
Abstract
Key message
A UDP-glucose pyrophosphorylase gene ( LgUGPase ) was identified from Larix gmelinii, and its function in enhancing vegetative growth and cellulose biosynthesis was confirmed by analyzing transgenic Arabidopsis thaliana overexpressed LgUGPase .
Abstract
UDP-glucose pyrophosphorylase (UGPase), an important regulatory enzyme in carbohydrate metabolism, catalyzes the reversible production of glucose 1-phosphate and the conversion of uridine triphosphate to uridine diphosphate glucose and pyrophosphate. In this study, a larch UGPase (LgUGPase) gene was isolated from Larix gmelinii. The 1,443-bp open reading frame encodes a protein of 480 amino acids with a predicted molecular weight of 53.7 kDa and shows striking sequence similarity to UGPase proteins from Pinus taeda and Picea sitchensis. Semiquantitative reverse transcription-polymerase chain reaction showed that the LgUGPase gene was expressed primarily in the larch stem in addition to its root and leaf. Southern blot analysis indicated that LgUGPase is encoded by two genes in the L. gmelinii genome. Overexpression of LgUGPase enhanced vegetative growth in transgenic Arabidopsis and increased the contents of soluble sugars and cellulose, and thickened parenchyma cell walls. These results revealed that L. gmelinii UGPase participates in sucrose/polysaccharide metabolism and cell wall biosynthesis, suggesting that LgUGPase may be a good candidate gene for improvement of fiber cell development in plants.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
UDP-glucose pyrophosphorylase (UGPase) provides important enzymatic activity in carbohydrate metabolism that is found in all prokaryotic and eukaryotic organisms and catalyzes the reversible production of glucose 1-phosphate (Glc-1-P) and the conversion of uridine triphosphate to uridine diphosphate glucose (UDPG) and pyrophosphate (PPi) depending on the metabolic status of the tissue. In photosynthetic source tissues, UGPase with sucrose phosphate synthase is primarily involved in the synthesis of sucrose, the major sugar for photoassimilate export (Kleczkowski et al. 2004). In nonphotosynthetic sink tissues, which are to some extent dependent on imported carbon resources, UGPase is linked to sucrose degradation pathways through the conversion of UDPG produced by sucrose synthase (SuSy) to Glc-1-P to meet the demands of metabolic processes (Winter and Huber 2000). UDPG can be transformed into ADP-glucose by combined activities of UGPase and ADP-glucose pyrophosphorylase or catabolized through the glycolysis pathway and finally stored as starch (Kleczkowski et al. 1994). UDPG, the substrate/product of UGPase, is the key precursor for synthesizing sucrose, mannose, cellulose, cutin, glucoprotein, glycolipid, and carbohydrate (Kleczkowski et al. 2004).
UGPases have been identified in various plants, including Solanum tuberosum (Katsube et al. 1990), Hordeum vulgare (Eimert et al. 1996), Musa acuminata (Pua et al. 2000), Astragalus membranaceus (Wu et al. 2002), Oryza sativa (Abe et al. 2002), Cucumis melo (Dai et al. 2006), Populus tremula × tremuloides (Meng et al. 2007), Bambusa oldhamii (Weng et al. 2009), and Gossypium hirsutum (Wang et al. 2011), and the features and functions of some UGPase orthologs have been investigated. UGPase proteins form two distinct classes, UGPase-A and UGPase-B, which share the same catalytic function, although the two classes have little or no amino acid sequence and crystal structure identity (Kleczkowski et al. 2010, 2011). Plants generally have two copies of the UGPase A gene, whereas UGPase B occurs as a single-copy gene (Kleczkowski et al. 2004; Kotake et al. 2004; Chang et al. 2005; Schnurr et al. 2006; Meng et al. 2007; Okazaki et al. 2009). Earlier studies on the biological function of plant UGPase genes was focused on whether it was a rate-limiting step in carbohydrate metabolism. In developing potato tubers, UGPase activity could be reduced as much as 96 % by an antisense approach, although no change in carbohydrate metabolism was found in the antisense-suppression plants (Zrenner et al. 1993). UGPase activity was greatly inhibited in transgenic potatoes, but no substantial decrease in sugar content was found in stored tubers (Spychalla et al. 1994; Borovkov et al. 1996). These results showed that UGPase is not a rate-limiting enzyme in the carbohydrate synthesis pathway. In AtUGPase 1 antisense-suppression Arabidopsis thaliana (L.) plants, UGPase activity decreased by 30 % and led to a large decrease in carbohydrate content, but without any detectable changes in growth parameters (Johansson 2003). A double knockout mutant of AtUGPase1 and AtUGPase2 in Arabidopsis maintained normal growth and development, while the UGPase activity was reduced by 85 %, resulting in a significant decrease in output under field conditions (Meng et al. 2009b). Plant growth defects and male sterility, however, were found in the AtUGPase1 and AtUGPase2 double mutant (Park et al. 2010). These results indicated that UGPase is a critical factor in carbohydrate metabolism and is rate limiting in vegetative and reproductive phases in A. thaliana.
In plants, UGPase participates not only in carbohydrate metabolism but is also involved in cell wall biosynthesis. UDPG acts as a precursor for the synthesis of the carbohydrate moiety of glycolipids, glycoproteins, and cell wall components including callose, pectin, and cellulose (Amor et al. 1995; Delmer and Amor 1995; Gibeaut 2000; Dong 2004). Cellulose deposition is the main process in cell wall formation. Cellulose is formed from UDPG that is synthesized by reactions catalyzed by three enzymes: UGPase, UDP-sugar pyrophosphorylase, and sucrose synthase. Complementation analysis of cellulose-negative mutants confirmed that Acetobacter xylinum UGPase is involved in cellulose biosynthesis (Valla et al. 1989). Two O. sativa orthologs, OsUGPase1 and OsUGPase2, were shown to play critical roles in plant growth and reproduction (Chen et al. 2007a, b; Woo et al. 2008; Mu et al. 2009). The expression of A. xylinum UGPase in poplar demonstrated the role of UGPase in late cell expansion and secondary cell wall formation (Hertzberg et al. 2001; Coleman et al. 2007). Transformation experiments confirmed that overexpression of UGPase not only increased cellulose deposition, but also enhanced growth rates of transgenic plants. Overexpression of A. xylinum UGPase in tobacco and hybrid poplar resulted in significant changes in plant height (Coleman et al. 2006, 2007). Overexpression of G. hirsutum UGPase in Arabidopsis increased the growth rate and the contents of cellulose relative to control lines (Wang et al. 2011).
