
Abstract
Requirement for antibiotic-resistance selection markers and difficulty in identifying transgenes with the highest expression levels remain the major obstacles for rapid production of recombinant proteins in plants. An alternative approach to producing transgenic plants free of antibiotic-resistance markers is the phenotypic-based selection with root-proliferation genes (rol genes) of Agrobacterium rhizogenes. By using Agrobacterium tumefaciens harboring the pRYG transformation vector with a cluster of rol genes linked to a heterologous gene of interest, we have developed a rapid transformation tool using hairy root formation as a selection marker. The expression of β-glucuronidase in newly induced transgenic tobacco roots could be detected as early as 12 days after inoculation. Higher levels of transgene expression in the roots correlated positively with the rates of root elongation on hormone-free medium and thus could be used for positive selection. When tobacco plants were transformed with pRYG harboring the expression cassette for secreted alkaline phosphatase (SEAP), the release of SEAP from roots of the fully regenerated transgenic plants could be quantified at rates as high as 28 μg/g root dry weight per day.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Transformation systems usually rely on a selectable marker gene to isolate and propagate a rare transformation event from the non-transformed plant tissue. Traditionally, this is achieved by co-expression of the gene of interest together with the antibiotic or herbicide resistance marker, which remains in the genome of a transgenic plant after the selection and regeneration processes are completed. While the product of the antibiotic-resistance gene does not interfere with plant development directly, the use of appropriate selective agents may significantly decrease the proliferation and regeneration capabilities of the transformed cells. In addition, the potential release of the antibiotic selection markers into the environment creates safety and ethical concerns, further hampering the agricultural use of many transgenic crops. Antibiotic resistance genes can be removed from the genome of the transgenic plant by a number of excision techniques, although each method has limitations that need to be considered (reviewed in Ebinuma et al. 2001). An alternative approach to producing transgenic plants free of antibiotic resistance markers is phenotypic-based selection using regeneration-promoting genes (reviewed in Zuo et al. 2002). This system may improve the overall efficiency of plant transformation by abandoning the need for antibiotic selection (Kunkel et al. 1999). Using this technique, Endo et al. (2002) have developed a single-step transformation system for rice, where marker-free transgenic rice plants were regenerated directly from the ipt-induced rice embryogenic tissues.
Similar to the widely used Agrobacterium tumefaciens, A. rhizogenes stably transfers a portion of its Ri plasmid into a plant cell at the time of infection, thereby inducing the rapid proliferation of hairy roots on stem and leaf sections of many plant species (Chilton 1982). This unique rooting response is caused by the expression of rol genes located within the infectious T-DNA region of A. rhizogenes (Slightom et al. 1986). When cloned in a cosmid and conjugated into an avirulent strain of A. rhizogenes, this region was capable of inducing a root proliferation response comparable to that induced by the wild-type A. rhizogenes strain (Vilaine and Casse-Delbart 1987). Since each transformation event can be easily distinguished as an independent hairy root clone developing on the hormone-free medium without any selective agents (Tepfer 1984), the system provides a valuable alternative approach for antibiotic-free plant transformation and regeneration.
Several techniques for the simultaneous introduction of rol genes and a gene of interest into the plant genome have been reported over the years. Simpson et al. (1986) used a binary system based on wild-type A. rhizogenes culture harboring a binary plant transformation vector to generate a hairy root clone expressing a target gene. However, this system is inefficient and requires an antibiotic selection to propagate the rare co-delivery event (Nicoll et al. 1995; Damiani et al. 1998). Co-transformation using a mixture of A. rhizogenes and A. tumefaciens cultures harboring the binary expression vector (Torregroza and Bouquet 1997) did not overcome these limitations, while the direct integration of the expression cassette into A. rhizogenes T-DNA via homologous recombination (Stougaard et al. 1987) proved cumbersome and inefficient. Finally, a removal MAT vector system combined with a positive rooting marker consisting of four genes—rolA, rolB, rolC, and rolD (Ebinuma et al. 1997)—was exploited for the transformation and regeneration of marker-free transgenic snapdragon plants, with only 3% of regenerated shoots reported suitable for the study (Cui et al. 2001).
We report here on a novel, antibiotic-free transformation strategy that results in rapid and efficient expression and secretion of recombinant proteins from the hairy roots only 2 weeks after the initial infection. We show that the hairy root phenotype, which is regarded as being undesirable in standard transformation experiments (Ebinuma et al. 1997; Cui et al. 2001), is beneficial if the goal of the transformation is to rapidly produce and analyze significant quantities of the recombinant protein.
