrast to the previous finding of photosynthesis being shutdown in response to Xam (López et al. 2005). This could be explained by the differences in the systems studied. In Lopez et al., a cassava variety resistant to various Xam strains (MBRA685) was confronted to a highly virulent strain (CIO151), and the response was similar to “classical” R-gene-mediated responses (López et al. 2005; Zhu et al. 2012). On the other hand, in the present study, a susceptible variety (MCOL1522) was confronted with a non-pathogenic (ORST4) and a pathogenic strain [ORST4(TALE1 Xam )]. It is then possible that the more susceptible variety activates a weaker or later response to the bacterium.
To counter these above described early responses, pathogenic bacteria translocate effector proteins via the T3SS. In our case, TALE1 Xam manages to overcome plant defenses probably by inducing gene expression changes that will favor colonization causing the observed CBB symptoms (Castiblanco et al. 2013). The RNA-seq analysis revealed that a relatively small set of genes appears to be differentially expressed specifically in response to TALE1 Xam . A similar behavior can be found when analyzing available data in similar systems: In the rice-Xanthomonas oryzae pv. oryzae (Xoo) interaction, fewer genes are found as differentially expressed when comparing plants inoculated with a wild-type (wt) Xoo strain and a Xoo mutant for a single TALE, than when the comparison is made between wt-inoculated and mock-inoculated plants, suggesting that individual contributions of TALEs to transcriptomic changes are small (derived for Plexdb analyses, data not shown). This is true for the effectors PthXo1 from Xoo strain PXO99A and for TalC from Xoo strain BAI3 [Experiments GSE36272 and GSE19844, MAS5 normalization, logFC >4, treatments compared in PlexDB (Dash et al. 2012)]. This is not surprising given that TALEs in most cases appear to directly target only one or a few genes (Pérez-Quintero et al. 2013).
When comparing the expression data to predictions of TALE binding sites we found an interesting virulence target candidate: HsfB3. This heat shock transcription factor satisfied criteria corresponding to known targets: it displays the highest score when screening the genome for binding sites and is among the highest induced genes in presence of TALE1 Xam (Pérez-Quintero et al. 2013). HsfB3 encodes a transcription factor that likely acts as a transcriptional repressor by binding to heat shock elements in the upstream region of genes (Scharf et al. 2012). Some transcription factors have been previously found as targets for TALEs (Sugio et al. 2007), although none with possible repression activity. It is still unclear how their induction contributes to virulence. It is possible that transcription factors constitute network interaction hubs in the plant, which are suggested to be targeted by bacterial effectors (Mukhtar et al. 2011). Little is also known about the function of the HSFB3 family in plants. They are differentially expressed in response to biotic and abiotic stress (Miller and Mittler 2006; Swindell et al. 2007; Scharf et al. 2012; Soares-Cavalcanti et al. 2012). A recent study reported an Hsf-like protein as a molecular switch between development and defense in Arabidopsis (Pajerowska-mukhtar et al. 2012). Future work will focus on characterizing the contribution of HSFB3 and/or other TALE1 Xam target candidates on the cassava susceptibility to Xam.
Altogether our results point to an early defense mechanism in cassava that relies on the disruption of JA signaling and the production of phenylpropanoid derivatives. This defense seems to be still active in response to a pathogenic strain containing the effector TALE1 Xam , which suggests that the effector manages to promote bacterial growth by activating susceptibility genes and not by suppressing defense responses.
Materials and methods
Plant material
Cassava plants from cultivar MCOL1522 (susceptible to Xam) were obtained from the CIAT germplasm collection. Plants were propagated, multiplied and maintained under greenhouse conditions (day/night temperature 28/19 °C, photoperiod 12 h, and relative humidity = 80 %). 8-week-old plants were used for all assays.
Bacterial strains
Xam strain ORST4 [CFBP1851Δp44 in (Castiblanco et al. 2013)] is a non-pathogenic mutant lacking TALE1 Xam (Castiblanco et al. 2013). For functional studies this strain was previously complemented with TALE1 Xam [ORST4(TALE1 Xam ) in this study] which is expressed from the npt2 promoter from the plasmid pBAV226 (Castiblanco et al. 2013; Vinatzer et al. 2006). Both strains were streaked onto YPGA medium (5 % yeast extract, 5 % peptone, 5 % glucose and 15 % agar) and incubated at 28 °C for growth.