UGPase sequences from Pinus taeda and Picea sitchensis were registered in GenBank, but the features and functions of UGPase from gymnosperms have not been reported. Gymnosperms, conifer trees in particular, have a high content of fiber cells; therefore, study of UGPase from conifer trees may provide new insights to improve fiber quality and cellulose biosynthesis. Dahurian larch, Larix gmelinii (Rupr.), one of the three major timber conifers in northeast China, has high wood density, strong mechanical properties, and a clear wood texture. In addition, larch fiber cells are long and the nonfiber cell content is low, so L. gmelinii has important economic value in the construction, decoration, paper production, and chemical fiber industries. Therefore, investigating the role of LgUGPase in cellulose biosynthesis is important to improve growth and timber qualities by controlling LgUGPase gene expression. In this study, a novel UGPase gene was isolated from L. gmelinii and its critical role in carbohydrate metabolism and cell wall biosynthesis was evaluated in transgenic Arabidopsis.
Materials and methods
Plant materials
Mature seeds of L. gmelinii were provided by the Ganhe Forestry Bureau Seed Orchard, Inner Mongolia, and were stored in plastic bags at 4 °C for 6–12 months until being used for experiments. The seeds were sown in pots with a wet mixture of soil and vermiculite (1:1 w/w) for germination, and 4-week-old plants were used to isolate DNA and RNA.
Seeds of A. thaliana, Nossen ecotype, were sterilized with 70 % ethanol and 2 % NaClO solution and then sown on MS medium (Murashige and Skoog 1962). After vernalization at 4 °C for 3 days, the seeds were cultured under a photoperiod of 16 h light/8 h dark at 22 °C. Ten-day-old A. thaliana seedlings were transplanted into pots with nutrient soil and grown under the same conditions.
Isolation of the LgUGPase gene
Total RNA was extracted from 4-week-old L. gmelinii plants using the cetyltrimethylammonium bromide (CTAB) method (Chang et al. 1993). L. gmelinii tissue (1.0 g) was ground in liquid nitrogen and transferred to 10 ml of 2× CTAB buffer (Chang et al. 1993). The mixture was incubated at 65 °C for 30 min and then treated with an equal volume of chloroform:isoamyl alcohol (24:1 v/v). After centrifugation at 3,500×g for 20 min, the supernatant was collected and the RNA was precipitated by adding 1/4 volume of 10 M lithium chloride (LiCl) and incubating at −20 °C for 2 h. The RNA pellet was collected, dissolved in TE buffer, and then sequentially treated with equal volumes of TE-saturated phenol:chloroform:isoamyl alcohol (25:24:1 by vol.) and chloroform:isoamyl alcohol (24:1 v/v). The RNA was precipitated again by adding 1/4 volume of 10 M LiCl at −20 °C for 2 h and then dissolved in TE buffer.
An LgUGPase cDNA corresponding to an open reading frame (ORF) was obtained by reverse transcription-polymerase chain reaction (RT-PCR) using a Takara RT-PCR Kit with Takara Ex Taq HS (Takara Bio, Dalian, China) and specific flanking primers designed using the P. taeda UGPase cDNA sequence (EF619969.1): UGPase-forward, 5′-ATGGCTGCA GCACCAGCAGTTGC-3′, base pairs 108–131; and UGPase-reverse, 5′-CTAGTTCACAATATCATCAGGACTGCTCACTACC-3′, base pairs 1,518–1,551. First-strand cDNA was synthesized from 1 μg of total RNA at 42 °C for 60 min with 1 μl of oligo-dT-Adaptor (Takara Bio). The PCR reaction was carried out with 2.5 μl of 10× Ex Taq buffer (Takara Bio), 2 μl of 2.5 mM dNTPs, 2.5 U of Takara Ex Taq HS DNA polymerase (Takara Bio), 1 μl of 10 μM primers (UGPase-forward and UGPase-reverse), and 1 μl of cDNA in a 25-μl volume under the following conditions: 35 cycles at 94 °C for 30 s, 58 °C for 30 s, and 72 °C for 1 min followed by 10 min at 72 °C for a final extension. The 1,443-bp amplification product was cloned into the pEASY-T1 Cloning Vector (TransGen Biotech, Beijing, China) according to the manufacturer’s instructions. DNA sequencing was performed by BGI (Beijing, China) and analyzed with BLAST.
Phylogenetic analysis
Phylogenetic analysis was performed with mature protein sequences of UGPases from various plants using the ClustalW and MEGA 5 programs. GenBank accession numbers of the UGPases used for the phylogenetic analysis are as follows: P. sitchensis (ABR17271.1), P. taeda (ABR15472.1), Zea mays (DAA62613.1, DAA62614.1, AFW82285.1), O. sativa (ABD57308.1, AAF62555.1, EEE64040.1), H. vulgare (CAA62689.1, BAJ85276.1), Glycine max (XP_003538374.1, XP_003552278.1), Pyrus pyrifolia (BAB88218.1, BAB88217.1) Prunus persica (AGH25528.1, EMJ20759.1), Populus trichocarpa (XP_002315147.1), P. tremula × tremuloides (AAP86317.1, ABB88893.1), B. oldhamii (AAO48422.1), G. hirsutum (ADO32901.1), Solanum lycopersicum (XP_004250240.1, XP_004250241.1, XP_004240914.1), S. tuberosum (AAL99194.1, AAL99198.1), and A. thaliana (AAL15254.1, AAK25954.1, NP_567031.1).
Southern blot analysis
Total genomic DNA was isolated from 4-week-old L. gmelinii and Arabidopsis plants using the CTAB method (Murray and Thompson 1980). Probes of about 0.5 kb corresponding to the coding region of the LgUGPase and NPT II genes were labeled using a PCR DIG Probe Synthesis Kit (Roche Molecular Biochemicals, Indianapolis, IN, USA) with the following PCR primer sets: P-F (5′-CTCCTGGACAAGCTTGTTGTGCT-3′, base pairs 252–275) and P-R (5′-GTTTTGGGAGTGACCTCCATGCA-3′, base pairs 756–779), NPT II-F (5′-TTGTCACTGAAGCGGGAAGG-3′) and NPT II-R (5′-CGGCGATACCGTAAAGCAC-3′), respectively. RT-PCR and genomic-PCR were performed with the P-F and P-R primers to confirm that no intron was present in the probe region. For Southern blotting, genomic DNA (10 μg for L. gmelinii, 30 μg for Arabidopsis) was digested with XbaI or BamHI, subjected to electrophoresis, transferred to membranes, and hybridized with the LgUGPase or NPT II probe (Fig. 4a). The hybridized products were detected with a nonradioactive digoxigenin luminescence detection system (Boehringer Mannheim, Mannheim, Germany).