Materials and methods
Vector construction
The rolABC gene cluster (harboring the rolA, rolB, and rolC genes) was cut out of the pUC9-rolABC plasmid as a 4.3-kb fragment (Schmulling et al. 1988) by EcoRI (complete)-PstI (partial) digestion and ligated into the EcoRI-PstI sites of pGreen (Hellens et al. 2000) to construct pRYG, the basic transformation vector for induction of the hairy root proliferation response. The β-glucuronidase (GUS) expression cassette including the mas2′ promoter, the GUS gene interrupted with an intron sequence, and the nos terminator was subcloned as the PvuII fragment from pE1226 (provided by Stanton Gelvin, Purdue University) into the EcoRI-PstI blunted sites of pLit-aps (Borisjuk et al. 2000) downstream from the aps element to produce pLit-aps-mas-GUS. From here, the complete GUS expression cassette including aps was ligated as the KpnI-BsiWI (blunted) fragment into the KpnI-EcoRI (blunted) sites of pRYG to produce pRYG(GUS). The blunted HindIII fragment, containing the SEAP gene with its own secretion signal peptide and the nos terminator from pNB-35S-SEAP (Borisjuk et al. 1999), was ligated into the blunted XbaI-XhoI site of pLit-aps-mas-GUS, replacing the GUS gene with that of SEAP to construct pLit-aps-mas-SEAP. From here, the KpnI-BsiWI (blunted) fragment harboring the entire SEAP expression cassette was ligated into the KpnI-EcoRI (blunted) sites of pRYG to produce pRYG(SEAP). KpnI-EcoRI (blunted) fragment from pLit-aps-mas-GUS was also ligated into the KpnI-HindIII (blunted) sites of pBin19-aps (Borisjuk et al. 2000), replacing the aps element in that construct with a complete GUS expression cassette to produce pBin-mas-GUS.
pRYG-bed plant tansformation
Mature tobacco seeds (Nicotiana tabacum L. cv. Wisconsin) were surface sterilized in 70% (v/v) ethanol and 10% (v/v) sodium hypochloride and then washed thoroughly in sterilized water. The seeds were germinated and subcultured on half-strength MS medium (Murashige and Skoog 1962) supplemented with 15 g sucrose and 7 g agar (pH 5.8). All reagents were obtained from Sigma-Aldrich (St. Louis, Mo.) unless otherwise stated. Approximately 1-cm2 sections of aseptic leaves of 2-month-old plants containing the vascular tissue of the major vein were excised using a sterile surgical blade that had been immersed into a 2-day-old culture of A. tumefaciens EHA105 harboring the appropriate transformation vector. The leaf sections were then transferred to half-strength MS medium for infection (25°C, 48 h in the dark). The bacterial culture was eliminated on MSR medium (half-strength MS supplemented with 400 mg/l carbenicillin) under standard conditions [28°C, 16/8-h (light/dark) photoperiod, irradiance of 150 mE/m2 per second supplied by a mixture of fluorescent and incandescent lamps]. The leaf sections were subsequently transferred every 4 days to fresh MSR medium with no hormone supplementation. In vivo infection of 3-month-old plants was performed under standard greenhouse conditions. Identically prepared bacterial cultures were inoculated at the mid-section of the tobacco stems using an aseptic syringe and needle (4 ml inoculum/8 cm of plant stem). The infected areas were immediately enclosed within a plastic tube sealed off by Parafilm (Fig. 3C) to provide a high humidity atmosphere favorable for root formation (Gaume et al. 2003). In all experiments, the hairy root proliferation response was observed 9–14 days after bacterial inoculation. Control inoculations with A. tumefaciens EHA105 cultures harboring the pGreen vector alone were performed following the identical procedure, and no root development was observed on control plants.
Binary system transformation and root analysis
In vivo infection of 3-month-old plants using the binary transformation system was performed as described above using the A. rhizogenes A4 bacterial culture harboring pBin19-mas-GUS or the wild-type A4 culture as a negative control. In these experiments, hairy roots developed 9–12 days post-infection. Three- to five-day-old roots were rinsed thoroughly in sterile water and stained for GUS activity as described below.