Stem inoculation
A single colony was grown in liquid YPG medium and incubated at 28 °C for 18 h. Inocula were adjusted to an OD600nm of 0.2, corresponding to about 108 colony forming units. Stems were inoculated by puncturing with a sterile toothpick at the third internode from the apical region. 10 ml of adjusted inoculum were loaded onto the puncture. Two biological replicates were performed.
RNA extraction
Three centimeters (2 cms above and one below the inoculation point) were collected at 0, 5, and 7 days post inoculation (dpi). Collected samples were immediately submerged in liquid nitrogen and then stored at −80 °C. 100 mg of ground tissue were incubated in lysis buffer (2 % beta-mercaptoethanol, 2 % PVP, 1 % SDS, 25 mM EDTA, 100 mM NaCl and 100 mM tris HCl pH 7.5). RNA extraction was carried out with acid phenol mixed with chloroform and isoamyl alcohol (25:24:1) and precipitated using 5 M NaCl and ethanol. Samples were treated with DNAse I following manufacturer’s instructions (Ambion). The DNA-free total RNA was then visualized by agarose gel electrophoresis and quantified using a NanoDrop (thermo scientific).
Construction and sequencing of RNAseq libraries
Protocol for library construction was adapted from (Zhong et al. 2011). Six RNAseq libraries were constructed from 7 µg of DNA-free mRNA purified by oligo dT25 dynabeads (invitrogen). cDNA was synthesized by employing Superscript III Reverse Transcriptase (Life Technologies). cDNA quality and quantity were assessed using an Agilent Bioanalyzer on a high sensitivity DNA chip (Agilent Technologies). Equal amounts (20–50 ng) of the six indexed libraries were pooled and sequenced together in two lanes of an Illumina flow cell to give 100 + 100 paired-end reads.
Bioinformatics and statistical analyses
Quality of the libraries was verified using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/, V0.10.1), reads with more than 80 % of bases with Phred quality values lower than 20 were excluded. Reads were mapped against the cassava genome using tophat v2.0.6 (default parameters) (Trapnell et al. 2012) and FPKM (fragments per kilobase of exon per million fragments mapped) values for annotated cassava genes were obtained with cufflinks v2.0.2 (Trapnell et al. 2012). Differentially expressed genes were identified by pairwise comparisons of treatments (i.e., ORST4_5dpi vs. ORST4_0dpi) taking into account both technical replicates using NOISeqBIO 2.6.0 (norm = “n”, factor = “time”, lc = 1, r = 50, adj = 1.5, a0per = 0.9, q = 0.9995, θ > 4) (Tarazona et al. 2011).
FPKM values were used to calculate distances (dist, method = euclidean) and perform hierarchical clustering (pvclust, nboot = 10,000) (Suzuki and Shimodaira 2006), as well as for principal component analyses (prcomp, default parameters) in R. Analysis of overrepresentation of GO terms were performed using AgriGO v1.2 (hypergeometric test with Benjamini–Hochberg–Yekutieli p value correction) (Du et al. 2010) using available GO annotations for the last version of the cassava genome (Prochnik et al. 2012). For overrepresentation of KEGG and BioCyc pathways cassava genes were used in Blastx analysis against genes of Arabidopsis thaliana using blastx 2.2.25+ (E value = 0.01, -best_hit_overhang = 0.5, -max_target_seqs = 2) and the A. thaliana annotations were used in Kobas 2.0 (hypergeometric test with Benjamini–Hochberg–Yekutieli p value correction) (Xie et al. 2011).
Genes were clustered according to Log2Fold-change values in all relevant comparisons using k-means (centers = 10, nstart = 100), the number of centers (k) was determined using the “elbow” method by looking at the percentage of the variance explained and the within cluster sum of squares against different K’s.