Plant transformation of Arabidopsis and screening
To investigate the function and expression characteristics of LgUGPase, the plasmid pBI101-LgUGPase was constructed with the pBI101-35::Gus-Hm vector containing an intronless version of the GUS gene from pIG121-Hm (Akama et al. 1992). A fragment of the hygromycin phosphotransferase gene with a cauliflower mosaic virus (CaMV) 35S promoter as well as the GUS gene with a nopaline synthase terminator was excised from pBI101-35::Gus-Hm by digestion with XbaI and BamHI. The LgUGPase ORF was amplified with LgUGPase-XbaI-F (5′-TATCTAGAATGGCTGCAGCACCAGCAGTTGC-3′, base pairs 1–23), which contains an XbaI site, and LgUGPase-BamHI-R (5′-GCGGATCCCTAGTTCACAATATCATCAGGACTGC-3′, base pairs 1,417–1,443) which contains a BamHI site. The PCR product was digested with XbaI and BamHI and inserted between the CaMV 35S promoter and the nopaline synthase terminator of the digested pBI101-35::Gus-Hm vector to generate pBI101-LgUGPase (Fig. 4a).
The pBI101-LgUGPase construct was introduced into Agrobacterium tumefaciens strain LBA4404 by the freeze–thaw method, and Arabidopsis plant transformation was performed with the simplified in-plant infiltration method described by Kim et al. (1999). Transgenic lines were obtained by screening on MS medium with kanamycin (40 mg l−1) and confirmed by PCR. The PCR was performed using vector-specific primers NPT II-F and NPT II-R under the following conditions: 94 °C denaturation for 10 min, 33 cycles at 94 °C for 1 min, 55 °C for 30 s, 72 °C for 1 min, and a final extension at 72 °C for 10 min. The selected kanamycin-resistant individuals were self-pollinated to establish strains that were homozygous for the transgene. Homozygous lines (T3) were confirmed by analyzing the rate of kanamycin-resistant individuals in their progenies (T4), which showed 100 % kanamycin resistance in their progenies (T4). Wild-type (WT) seeds from mother plants that were grown together with T2 transgenic plants under comparable conditions were used as control. All analyses were carried out with T3 transgenic plants of each homozygous line. Physical characteristics such as area, length, and width of the fifth leaf from WT and transgenic Arabidopsis plants were measured with an LI-3000C Portable Area Meter (Li-Cor, Lincoln, NE, USA).
RT-PCR
To investigate LgUGPase expression, RT-PCR was performed with the LgUGPase-specific P-F and P-R primers. The LgActin gene (AB523401, LgACT-F: 5′-CTTCACCACAACCGCAGAG-3′, base pairs 809–827; LgACT-R: 5′-TGAACAGCCGAAGACCCA-3′, base pairs 1,479–1,496; Zhu et al. 2011) and AtActin 2 gene (Atactin2-F: 5′-CTGGATTCTGGTGATGGTGTGTCT-3′; Atactin2-R: 5′-GAACCACCGATCCAGACACTGTAC-3′; Lin et al. 2008) were used as internal controls. RNA was prepared from 40-day-old L. gmelinii plants or 3-week-old transgenic Arabidopsis plants using the CTAB method (Chang et al. 1993). The cDNAs were synthesized from 1 μg of total RNA at 42 °C for 60 min with 1 μl of oligo-dT adapter primer using a RT-PCR kit (Takara Bio). The PCR reaction was carried out with 2.5 μl of 10× Ex Taq buffer (Takara Bio), 2 μl of 2.5 mM dNTPs, 2.5 U of Takara Ex Taq HS DNA polymerase (Takara Bio), 1 μl of 10 μM primers, and 1 μl of cDNA in a 25-μl volume under the following conditions: 35 cycles at 94 °C for 30 s, 58 °C for 30 s, and 72 °C for 1 min followed by 10 min at 72 °C for a final extension.
UGPase activity assay
Tissue extraction and UGPase activity assays were performed as described by Sowokinos (1976) with some modifications (Ciereszko et al. 2001). The fifth rosette leaves of 40-day-old WT and transgenic Arabidopsis were ground to powder with liquid nitrogen. The proteins were extracted in medium containing 100 mM tris(hydroxymethyl)-aminomethane hydrochloride (Tris–HCl) buffer (pH 7.5), 5 mM magnesium chloride (MgCl2), 1 mM ethylene diamine tetraacetic acid (EDTA), 2 mM dithiothreitol, 0.05 % (v/v) Triton X-100, and 1 % (w/v) polyvinylpolypyrrolidone with a sample-to-buffer ratio of 1:5 (w/v). The homogenate was centrifuged at 14,000g for 10 min and the resulting supernatant was used to assay UGPase activity. Reaction mixtures contained 100 mM Tris–HCl (pH 7.5), 5 mM MgCl2, 0.8 mM UDP-glucose, 4 units each of phosphoglucomutase and glucose-6-phosphate dehydrogenase (both from Boehringer Mannheim), and 20 μl of extract. The reaction was initiated with 1 mM PPi, and reduction of nicotinamide adenine dinucleotide was monitored at 340 nm using TU-1810 spectrophotometer (Beijing Purkinje Instrument Co., Beijing, China).
Carbohydrate analysis
Sucrose, glucose, fructose, and starch contents were measured in the fifth rosette leaf of 40-day-old Arabidopsis plants at 3 h after illumination. The samples were ground into powder and extracted twice with 80 % ethanol at 80 °C for 30 min and further washed with 50 % ethanol at 80 °C for 30 min. After centrifugation (24,000×g, 30 min, 4 °C), sucrose, glucose, and fructose in the supernatant were measured by enzymatically determining the reduction in nicotinamide adenine dinucleotide phosphate absorbance at 340 nm after successively adding the coupling enzymes glucose 6-P-dehydrogenase, hexokinase, phosphoglucose isomerase, and invertase (Sekin 1978). The residual pellets were incubated at 60 °C for 3 h with α-amylase and amyloglucosidase, and the liberated glucose represented the starch content.
Determination of lignin, cellulose, and hemicellulose content
Lignin content was determined in stems of 40-day-old Arabidopsis using a modified micro-Klason method (Huntley et al. 2003). Dried stems (200 mg) of WT and transgenic Arabidopsis were ground into powder, passed through a 40-mesh screen, and extracted with acetone in a Soxhlet apparatus for 6 h. Then 100 mg of acetone extract was treated with 3 ml 72 % sulfuric acid (H2SO4) for 2 h at 20 °C, diluted with 112 ml of deionized water, and steamed for 1 h. The mixture was rinsed with hot water, then dried and weighed. The percentage of lignin equaled the weight of the extracted material divided by the total weight multiplied by 100.
The cellulose content in WT and transgenic Arabidopsis was measured according to the H2SO4 digestion method (Updegraff 1969). The stems (200 mg) of 40-day-old Arabidopsis were ground into powder in liquid nitrogen and extracted with 70 % ethanol for 1 h at 70 °C; the remaining ethanol insoluble residues were lyophilized and weighed. The powder (100 mg) of cell wall and anthrone was hydrolyzed with 67 % H2SO4 (v/v) at 100 °C for 16 min. After cooling on ice, the sample was measured using a TU-1810 spectrophotometer (Beijing Purkinje Instrument Co., Beijing, China) at 650 nm.