Selection and analysis of pRYG(SEAP)-transformed roots
For the experimental data reported in Fig. 5C, 3-day-old hairy roots attached to the leaf section were transferred to half-strength MS liquid medium (no agar) and cultivated on a rotary shaker (80 rpm, 20 ml media in 200-ml Erlenmeyer flasks) in the dark for 7 days. Portions of the fast-growing roots were excised and placed on top of a PVDF membrane with a little pressure applied to ensure the complete contact of the root surface with the membrane. Once the root tissue was removed, the dried membrane was heat-treated and stained for SEAP activity, as described below.
For the experimental data shown in Fig. 5D, E, 3-day-old hairy roots attached to the leaf section were positioned on the surface of the MSR medium and cultivated in a vertical orientation for 3 days. The observed roots were separated into slow- and fast-growing populations and transferred to fresh MSR plates for an additional 4 days of cultivation. At this point, the final root length of both populations was recorded, and 1-cm tip sections of the roots were excised along with the underlining block of medium. Blocks were allowed to air-dry overnight at 25°C, and the resulting gel film with root sections was stained for the SEAP activity as described below. For data presented in Table 1, total soluble protein was extracted from the individual root clones together with the supporting agar block by grinding in PBS buffer and subsequent centrifugation. Determination of protein quantity was done spectrophotometrically according to Bradford (1976), and the quantification of SEAP was performed as described below.
Plant regeneration and SEAP secretion
In all experiments where plant regeneration was performed, fast-growing root clones were cut into 1-cm-long sections and transferred to MSRR medium (MSR supplemented with 2 mg/l benzylaminopurine). Seven weeks after the initial infection, independent regenerated shoots were excised and cultivated on MSR medium. SEAP-transformed shoots with enhanced rooting response were transferred to the greenhouse under hydroponic conditions after 3 weeks, and SEAP secretion was measured as described below during a 24- period in 100 ml of 1 mM CaCl2 solution supplied to the plant roots.
GUS detection and SEAP quantification
In all experiments, GUS activity was visualized according to the published procedure of Jefferson et al. (1987). Detection of SEAP activity in plant roots (Fig. 5A, B), on the nitrocellulose membrane (Fig. 5C), and on the surface of the dried agar blocks (Fig. 5D, E) was performed by incubating the samples overnight at 37°C in a thin layer of BCIP-phosphatase substrate (Alkaline Phosphatase Isoenzymes assay; Sigma). Quantification of secreted SEAP in the hydroponic medium was performed using a chemiluminescence assay (Clontech, Palo Alto, Calif.) as described by Borisjuk et al. (1999). In order to minimize the background enzymatic activity associated with the presence of native phosphatases, all samples were treated at 65°C for 30 min prior to visualization and quantification of the SEAP activity. GUS and SEAP visualization was recorded using an Eclipse TE200 inverted microscope (Nikon, Tokyo) or Camedia C3030 camera (Olympus, Tokyo).
Results and discussion
A. rhizogenes-based binary transformation system
The original pBin19 vector carries the neomycin phosphotransferase II (nptII) gene under the control of the nopaline synthase (nos) promoter and nos terminator. For this particular study, a GUS expression cassette was cloned downstream of the nptII gene. The resulting vector (pBin-mas-GUS) was electroporated into A. rhizogenes strain A4, thus creating a binary transformation system (Fig. 1A). This system contained two independent T-DNA regions—the original A. rhizogenes T-DNA harboring the set of rol genes, and a binary T-DNA region of the pBin-mas-GUS vector. Upon successful inoculation, either T-DNA region (or both) can be transferred into the plant genome. Since induction of the hairy roots expressing GUS can be observed only as a result of the co-transformation event, this system can be particularly useful for the evaluation of the efficiency of a binary transformation system when no selection agent is applied.