Binding sites for transcription factors were predicted using plantTFBS v1.0 (default parameters) (Megraw and Hatzigeorgiou 2010) using the 1 kb region upstream the translation start site of annotated genes in cassava. Hypergeometric tests with Benjamini–Hochberg–Yekutieli p value correction were made in R to assess overrepresentation (phyper with default parameters, p. adjust method = BY).
Binding sites for TALE1 Xam and other TALEs were predicted with Talvez v3.1 (default parameters) (Pérez-Quintero et al. 2013) and Talgetter (default parameters) (Grau et al. 2013) using the 1 kb region upstream the translation start site of annotated genes in cassava. The top 200 predicted targets with best score were kept for analyses.
qRT-PCR
PCR mixtures were prepared using Full VelocityR SYBRR Green QPCR Master Mix (Stratagene). PCR was performed on an Mx3005P thermal cycler (Stratagene), with the following cycling program: 95 °C for 5 min, followed by 40 cycles of 10 s at 95 °C and 30 s at 58 °C. Analysis was performed using the delta–delta Ct method (Pfaffl 2001). The gene expression levels obtained by qRT-PCR were normalized, using the Ct obtained for the constitutive 18S RNA. All experiments were performed in triplicate. Primers for qRT-PCR are listed on supplementary Table 6.
References
Abramovitch RB, Anderson JC, Martin GB (2006) Bacterial elicitation and evasion of plant innate immunity. Nat Rev Mol Cell Biol 7:601–611. doi:10.1038/nrm1984
Antony G, Zhou J, Huang S, Li T, Liu B, White F, Yang B (2010) Rice xa13 recessive resistance to bacterial blight is defeated by induction of the disease susceptibility gene Os-11N3. Plant cell 22:3864–3876. doi:10.1105/tpc.110.078964
Bart R, Cohn M, Kassen A, McCallum EJ, Shybut M, Petriello A, Krasileva K, Dahlbeck D, Medina C, Alicai T, Kumar L, Moreira LM, Rodrigues Neto J, Verdier V, Santana MA, Kositcharoenkul N, Vanderschuren H, Gruissem W, Bernal A, Staskawicz BJ (2012) High-throughput genomic sequencing of cassava bacterial blight strains identifies conserved effectors to target for durable resistance. Proc Natl Acad Sci USA 109(28):E1972–E1979. doi:10.1073/pnas.1208003109
Boch J, Scholze H, Schornack S, Landgraf A, Hahn S, Kay S, Lahaye T, Nickstadt A, Bonas U (2009) Breaking the code of DNA binding specificity of TAL-type III effectors. Science 326:1509–1512. doi:10.1126/science.1178811
Bogdanove AJ, Schornack S, Lahaye T (2010) TAL effectors: finding plant genes for disease and defense. Curr Opin Plant Biol 13:394–401. doi:10.1016/j.pbi.2010.04.010
Castiblanco LF, Gil J, Rojas A, Osorio D, Perez Quintero AL, Koebnik R, Szurek B, López C, Restrepo S, Verdier V, Bernal AJ (2013) TALE1 from Xanthomonas axonopodis pv. manihotis acts as a transcriptional activator in plant cells and is important for pathogenicity in cassava plants. Mol Plant Pathol 14:84–95. doi:10.1016/j.pbi.2010.04.010
Chen LQ, Hou B, Lalonde S, Takanaga H, Hartung ML, Qu X, Guo W, Kim JG, Underwood W, Chaudhuri B, Chermak D, Antony G, White FF, Somerville SC, Mudgett MB, Frommer WB (2010) Sugar transporters for intercellular exchange and nutrition of pathogens. Nature 468:527–532. doi:10.1038/nature09606
Chisholm ST, Coaker G, Day B, Staskawicz BJ (2006) Host-microbe interactions: shaping the evolution of the plant immune response. Cell 124:803–814. doi:10.1016/j.cell.2006.02.008
Dash S, Van Hemert J, Hong L, Wise RP, Dickerson JA (2012) PLEXdb: gene expression resources for plants and plant pathogens. Nucleic Acids Res 40:D1194–D1201. doi:10.1093/nar/gkr938