Hemicelluloses were sequentially extracted following the standard method of Van Soest (1963, 1967) modified for the use of small sample volumes and starch-rich material (Schädel et al. 2010). Dried 40-day-old transgenic and WT Arabidopsis stems (50 mg) were ground to powder and extracted with a heat-stable α-amylase (from Bacillus licheniformis; Sigma, Buchs, Switzerland) for 30 min at 85 °C to remove starch. The starch-free pellet was dissolved in a neutral detergent (sodium tetraborate decahydrate, EDTA, sodium dodecyl sulfate, triethylene glycol, sodium phosphate, and distilled water) and boiled for 60 min in a water bath to extract proteins, low molecular weight carbohydrates, lipids, and pectins, which were separated by centrifugation at 12,000 rpm and removed from the pellet. The pellet containing the cell wall fraction (cellulose, hemicelluloses, and lignin) was washed (2× hot deionized water, 1× acetone, 1× deionized water) and dried overnight before weighing. The dried pellet was mixed with an acid detergent containing 1 NH2SO4 and 55 mmol l−1 hexadecyl trimethylammonium bromide and boiled for 60 min to extract and hydrolyze hemicelluloses. The remaining cell wall fraction (cellulose and lignin) was washed with deionized water and acetone before drying and weighing. Total hemicellulose concentrations were calculated as the gravimetric difference between “total cell wall fraction” and “cellulose and lignin fraction.”
Histological observation
The leaves and stems of 40-day-old transgenic and WT Arabidopsis were dissected into 5 × 10-mm segments and fixed in FAA (63 % ethanol, 5 % acetic acid, and 5 % formaldehyde) overnight at room temperature (RT). After fixation, the samples were dehydrated using a series of 50, 70, 80, 90, 95, and 100 % ethanol for 2 h at RT for each step followed by ethanol:xylene 1:1 and then 100 % xylene for 2 h at RT for each step. The samples were subsequently embedded in xylene:paraffin 1:1 and then 100 % paraffin for 2 days at 67 °C for each step. Xylene was subsequently replaced with paraffin wax. Sections (8 μm thick) were cut with a glass a rotary microtome (Microm HM 325; Thermo Scientific, Suzhou, China). Paraffin ribbons were floated on 4 % formalin on a 42 °C warming plate and excess formalin was removed. Slides were dried in a 42 °C oven and deparaffinized in two xylene treatments for 15 min each. Slides were then brought to 70 % ethanol using a graded ethanol series (xylene:ethanol 1:1; and 100, 95, and 70 % ethanol each for 10 min). Johansen’s Safranin O and Fast Green were used as staining procedures (Johansen 1940) as in Ruzin (1999). The sections were observed with a microscope (Eclipse 80i; Nikon, Tokyo, Japan) and photographs were taken with a Nikon DS-Ri1 digital camera.
Results
Isolation of UGPase gene from L. gmelinii
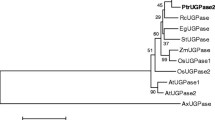
An ORF corresponding to an LgUGPase cDNA was amplified by RT-PCR with primers designed using the P. taeda UGPase cDNA sequence (Fig. 1). The 1,443-bp LgUGPase ORF encodes a protein of 480 amino acids with a theoretical molecular mass of 53.7 kDa and a pI of 5.41. A putative N-glycosylation motif (N-Q-S) was identified in amino acids 170–172, which is thought to be related to the membrane-binding activity of UGPase. A nucleotide-binding (NB) loop, T-M-G-C, related to substrate binding, was identified at amino acids 101–104 including the key amino acid Cys-104, which has an important role in PPi binding. Conserved amino acids that play a primary role in maintaining UGPase catalytic activity and substrate binding (Abe et al. 2002; Meng et al. 2009a; Wang et al. 2011) were identified at Try-196, Try-307, Lys-265, Lys-331, and Lys-370 in the LgUGPase amino acid sequence. BLAST analysis showed that the deduced LgUGPase amino acid sequence had high identity with UGPase proteins from P. taeda (94 %), P. sitchensis (92 %), Populus deltoides (80 %), Z. mays (80 %), G. max (78 %), and A. thaliana (76 %). Phylogenetic analyses of UGPases from P. sitchensis, P. taeda, Z. mays, O. sativa, H. vulgare, G. max, P. pyrifolia, P. persica, P. trichocarpa, P. tremula × tremuloides, B. oldhamii, G. hirsutum, S. lycopersicum, S. tuberosum, and A. thaliana revealed that these plant UGPases divide into two main branches, UGPase-A and UGPase-B. LgUGPase belongs to a gymnosperm UGPase A clade that includes P. taeda and P. sitchensis (Fig. 2). These results suggested that LgUGPase was a class A UGPase with features and functions similar to those of P. taeda and P. sitchensis.

Nucleotide and deduced amino acid sequences of Larix gmelinii UGPase. Nucleotides and amino acids are numbered on the left and right, respectively. The termination codon is indicated by an asterisk. A putative N-glycosylation motif is underlined (N-Q-S, aa 170–172). Key amino acids for a nucleotide-binding (NB) loop (T-M-G-C, aa 101–104) are indicated with a wavy line and Cys-104 is boxed. Conserved amino acid residues (Try-196, Try-307, Lys-265, Lys-331, and Lys-370) are circled

Phylogenetic tree generated from deduced UGPase amino acid sequences from Larix gmelinii, Picea sitchensis (ABR17271.1), Pinus taeda (ABR15472.1), Zea mays (DAA62613.1, DAA62614.1, AFW82285.1), Oryza sativa (ABD57308.1, AAF62555.1, EEE64040.1), Hordeum vulgare (CAA62689.1, BAJ85276.1), Glycine max (XP_003538374.1, XP_003552278.1), Pyrus pyrifolia (BAB88218.1, BAB88217.1), Prunus persica (AGH25528.1, EMJ20759.1), Populus trichocarpa (XP_002315147.1), Populus tremula × tremuloides (AAP86317.1, ABB88893.1), Bambusa oldhamii (AAO48422.1) Gossypium hirsutum (ADO32901.1), Solanum lycopersicum (XP_004250240.1, XP_004250241.1, XP_004240914.1), Solanum tuberosum (AAL99194.1), and Arabidopsis thaliana (AAL15254.1, AAK25954.1, NP_567031.1). To estimate the reliability of the branches of the tree, the bootstrap procedure in CLUSTALW was used (1,000 replications). Full-length UGPase amino acid sequences from various plants were used to construct the phylogenetic tree with the neighbor-joining method
To examine the transcription pattern of LgUGPase, total RNA was extracted from roots, stems, and leaves of 40-day-old L. gmelinii, and semiquantitative RT-PCR was performed with LgUGPase-specific primers. The LgUGPase gene was expressed in all larch plant organs with higher expression in stems than in leaves or roots (Fig. 3a).