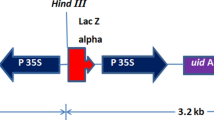
Schematic representation of transformation vectors used in this study. A pBin-mas-GUS. nos-NPTII kanamycin selection marker, mas2′ mannopine synthase promoter, aps amplification-promoting sequence. B pRYG. rolA, rolB, rolC are the cluster of rol genes responsible for root induction. C pRYG(GUS). The GUS gene is interrupted with an intron sequence shown as an orange-shaded box. D pRYG(SEAP). The yellow-shaded box depicts the SEAP original secretion signal peptide. Solid black triangles denote the location of the left and right T-DNA borders, respectively
If no exogenous auxins were supplied to the medium, only cells transformed with the A. rhizogenes T-DNA developed into auxin-independent root clones 12 days after inoculation. These roots displayed a classic hairy root proliferation response (Fig. 2A)—i.e., a set of phenotypic characteristics typical of the roots expressing rol genes (Schmulling et al. 1988). A second T-DNA originating from the pBin-mas-GUS transformation vector and harboring the GUS expression cassette was rarely co-transformed. The efficiency of co-delivery in the absence of selective pressure was low; approximately 5% of induced hairy roots expressed GUS at the level of visual detection (Fig. 2B). Similar co-transformation frequencies (4–7%) were observed in the hairy roots induced on aseptic tobacco leaf sections in vitro (data not shown). Although kanamycin selection, not easily applicable to tobacco stems, would have potentially eliminated the hairy roots that did not carry both nptII and GUS transgenes, a significant reduction in the total number of hairy roots would also follow. While not critical for tobacco transformation, this may become a major limitation step in the transformation and regeneration of many recalcitrant species. In addition, the stable introduction of antibiotic resistance genes into the plant genome is cumbersome and increases the risk of horizontal gene transfer (Kay et al. 2002).

Hairy roots induced on a tobacco stem in vivo using an Agrobacterium rhizogenes binary transformation system. A A 3-month-old tobacco stem developing a typical hairy root proliferation response 12 days after inoculation with an A. rhizogenes culture harboring the pBin-mas-GUS vector. B Less than 5% of newly developed roots showed visible GUS staining 15 days after inoculation. No exogenous hormones were added at the time of inoculation, and no kanamycin selection was applied
Efficiency of rol genes as selection markers
Vilaine and Casse-Delbart (1987) and Schmulling et al. (1988) reported that a cluster of three rolA, rolB, and rolC genes (rolABC) within the 4.3-kb region of the Ri plasmid is sufficient to induce a typical root proliferation response on A. rhizogenes-infected plants in vitro when no exogenous auxins are supplied to the cultivation medium. These observations provided the foundation for investigating the use of the rolABC cluster as a phenotypic marker for antibiotic resistance-free transformation. We hypothesized that if the rolABC cluster is linked to the gene of interest on the same transformation vector, it may become possible to develop a generic, single-step transformation system for the rapid expression and secretion of multiple protein targets. Unlike Cui et al. (2001), we decided to avoid using the rolD gene as a part of the selective marker. Earlier, Constantino et al. (1994) reported that rolD may encode a transportable factor that induces early flowering in tobacco. Therefore, the presence of rolD significantly shortens the life-time potential of a transgenic plant (Mauro et al. 1996) and may become an obstacle to a long-time secretion of recombinant proteins from plant roots.
The rolABC cluster was cloned into the pGreen plant transformation vector (Hellens et al. 2000) to generate a basic backbone vector, pRYG (Root Your Gene, Fig. 1B). Using the standard A. tumefaciens-mediated transformation protocol, we investigated the ability of this vector to induce hairy roots on the aseptic leaf sections of tobacco plants in vitro (Fig. 3A, B) or on stems of greenhouse-grown plants in vivo (Fig. 3C, D). No exogenous auxins were added throughout the time course of the experiment. Under these conditions no new roots developed on the control leaf sections (Fig. 3A, top) nor on the plant stems (Fig. 3D, top) mock-inoculated with the A. tumefaciens culture harboring the pGreen vector alone. To the contrary, plant tissues infected with A. tumefaciens containing pRYG showed a remarkable proliferation of roots both in vitro and in vivo 12 days after inoculation (Fig. 3A, D). In all experiments, the development of new roots was observed at the inoculation area only, with the majority of roots emerging from vascular tissue, including the major and secondary veins of the leaf sections. Since the rolABC cluster does not participate in auxin biosynthesis directly, but rather increases the sensitivity of plant tissues to the endogenous auxin levels (Cardarelli et al. 1987), a simple hormone-free MS medium was sufficient to select for a transformation event. Considering that no auxin-synthesizing genes are transferred into the plant genome, these newly developed roots should require an external auxin source to support their growth in vitro once the depot of in situ auxin is depleted. To maintain the optimum growth of the transformed roots generated on the leaf sections, we routinely added indole-3-acetic acid to the cultivation medium after the hairy roots were established—usually 3 weeks post-inoculation (Fig. 3B).