Deslandes L, Rivas S (2012) Catch me if you can: bacterial effectors and plant targets. Trends Plant Sci 17:644–655. doi:10.1016/j.tplants.2012.06.011
Doyle EL, Stoddard BL, Voytas DF, Bogdanove AJ (2013) TAL effectors: highly adaptable phytobacterial virulence factors and readily engineered DNA-targeting proteins. Trends Cell Bio 23:390–398. doi:10.1016/j.tcb.2013.04.003
Du Z, Zhou X, Ling Y, Zhang Z, Su Z (2010) agriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Res 38:W64–W70. doi:10.1093/nar/gkq310
Edgar R, Domrachev M, Lash AE (2002) Gene expression omnibus: nCBI gene expression and hybridization array data repository. Nucleic Acids Res 30:207–210. doi:10.1093/nar/30.1.20
Glazebrook J (2005) Contrasting mechanisms of defense against biotrophic and necrotrophic pathogens. Annu Rev of Phytopathol 43:205–227. doi:10.1146/annurev.phyto.43.040204.135923
Grau J, Wolf A, Reschke M, Bonas U, Posch S, Boch J (2013) Computational predictions provide insights into the biology of TAL effector target sites. PLoS Comput Biol 9:e1002962. doi:10.1371/journal.pcbi.1002962
Hématy K, Cherk C, Somerville S (2009) Host-pathogen warfare at the plant cell wall. Curr Opin Plant Biol 12:406–413. doi:10.1016/j.pbi.2009.06.007
Hilaire E, Young SA, Willard LH, McGee JD, Sweat T, Chittoor JM, Guikema JA, Leach JE (2001) Vascular defense responses in rice: peroxidase accumulation in xylem parenchyma cells and xylem wall thickening. Mol Plant Microbe Interact 14:1411–1419. doi:10.1094/MPMI.2001.14.12.1411
Jorge V, Fregene M, Duque MC, Bonierbale MW, Tohme J, Verdier V (2000) Genetic mapping of resistance to bacterial blight disease in cassava (Manihot esculenta Crantz). Theor Appl Genet 101:865–872. doi:10.1007/s001220051554
Jorge V, Fregene M, Vélez CM, Tohme DJ, Verdier V (2001) QTL analysis of field resistance to Xanthomonas axonopodis pv. manihotis in cassava. Theor Appl Genet 102:564–571. doi:10.1007/s001220051683
Katsir L, Schilmiller AL, Staswick PE, He SY, Howe GA (2008) COI1 is a critical component of a receptor for jasmonate and the bacterial virulence factor coronatine. Proc Natl Acad Sci USA 105:7100–7105. doi:10.1073/pnas.0802332105
Kay S, Hahn S, Marois E, Hause G, Bonas U (2007) A bacterial effector acts as a plant transcription factor and induces a cell size regulator. Science 318:648–651. doi:10.1126/science.1144956
Kpémoua K, Boher B, Nicole M, Calatayund P, Geiger JP (1996) Cytochemistry of defense responses in cassava infected by Xanthomonas campestris pv. manihotis. Can J of Microbiol 42:1131–1143. doi:10.1139/m96-145
Kunkel BN, Brooks DM (2002) Cross talk between signaling pathways in pathogen defense. Curr Opin Plant Biol 5:325–331. doi:10.1016/S1369-5266(02)00275-3
López CE, Bernal AJ (2012) Cassava bacterial blight: using genomics for the elucidation and management of an old problem. Trop Plant Biol 5:117–126. doi:10.1007/s12042-011-9092-3
López CE, Acosta IF, Jara C, Pedraza F, Gaitán-Solís E, Gallego G, Beebe S, Tohme J (2003) Identifying resistance gene analogs associated with resistances to different pathogens in common bean. Phytopathology 93:88–95. doi:10.1094/PHYTO.2003.93.1.88
López C, Soto M, Restrepo S, Piégu B, Cooke R, Delseny M, Tohme J, Verdier V (2005) Gene expression profile in response to Xanthomonas axonopodis pv. manihotis infection in cassava using a cDNA microarray. Plant Mol Biol 57:393–410. doi:10.1007/s11103-004-7819-3