Molecular analysis of LgUGPase gene. a Expression of the LgUGPase gene in leaves (L), stems (S), roots (R), and total plants (T) of 40-day-old Larix gmelinii. The LgActin gene was used as an internal control. The relative expression value was evaluated by comparing signal densities of LgUGPase and LgActin. b Genomic Southern analysis showing the hybridization pattern of the LgUGPase gene. Genomic DNA was digested with the restriction enzymes XbaI (X) or BamHI (B) and hybridized with an LgUGPase probe
Southern hybridization was performed to investigate copy number of the UGPase gene in the L. gmelinii genome (Fig. 3b). Larch genomic DNA was digested with XbaI or BamHI restriction enzymes, which have no corresponding restriction sites in the LgUGPase cDNA region. The digested genomic DNA was hybridized with an LgUGPase probe (Fig. 4a). The absence of an intron in the probe region was confirmed by RT-PCR and genomic-PCR (data not shown). Two bands were detected in each DNA sample suggesting that the LgUGPase gene is present as two copies.

Molecular analysis of LgUGPase-transformed transgenic Arabidopsis lines. a T-DNA region of the vector pBI101-LgUGPase used to produce transgenic LgUGPase plants. 35Sp, cauliflower mosaic virus 35S promoter; NOSp, nopaline synthase promoter; NOSt, nopaline synthase terminator; NTP II, neomycin phosphotransferase II gene; BR, right border; BL, left border. The locations of probe used for Southern hybridization are indicated by black lines. b Southern blot analysis of non-transformed control plants (WT), four transgenic lines (UGPase1-4) and plasmid (pBI101-LgUGPase). DNA was digested with the restriction enzymes XbaI and hybridized with a NPT II probe. c RT-PCR analysis of expression levels of the LgUGPase gene in WT and transgenic plants. The AtActin gene was used as an internal control. d The UGPase activity of WT and transgenic Arabidopsis (UGPase1-4) lines. Mean values (±SE) were calculated from five plants per line
Phenotype analysis of LgUGPase transgenic Arabidopsis
To further analyze features and functions of LgUGPase, the plasmid pBI101-LgUGPase containing the LgUGPase cDNA driven by the CaMV35S promoter was constructed and introduced into Arabidopsis via Agrobacterium-mediated transformation (Fig. 4a). Stable insertion of the transgene in each transgenic line was confirmed by Southern hybridization, showing the integration of the introduced DNA into different sites of the transgenic Arabidopsis genome with a single-copy insertion (Fig. 4b). LgUGPase transgene expression was confirmed by RT-PCR in four homozygous transgenic lines produced by self-pollination (Fig. 4c). UGPase activity was measured in the four transgenic lines using fifth rosette leaf of 40-day-old Arabidopsis plants (Fig. 4d). Compared to the WT (5.00 μmol g−1 min−1), the activity of UGPase increased significantly in the four LgUGPase transgenic lines (6.88–7.73 μ mol g−1 min−1). The transgenic lines, UGPase 1 and UGPase 3, showed higher UGPase activity than UGPase 2 and UGPase 4 (Fig. 4d).
Transgenic Arabidopsis overexpressing LgUGPase displayed distinctive phenotypes compared to the WT at different developmental stages. No significant difference between WT and transgenic plants was observed in germination. WT and transgenic Arabidopsis seedlings 3 days after sowing in MS medium are shown in Fig. 5a. Transgenic plants displayed a faster growth rate than WT plants (Fig. 5b–d). After transplanting to soil, transgenic plants showed greater heights and growth rates compared to control plants. Overexpression of LgUGPase gave enhanced vegetative growth in 3-, 4-, and 5-week-old transgenic plants compared to WT control plants (Fig. 5e–i). To precisely analyze the differences in phenotypes between transgenic and WT plants, the physical characteristics of 40-day-old plants were measured, including the area, length, and width of the fifth rosette leaf, the number of cauline branches, internode distances, and plant height (Table 1). All of the transgenic lines were taller than the WT control plants after 40 days of growth. No significant differences in leaf length or cauline branch number were noted, but significant differences in internode distance, leaf area, and leaf width were detected between WT and LgUGPase transgenic plants (Table 1).

Effects of LgUGPase overexpression on transgenic Arabidopsis. Transgenic lines (below the red line) and wild type (WT; above the red line) cultivated on MS medium at 3 (a), 7 (b), 10 (c), and 14 days (d) after sowing. Three-week-old transgenic plants (f) showed significantly more growth than WT plants (e). Four-week-old transgenic plants (h) showed much faster growth rate than WT plants (g). Transgenic plants (left) were much taller than WT plants (right) at 5 weeks after sowing (i). Compared with the WT (j, k, n, and o), the parenchyma cell wall was thicker in the fifth leaf (j–m) and stem (n–q) of the LgUGPase-transformed lines (l, m, p, and q). The regions contained in the white boxes indicated by arrows in j, l, n, and p are enlarged in k, m, o, and q, respectively (color figure online)
Three types of soluble sugars, sucrose, fructose and glucose, and starch were measured in the fifth rosette leaf of WT and transgenic Arabidopsis plants grown for 40 days (Table 2). The contents of sucrose and total soluble sugars increased significantly in the four LgUGPase transgenic lines compared to the WT, while significant increases in glucose and fructose were detected in three of the four transgenic lines. No significant change in starch levels occurred in the fifth rosette leaf of the four transgenic lines compared to the control plants (Table 2).
Lignin, cellulose, and hemicellulose contents were measured in the stems of 40-day-old WT and transgenic plants (Table 3). Lignin content, as determined by a modified micro-Klason method, was not significantly different between the transgenic lines (27.34–28.11 %) and WT plants (28.60 %). Cellulose content in stems of the four transgenic lines was significantly higher (21.05–25.29 %) than in the control lines (17.95 %). No significant difference in hemicellulose amount was found between the stems of the WT and transgenic plants.
Paraffin sections of leaf veins and stems from 40-day-old WT and transgenic Arabidopsis were used to observe parenchyma cells (Fig. 5j–q). Thicker cell walls were noted in parenchyma cells in leaf veins (Fig. 5l, m) and stems (Fig. 5p, q) of the LgUGPase-transformed lines compared to WT plants (Fig. 5j, k, n, o). These results revealed that the LgUGPase gene plays a role in controlling the biosynthesis of secondary cell walls.
Discussion
A UGPase gene was isolated from L. gmelinii, and its functions and features were analyzed. Based on the alignment of UGPases from P. taeda and other plants, functional consensus motifs previously identified in various other eukaryotic organisms were found in the deduced LgUGPase sequence. These conserved motifs, such as the NB loop and N-Q-S, have been demonstrated to be essential for substrate binding and catalytic activity. Recent research on H. vulgare UGPase demonstrated the functions of the NB loop using domain deletion approaches that resulted in complete inactivation of the enzyme (Meng et al. 2009a), and Cys-99 was shown to affect PPi binding (Martz et al. 2002). The functionally important amino acids of the NB loop corresponded to amino acids 101–104 of L. gmelinii UGPase, and Cys-104 may be related to substrate binding. Another plant UGPase motif N-X-S/T (where X represents any amino acid other than proline) is involved in membrane-binding activity. The S. tuberosum UGPase has two putative glycosylation sites at amino acid positions 168 (N-Q-S) and 307 (N-L-S). Two similar motifs were observed at amino acids 162 (N-Q-S) and at amino acids 201 (N-N-S) in O. sativa UGPase (Kleczkowski 1994), but the UGPase from cotton has only one potential glycosylation site at amino acids 157 (N-Q-S) (Wang et al. 2011). LgUGPase has only one potential glycosylation site at amino acids 170 (N-Q-S). Key Lys residues in potato UGPase were demonstrated by site-directed mutagenesis to be important with Lys-367 potentially being essential for catalytic activity, and Lys-263 and Lys-329 for binding MgPPi or Glc-1-P substrate (Katsube et al. 1990). Abe et al. (2002) reported that Lys-257, Lys-323, and Lys-361 in O. sativa UGPase are positioned similarly to the three Lys residues in potato UGPase and have similar functions. Meng et al. (2009a) demonstrated that a Lys-260 mutant had low activity and a high Km for PPi, and that Trp-191 and Trp-302 were involved in UDPG binding. L. gmelinii UGPase has residues at Lys-265, Lys-331, Lys-370, Try-196, and Try-307 analogous to O. sativa and potato. These results suggested that Lys-370 may be responsible for the catalytic activity of LgUGPase and that Lys-265, Lys-331, Try-196, and Try-307 are involved in substrate binding.
Tissue-specific expression of G. hirsutum UGPase was highest in roots followed by stems and then leaves (Wang et al. 2011). However, LgUGPase was expressed highest in stems followed by leaves and was lowest in roots (Fig. 3a), suggesting a role for LgUGPase in fiber cell formation.
From early studies, UGPase genes were thought to be present only as a single gene in plants. For example, only one type of UGPase transcript was found in H. vulgare (Eimert et al. 1996). However, in S. tuberosum, two copies of UGPase were reported that resulted from allelic polymorphism (Sowokinos et al. 1997). The O. sativa genome contains two UGPase paralogs: OsUGPase1 on chromosome 9 (Abe et al. 2002) and OsUGPase2 on chromosome 2 (Mu 2002). Kleczkowski et al. (2004) and Kiyosumi et al. (2002) found two highly identical UGPase genes belonging to the UGPase-A class in Arabidopsis, aspen, Japanese pear, and slime mold. Recent research suggests that plants generally have two UGPase-A genes to encode UGPase-A protein and a single gene encoding UGPase-B protein (Kleczkowski et al. 2010). Southern blot analysis suggested that the LgUGPase is present as two copies (Fig. 3b) that encode a UGPase-A protein in accordance with work on UGPase copy number in plants by Kleczkowski et al. (2010).
Overexpression LgUGPase under the control of a single CaMV 35S promoter enhanced vegetative growth of transgenic A. thaliana (Fig. 5a–i). This result is consistent with previous work showing that overexpression of A. xylinum UGPase under the control of the CaMV 35S promoter increased transgenic tobacco plant height (Coleman et al. 2006). Overexpression of cotton UGPase in A. thaliana increased plant height (2.6–5.8 cm), but there was no significant increase in leaf area, internode distance, or cauline branching (Wang et al. 2011). The overexpression of LgUGPase in 40-day-old A. thaliana plants similarly resulted in a significant increase in height (4.82–6.56 cm) and also in increased leaf area (1.00–1.04 cm2), leaf width (0.24–0.36 cm), and internode distance (2.14–2.86 cm), but no significant increase in cauline branching was detected (Table 1). LgUGPase was more effective in enhancing transgenic Arabidopsis vegetative growth, suggesting that UGPase plays an important role in controlling the strength of sink tissues.
Soluble carbohydrate content was increased by UGPase overexpression, suggesting that UGPase is a critical factor in carbohydrate metabolism (Coleman et al. 2006, 2007; Park et al. 2010; Wang et al. 2011). Overexpression of A. xylinum UGPase increased the contents of glucose and fructose in transgenic tobacco (Coleman et al. 2006) and significantly increased the contents of soluble carbohydrates (sucrose, glucose, and fructose) in transgenic poplar leaves (Coleman et al. 2007). The contents of sucrose, glucose, fructose, and starch were elevated in the leaf tissues of transgenic Arabidopsis overexpressing GhUGPase from cotton relative to WT plants (Wang et al. 2011). In this study, soluble carbohydrates including sucrose, glucose, and fructose, were significantly higher in the rosette leaves of LgUGPase transgenic Arabidopsis (Table 2), indicating that LgUGPase may modify carbon allocation in favor of soluble sugars. However, no difference in starch content was found between WT and LgUGPase transgenic plants, indicating that accumulated carbohydrate is probably not stored in the form of starch in leaves, but forms a pool of available soluble sugars to accelerate glycometabolism and enhance vegetative growth.
The main components of the cell wall are cellulose, hemicelluloses, and lignin. Previous studies showed that UGPase played an important role in cellulose biosynthesis (Coleman et al. 2006, 2007). Ruan et al. (2003) also confirmed that UGPase played an indispensable role in cotton fiber cell initiation and elongation and that SuSy was strongly associated with cellulose formation in cotton. Wang et al. (2011) showed that cellulose content increased significantly in GhUGPase-transformed lines (2.1–6.3 %). In this work, the cellulose content of LgUGPase overexpressing plants increased by 3.1–7.34 % (Table 3), showing that LgUGPase was more effective in increasing the cellulose content in transgenic plants. Our data on parenchyma cells in the leaf veins (Fig. 5l, m) and stems (Fig. 5p, q) of LgUGPase-transformed lines further support previous evidence that UGPase is involved in cell wall biosynthesis and that LgUGPase activity may be related to the activity of cellulose synthase complex in the cell wall. These findings suggest that LgUGPase may play an important role in the allocation of carbon, especially in cellulose biosynthesis, and that LgUGPase may be a good candidate gene to accelerate fiber cell development and to enhance vegetative growth in plants.