Lozano JC (1986) Cassava bacterial blight: a manageable disease. Plant Dis 70:1089–1093. doi:10.1094/PD-70-1089
Maddox CE, Laur LM, Tian L (2010) Antibacterial activity of phenolic compounds against the phytopathogen Xylella fastidiosa. Curr Microbiol 60:53–58. doi:10.1007/s00284-009-9501-0
Mak ANS, Bradley P, Cernadas R, Bogdanove AJ, Stoddard BL (2012) The crystal structure of TAL effector PthXo1 bound to its DNA target. Science 335:716–719. doi:10.1126/science.1216211
Megraw M, Hatzigeorgiou AG (2010) MicroRNA promoter analysis. Methods Mol Biol 592:149–161. doi:10.1007/978-1-60327-005-2_11
Miller G, Mittler R (2006) Could heat shock transcription factors function as hydrogen peroxide sensors in plants? Ann Bot 98:279–288. doi:10.1093/aob/mcl107
Moscou MJ, Bogdanove AJ (2009) A simple cipher governs DNA recognition by TAL effectors. Science 326:1501. doi:10.1126/science.1178817
Mukhtar MS, Carvunis AR, Dreze M, Epple P, Steinbrenner J, Moore J, Tasan M, Galli M, Hao T, Nishimura MT, Pevzner SJ, Donovan SE, Ghamsari L, Santhanam B, Romero V, Poulin MM, Gebreab F, Gutierrez BJ, Tam S, Monachello D, Boxem M, Harbort CJ, McDonald N, Gai L, Chen H, He Y, Vandenhaute J, Roth FP, Hill DE, Ecker JR, Vidal M, Beynon J, Braun P, Dangl JL (2011) Independently evolved virulence effectors converge onto hubs in a plant immune system network. Science 333:596–601. doi:10.1126/science.1203659
Muñoz-Bodnar A, Bernal A, Szurek B, López CE (2013) Tell me a tale of TALEs. Mol Biotechnol 53:228–235. doi:10.1007/s12033-012-9619-3
Navarro L, Zipfel C, Rowland O, Keller I, Robatzek S, Boller T, Jones JDG (2004) The transcriptional innate immune Rresponse to flg22. Interplay and overlap with Avr gene-dependent defense responses and bacterial pathogenesis. Plant Physiol 135:1113–1128. doi:10.1104/pp.103.036749.1
Pajerowska-mukhtar KM, Wang W, Tada Y, Oka N, Tucker CL, Fonseca JP, Dong X (2012) The HSF-like transcription factor TBF1 is a major molecular switch for plant growth-to-defense transition. Curr Biol 22:103–112. doi:10.1016/j.cub.2011.12.015
Pérez-Quintero AL, Rodriguez-R LM, Dereeper A, López C, Koebnik R, Szurek B, Cunnac S (2013) An improved method for TAL effectors DNA-binding sites prediction reveals functional convergence in TAL repertoires of Xanthomonas oryzae strains. PLoS One 8:e68464. doi:10.1371/journal.pone.0068464
Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:e45. doi:10.1093/nar/29.9.e45
Pieterse CMJ, Leon-Reyes A, Van der Ent S, Van Wees SCM (2009) Networking by small-molecule hormones in plant immunity. Nat Chem Biol 5:308–316. doi:10.1038/nchembio.164
Prochnik S, Marri PR, Desany B, Rabinowicz PD, Kodira C, Mohiuddin M, Rodriguez F, Fauquet C, Tohme J, Harkins T, Rokhsar DS, Rounsley S (2012) The cassava genome: current progress, future directions. Trop Plant Biol 5:88–94. doi:10.1007/s12042-011-9088-z
Qi T, Song S, Ren Q, Wu D, Huang H, Chen Y, Fan M, Peng W, Ren C, Xie D (2011) The Jasmonate-ZIM-domain proteins interact with the WD-Repeat/bHLH/MYB complexes to regulate Jasmonate-mediated anthocyanin accumulation and trichome initiation in Arabidopsis thaliana. Plant cell 23:1795–1814. doi:10.1105/tpc.111.083261
Restrepo S, Duque MC, Verdier V (2000) Characterization of pathotypes among isolates of Xanthomonas axonopodis pv. manihotis in Colombia. Plant Pathol 49:680–687. doi:10.1046/j.1365-3059.2000.00513.x
Robert-Seilaniantz A, Grant M, Jones JDG (2011) Hormone crosstalk in plant disease and defense: more than just jasmonate-salicylate antagonism. Annu Rev Phytopathol 49:317–343. doi:10.1146/annurev-phyto-073009-114447