Abbreviations
- CTAB:
-
Cetyltrimethylammonium bromide
- Glc-1-P:
-
Glucose 1-phosphate
- NB:
-
Nucleotide binding
- NPT II:
-
Neomycin phosphotransferase II gene
- ORF:
-
Open reading frame
- RT-PCR:
-
Reverse transcription-polymerase chain reaction
- SuSy:
-
Sucrose synthase
- UDPG:
-
Uridine diphosphate glucose
- UGPase:
-
UDP-glucose pyrophosphorylase
References
Abe T, Niiyama H, Sasahara T (2002) Cloning of cDNA for UDP glucose pyrophosphorylase and the expression of mRNA in rice endosperm. Theor Appl Genet 105:216–221
Akama K, Shiraishi H, Ohta S, Nakamura K, Okada K, Shimura Y (1992) Efficient transformation of Arabidopsis thaliana: comparison of the efficiencies with various organs, plant ecotypes and Agrobacterium strains. Plant Cell Rep 12:7–11
Amor Y, Haigler CH, Johnson S, Wainscott M, Delmer DP (1995) A membrane-associated form of sucrose synthase and its potential role in synthesis of cellulose and callose in plants. Proc Natl Acad Sci USA 92:9353–9357
Borovkov AY, McClean PE, Sowokinos JR, Ruud SH, Secor GA (1996) Effect of expression of UDP-glucose pyrophosphorylase ribozyme and antisense RNAs on the enzyme activity and carbohydrate composition of field-grown transgenic potato plants. Plant Physiol 147:644–652
Chang S, Puryear T, Caimey I (1993) A simple and efficient method for isolating RNA from pine trees. Plant Mol Bio Rep 11:113–116
Chang CW, Moseley JL, Wykoff D, Grossman AR (2005) The LPB1 gene is important for acclimation of Chlamydomonas reinhardtii to phosphorus and sulfur deprivation. Plant Physiol 138:319–329
Chen R, Zhao X, Shao Z, Zhu LL, He G (2007a) Multiple isoforms of UDP-glucose pyrophosphorylase in rice. Physiol Plant 129:725–736
Chen R, Zhao X, Shao Z, Wei Z, Wang Y, Zhu L, He G (2007b) Rice UDP-glucose pyrophosphorylase1 is essential for pollen callose deposition and its cosuppression results in a new type of thermosensitive genic male sterility. Plant Cell 19:847–861
Ciereszko I, Johansson H, Hurry V, Kleczkowski LA (2001) Phosphate status affects the gene expression, protein content and enzymatic activity of UDP-glucose pyrophosphorylase in wild-type and pho mutants of Arabidopsis. Planta 212:598–605
Coleman HD, Ellis DD, Gilbert M, Mansfield SD (2006) Up-regulation of sucrose synthase and UDP-glucose pyrophosphorylase impacts plant growth and metabolism. Plant Biotechnol J 4:87–101
Coleman HD, Canam T, Kang KY, Ellis DD, Mansfield SD (2007) Over-expression of UDP-glucose pyrophosphorylase in hybrid poplar affects carbon allocation. J Exp Bot 58:4257–4268
Dai N, Petreikov M, Portnoy V, Katzir N, Pharr DM, Schaffer AA (2006) Cloning and expression analysis of a UDP-galactose/glucose pyrophosphorylase from Melon fruit provides evidence for the major metabolic pathway of galactose metabolism in raffinose oligosaccharide metabolizing plants. Plant Physiol 142:294–304
Delmer DP, Amor Y (1995) Cellulose biosynthesis. Plant Cell 7:987–1000
Dong X (2004) Function investigation of Arabidopsis callose synthases and the signal transduction pathway. PhD dissertation, The Ohio State University
Eimert K, Villand P, Kilian A, Kleczkowski LA (1996) Cloning and characterization of several cDNAs for UDP-glucose pyrophosphorylase from barley (Hordeum vulgare) tissues. Gene 170:227–232
Gibeaut DM (2000) Nucleotide sugars and glucosyltransferases for synthesis of cell wall matrix polysaccharides. Plant Physiol Biochem 38:69–80
Hertzberg M, Aspeborg H, Schrader J, Andersson A, Erlandsson R, Blomqvist K, Bhalerao R, Uhlen M, Teeri TT, Lundeberg J, Sundberg B, Nilsson Peter, Sandberg G (2001) A transcriptional roadmap to wood formation. Proc Natl Acad Sci USA 98:14732–14737
Huntley SK, Ellis D, Gilbert M, Chapple C, Mansfield SD (2003) Significant increases in pulping efficiency in C4H–F5H-transformed poplars; improved chemical savings and reduced environmental toxins. J Agric Food Chem 51:6178–6183
Johansen DA (1940) Plant microtechnique. McGraw-Hill, NewYork
Johansson H (2003) Gene regulation of UDP-glucose synthesis and metabolism in plants. PhD dissertation, Umeå University
Katsube T, Kazuta Y, Mori H, Nakano K, Tanizawa K, Fukui T (1990) UDP-glucose pyrophosphorylase from potato tuber: cDNA cloning and sequencing. J Biochem 108:321–332
Kim G, Tsukaya H, Saito Y, Uchimiya H (1999) Changes in the shapes of leaves and flowers upon overexpression of the novel cytochromeP450 in Arabidopsis thaliana. Proc Natl Acad Sci USA 96:9433–9437
Kiyosumi D, Ishimizu T, Nakanishi T, Norioka S (2002) Pollen UDP-glucose pyrophosphorylase showing polymorphism well-correlated to the S genotype of Pyrus pyrifolia. Sex Plant Reproduction 14:315–323
Kleczkowski LA (1994) Glucose activation and metabolism through UDP-glucose pyrophosphorylase in plants. Phytochemistry 37:1507–1515
Kleczkowski LA, Geisler M, Ciereszko I, Johansson H (2004) UDP glucose pyrophosphorylase. An old protein with new tricks. Plant Physiol 134:912–918
Kleczkowski LA, Kunz S, Wilczynska M (2010) Mechanisms of UDP-glucose synthesis in plants. Crit Rev Plant Sci 29:191–203
Kleczkowski LA, Elisabeth F, Malgorzata W (2011) A common structural blueprint for plant UDP-sugar-producing pyrophosphorylases. Biochem J 439(3):375–379
Kotake T, Yamaguchi D, Ohzono H, Hojo S, Kaneko S, Ishida HK, Tsumuraya Y (2004) UDP-sugar pyrophosphorylase with broad substrate specificity toward various monosaccharide 1-phosphates from pea sprouts. J Biol Chem 279:45728–45736
Lin XF, Minamisawa N, Takechi K, Zhang WB, Sato H, Takio S, Tsukaya H, Takano H (2008) Isolation and characterization of the Larix gmelinii ANGUSTIFOLIA (LgAN) gene. Planta 228:601–608
Martz F, Wilczynska M, Kleczkowski LA (2002) Oligomerization status, with the monomer as active species, defines catalytic efficiency of UDP-glucose pyrophosphorylase. Biochem J 367:295–300
Meng M, Geisler M, Johansson H, Mellerowicz EJ, Karpinski S, Kleczkowski LA (2007) Differential tissue/organ-dependent expression of two sucrose and cold-responsive genes for UDP glucose pyrophosphorylase in Populus. Gene 389:186–195
Meng M, Fitzek E, Gajowniczek A, Wilczynska M, Kleczkowski LA (2009a) Domain-specific determinants of catalysis/substrate binding and the oligomerization status of barley UDP-glucose pyrophosphorylase. Biochim Biophys Acta 1794:1734–1742
Meng M, Geisler M, Johansson H, Harholt J, Scheller HV, Mellerowicz EJ, Kleczkowski LA (2009b) UDP-glucose pyrophosphorylase is not rate-limiting, but is essential in Arabidopsis. Plant Cell Physiol 50:998–1011
Mu H (2002) Screening of genes related to pollen development in a thermo-sensitive male sterile rice (Oryza sativa L.): cloning and characterization of UDP-glucose pyrophosphorylase. PhD dissertation, University of Hong Kong
Mu H, Ke JH, Liu W, Zhang CX, Yip WK (2009) UDP-glucose pyrophosphorylase 2 (OsUgp2), a pollen-preferential gene in rice, plays a critical role in starch accumulation during pollen maturation. Chin Sci Bull 54:234–243
Murashige T, Skoog F (1962) A revised medium for rapid growth and bioassays with tobacco tissue cultures. Physiol Plant 15:473–497
Murray MG, Thompson WF (1980) Rapid isolation of high molecular weight DNA. Nucleic Acids Res 8:4321–4326
Okazaki Y, Shimojima M, Sawada Y, Toyooka K, Narisawa T, Mochida K, Tanaka H, Matsuda F, Hirai A, Yokota Hirai M, Ohta H, Saito K (2009) A chloroplastic UDP-glucose pyrophosphorylase from Arabidopsis is the committed enzyme for the first step of sulfolipid biosynthesis. Plant Cell 21:892–909
Park JI, Ishimizu T, Suwabe K, Sudo K, Masuko H, Hakozaki H, Nou IS, Suzuki G, Watanabe M (2010) UDP-glucose pyrophosphorylase is rate limiting in vegetative and reproductive phases in Arabidopsis thaliana. Plant Cell Physiol 56:981–996
Pua EC, Lim SSW, Liu JZ (2000) Expression of a UDP-glucose pyrophosphorylase cDNA during fruit ripening of banana (Musa acuminata). Aust J Plant Physiol 27:1151–1159
Ruan YL, Llewellyn DJ, Furbank RT (2003) Suppression of sucrose synthase gene expression represses cotton fiber cell initiation, elongation, and seed development. Plant Cell 15:952–964
Ruzin SE (1999) Plant microtechnique and microscopy. Oxford University Press, Cambridge
Schädel C, Blöchl A, Richter A, Hoch G (2010) Quantification and monosaccharide composition of hemicelluloses from different plant functional types. Plant Physiol Biochem 48:1–8
Schnurr JA, Storey KK, Jung HJG, Somers DA, Gronwald JW (2006) UDP-sugar pyrophosphorylase is essential for pollen development in Arabidopsis. Planta 224:520–532
Sekin S (1978) Enzymatic determination of glucose, fructose and sucrose in tobacco. Tobacco Sci 23:75–77
Sowokinos JR (1976) Pyrophosphorylases in Solanum tuberosum: I. changes in ADP-glucose and UDP-glucose pyrophosphorylase activities associated with starch biosynthesis during tuberization, maturation, and storage of potatoes. Plant Physiol 57:63–68
Sowokinos JR, Thomas C, Burrell MM (1997) Allelic polymorphism of UDP-glucose pyrophosphorylase in potato cultivars and its association with tuber resistance to sweetening in the cold. Plant Physiol 113:511–517
Spychalla JP, Scheffler BE, Sowokinos JR, Bevan MW (1994) Cloning, antisense RNA inhibition and the coordinated expression of UDP-glucose pyrophosphorylase with starch biosynthetic genes in potato tubers. Plant Physiol 144:444–453
Updegraff DM (1969) Semi-micro determination of cellulose in biological materials. Anal Biochem 32:420–424
Valla S, Coucheron DH, Fjaervik E, Kjosbakken J, Weinhouse H, Ross P, Amikam D, Benziman M (1989) Cloning of a gene involved in cellulose biosynthesis in Acetobacter xylinum: complementation of cellulose-negative mutants by the UDPG pyrophosphorylase structural gene. Mol Gen Genet 217:26–30
Van Soest PJ (1963) Use of detergents in analysis of fibrous feeds. 2. A rapid method for determination of fiber and lignin. J Assoc Off Anal Chem 46:829–835
Van Soest PJ, Wine RH (1967) Use of detergents in analysis of fibrous feeds. 4. Determination of plant cell-wall constituents. J Assoc Off Anal Chem 50:50–55
Wang QH, Zhang X, Li FG (2011) Identification of a UDP-glucose pyrophosphorylase from cotton involved in cellulose biosynthesis in Arabidopsis thaliana. Plant Cell Rep 30:1303–1312
Weng CJ, Deng JY, Lin DG, Jeang CL (2009) Cloning, expression and characterization of UDP-glucose pyrophosphorylase from shoots of Bambusa oldhamii. J Biochem. doi:10.1093/jb/mvp084
Winter H, Huber SC (2000) Regulation of sucrose metabolism in higher plants: localization and regulation of activity of key enzymes. Crit Rev Biochem Mol Biol 35:253–289
Woo MO, Ham TH, Ji HS, Choi MS, Jiang W, Chu SH, Piao R, Chin JH, Kim JA, Park BS, Seo HS, Jwa NS, McCouch S, Koh HJ (2008) Inactivation of the UGPase1 gene causes genic male sterility and endosperm chalkiness in rice (Oryza sativa L.). Plant J 54:190–204
Wu XJ, Du M, Weng YQ, Liu D, Hu ZB (2002) UGPase of Astragalus membranaceus: cDNA clone, analyzing and expressing in Escherichia coli. Bot Sin 44:689–693
Zhu W, Lin X, Zhang W, Xu Y, Takano H (2011) Isolation and Sequence Analysis of Larix gmelinii Actin Gene. Biotechnol Bull 7:019
Zrenner R, Willmitzer L, Sonnewald U (1993) Analysis of the expression of potato uridinediphosphate-glucose pyrophosphorylase and its inhibition by antisense RNA. Planta 190:247–252
Acknowledgments
We would like to thank Professor Nakamura Kenzo in Chubu University for kindly providing pBI101-35::Gus-Hm vector. This study was supported by the National Natural Science Foundation of China (No. 31160143, No. 31060106, No. 30960030 and 31260168), Natural Science Foundation of Inner Mongolia (No. 2010BS0509 and 2009MS0505) and Chun-Hui project (No. Z2009-1-01009).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by R. Schmidt.
The nucleotide sequences reported in this paper have been submitted to the DNA data bank of Japan (DDBJ) under accession number AB830476.
Rights and permissions
About this article
Cite this article
Li, N., Wang, L., Zhang, W. et al. Overexpression of UDP-glucose pyrophosphorylase from Larix gmelinii enhances vegetative growth in transgenic Arabidopsis thaliana . Plant Cell Rep 33, 779–791 (2014). https://doi.org/10.1007/s00299-013-1558-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00299-013-1558-3