Rosenthal DM, Ort DR (2012) Examining cassava’s potential to enhance food security under climate change. Trop Plant Biol 5:30–38. doi:10.1007/s12042-011-9086-1
Santaella M, Suárez E, López C, González C, Mosquera G, Restrepo S, Tohme J, Badillo A, Verdier V (2004) Identification of genes in cassava that are differentially expressed during infection with Xanthomonas axonopodis pv. manihotis. Mol Plant Pathol 5:549–558. doi:10.1111/J.1364-3703.2004.00254.X
Scharf KD, Berberich T, Ebersberger I, Nover L (2012) The plant heat stress transcription factor (Hsf) family: structure, function and evolution. Biochim Biophys Acta 1819:104–119. doi:10.1016/j.bbagrm.2011.10.002
Soares-Cavalcanti NM, Belarmino LC, Kido E, Pandolfi V, Marcelino-Guimarães FC, Rodrigues F, Pereira G, Benko-Iseppon AM (2012) Overall picture of expressed heat shock factors in glycine max, Lotus japonicus and Medicago truncatula. Genet Mol Biol 35:247–259. doi:10.1590/S1415-47572012000200006
Staswick PE (2008) JAZing up jasmonate signaling. Trends Plant Sci 13:66–71. doi:10.1016/j.tplants.2007.11.011
Streubel J, Pesce C, Hutin M, Koebnik R, Boch J, Szurek B (2013) Five phylogenetically close rice SWEET genes confer TAL effector-mediated susceptibility to Xanthomonas oryzae pv. oryzae. New Phytol 200:808–819. doi:10.1111/nph.12411
Sugio A, Yang B, Zhu T, White FF (2007) Two type III effector genes of Xanthomonas oryzae pv. oryzae control the induction of the host genes OsTFIIAgamma1 and OsTFX1 during bacterial blight of rice. Proc Natl Acad Sci USA 104:10720–10725. doi:10.1073/pnas.0701742104
Suzuki R, Shimodaira H (2006) Pvclust: an R package for assessing the uncertainty in hierarchical clustering. Bioinformatics 22:1540–1542. doi:10.1093/bioinformatics/btl117
Swindell WR, Huebner M, Weber AP (2007) Transcriptional profiling of Arabidopsis heat shock proteins and transcription factors reveals extensive overlap between heat and non-heat stress response pathways. BMC Genomics 8:125. doi:10.1186/1471-2164-8-125
Tao Y, Xie Z, Chen W, Glazebrook J, Chang H, Han B, Zhu T, Zou G, Katagiri F (2003) Quantitative nature of Arabidopsis responses during compatible and incompatible interactions with the bacterial pathogen Pseudomonas syringe. Plant cell 15:317–330. doi:10.1105/tpc.007591.can
Tarazona S, García-Alcalde F, Dopazo J, Ferrer A, Conesa A (2011) Differential expression in RNA-seq: a matter of depth. Genome Res 21:2213–2223. doi:10.1101/gr.124321.111.Freely
Thomma BPHJ, Nürnberger T, Joosten MHAJ (2011) Of PAMPs and effectors: the blurred PTI-ETI dichotomy. Plant cell 23:4–15. doi:10.1105/tpc.110.082602
Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L (2012) Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7:562–578. doi:10.1038/nprot.2012.016
Vinatzer B, Teitzel GM, Lee MW, Jelenska J, Hotton S, Fairfax K, Jenrette J, Greenberg JT (2006) The type III effector repertoire of Pseudomonas syringae pv. syringae B728a and its role in survival and disease on host and non-host plants. Mol Microbiol 62:26–44. doi:10.1111/j.1365-2958.2006.05350.x
Wasternack C, Hause B (2013) Jasmonates: biosynthesis, perception, signal transduction and action in plant stress response, growth and development. An update to the 2007 review in annals of Botany. Ann Bot 111:1021–1058. doi:10.1093/aob/mct067
Wydra K, Zinsou V, Jorge V, Verdier V (2004) Identification of pathotypes of Xanthomonas axonopodis pv. manihotis in Africa and detection of quantitative trait loci and markers for resistance to bacterial blight of cassava. Phytopathology 94:1084–1093. doi:10.1094/PHYTO.2004.94.10.1084
Xie C, Mao X, Huang J, Ding Y, Wu J, Dong S, Kong L, Gao G, Li CY, Wei L (2011) KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res 39:W316–W322. doi:10.1093/nar/gkr483
Yadeta KA, Thomma BPHJ (2013) The xylem as battleground for plant hosts and vascular wilt pathogens. Front Plant Sci 4:97. doi:10.3389/fpls.2013.00097
Yang B, Sugio A, White FF (2006) Os8N3 is a host disease-susceptibility gene for bacterial blight of rice. Proc Natl Acad Sci USA 103:10503–10508. doi:10.1073/pnas.0604088103
Yilmaz A, Mejia-Guerra MK, Kurz K, Liang X, Welch L, Grotewold E (2011) AGRIS: the Arabidopsis gene regulatory information server, an update. Nucleic Acids Res 39:D1118–D1122. doi:10.1093/nar/gkq1120
Yu Y, Streubel J, Balzergue S, Champion A, Boch J, Koebnik R, Feng J, Verdier V, Szurek B (2011) Colonization of rice leaf blades by an African strain of Xanthomonas oryzae pv. oryzae depends on a new TAL effector that induces the rice nodulin-3 Os11N3 gene. Mol Plant-Microbe Interact 24:1102–1113. doi:10.1094/MPMI-11-10-0254
Zhong S, Joung JG, Zheng Y, Chen Y, Liu B, Shao Y, Xiang JZ, Fei Z, Giovannoni JJ (2011) High-throughput illumina strand-specific RNA sequencing library preparation. Cold Spring Harb Protoc 2011:940–949. doi:10.1101/pdb.prot5652
Zhu X, Thalor SK, Takahashi Y, Berberich T, Kusano T (2012) An inhibitory effect of the sequence-conserved upstream open-reading frame on the translation of the main open-reading frame of HsfB1 transcripts in Arabidopsis. Plant Cell Environ 35:2014–2030. doi:10.1111/j.1365-3040.2012.02533.x
Acknowledgments
AMB was financially supported by a PhD fellowship from DFS, IRD, Sibaghe-UM2 (France) and Ministerio de Agricultura y Desarrollo Rural (Colombia). Funding for BS, AB, CL come from Action Ecos Nord C11A02 and Ministerio de Agricultura y Desarrollo Rural (Colombia). Funding for ALPQ comes from the European Union through Erasmus Mundus Action 2 PRECIoSA Grant Agreement nr. 2012–2646/001–001–EMA2.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Communicated by Emmanuel Guiderdoni.
A. Muñoz-Bodnar and A. L. Perez-Quintero authors contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
299_2014_1667_MOESM1_ESM.tif
Supplementary Fig. 1. Bar plots showing the expression profiles of genes grouped in different clusters using k means. Numbers on top of the bars are the mean log FC for each cluster in the respective treatment (TIFF 16362 kb)
299_2014_1667_MOESM2_ESM.tif
Supplementary Fig. 2. Experimental validation of genes induced specifically by “+TALE1 Xam ”. Relative expression values were obtained by comparing each time point with the controls and using the 18S RNA as a reference gene, values in the graph correspond to (2−ΔCt 0dpi)/(2−ΔCt X dpi), where ΔCt = Ctgene − Ct18S. FPKM values for each gene are shown as a comparison (TIFF 2316 kb)
Rights and permissions
About this article

Cite this article
Muñoz-Bodnar, A., Perez-Quintero, A.L., Gomez-Cano, F. et al. RNAseq analysis of cassava reveals similar plant responses upon infection with pathogenic and non-pathogenic strains of Xanthomonas axonopodis pv. manihotis . Plant Cell Rep 33, 1901–1912 (2014). https://doi.org/10.1007/s00299-014-1667-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00299-014-1667-7