Induction of hairy root proliferation response by A. tumefaciens culture harboring pRYG. A Aseptic leaf sections developed hairy roots 12 days after inoculation in vitro on the hormone-free MSR medium. Leaf infection was performed using a sterile surgical blade immersed into a 48-h culture of A. tumefaciens harboring pGreen alone (negative control, top), wild-type A. rhizogenes (positive control, bottom left), and A. tumefaciens harboring pRYG (bottom right). B pRYG-induced roots retained the ability to grow in the hormone-free liquid medium 2 weeks after emergence. C Plastic tube system used to seal off the inoculation area to maintain the high humidity conditions required for root formation on tobacco stems in the greenhouse. D pRYG-based induction caused root proliferation response within 2 weeks post-inoculation (bottom); no roots appeared after pGreen-based mock inoculation (top)
Expression of GUS in tobacco hairy roots
In an attempt to estimate the efficiency of the pRYG transformation vector for target protein expression in hairy roots, we cloned GUS under the control of the root-specific mas2′ promoter (Leung et al. 1991) next to the rolABC cluster to produce pRYG(GUS). The aps element was subcloned in direct orientation immediately upstream of the mas2′ promoter region (Fig. 1C). This quantitative enhancer-like element positively influences both the transcription and copy number of nearby genes in either orientation (Borisjuk et al. 2000), putatively decreasing the positional effect often observed after transformation. When introduced into the tobacco genome via A. tumefaciens-mediated transformation, pRYG(GUS) induced the hairy root proliferation response similar to that observed with pRYG alone (Fig. 4). The variation in the inoculation technique, physiological status of the inoculated tissue, and/or other environmental conditions might have accounted for a significantly higher number of roots induced by the same amount of inoculum on the aseptic leaf sections.

Expression of GUS in tobacco hairy roots. A Uniform GUS expression in the 15-day-old hairy roots induced with pRYG(GUS) on aseptic tobacco leaf sections in vitro. B A representative portion of an in vivo-inoculated tobacco stem. The pRYG(GUS) vector induced hairy roots showing significantly higher variation in GUS staining among individual root clones. C Juvenile transgenic plant regenerated from a single pRYG(GUS) root clone preferentially expressed GUS in the root tissue. D, E Inheritance of the hairy root phenotype (D) and GUS gene (E) within the population of individual seedlings of the sexually produced T1 generation of the original transgenic line
GUS activity was visually detected as early as 1–5 days after the initial root appearance in 94% (144/150) and 79% (198/250) of newly developed roots that were induced in vitro (Fig. 4A) and in vivo (Fig. 4B), respectively. Bacterial expression cannot explain the observed GUS activity, since the GUS gene sequence was interrupted with an intron that can be correctly spliced out only in the eukaryotic expression system. Interestingly, while the roots that formed on the leaf sections showed very uniform GUS activity, those that formed on the stems exhibited much more variable spatial patterns and a greater magnitude of GUS expression. (Fig. 4A, B). The former can be at least partially explained by the aps effect, which in tobacco enhances transcription and reduces the positional effect (Borisjuk et al. 2000) in the leaf sections but not in the stems. The lack of the 100% transformation/ expression ratio in stem-produced hairy roots could possibly be explained by the positional effect, which is not circumvented by the aps effect, or by gene silencing and integration errors more pronounced in the in situ stem tissues.
Seven to eight weeks after inoculation, we obtained multiple regenerated shoots from 81% (71/88) of the root fragments transferred to the regeneration medium. Most of the regenerated shoots showed some degree of Ri symptoms, including the over-proliferation of roots and root hairs. From these, 8% (6/71) of the regenerated plants exhibited very few phenotypic abnormalities in the leaves and flowers and, when grown to maturity, they produced abundant seeds. In most of the regenerants, the strong GUS expression was observed in the roots and localized stem areas, as is typically expected from the mas2′ promoter (Fig. 4C). Although the mas2′ promoter is preferentially expressed in roots and wounded leaves (Teeri et al. 1989), a gradual downwards increase in expression beginning from the shoot apex down to the base was observed in the stems, leaves, and petioles (Langridge et al. 1989).
In order to study transgene expression in the next generation, the seeds of the above transgenic plants were surface-sterilized and germinated in vitro. A typical population of 14-day-old seedlings is shown in Fig. 4D. Transgenic seedlings were easily distinguished by their modified hairy root systems, as expected if the activity of the rol genes is retained in the next generation. Nearly all of the seedlings (94/121) with hairy roots tested positive for GUS expression, while the seedlings that developed a normal root system failed to show any GUS activity (Fig. 4E).
Extracellular targeting and secretion system based on pRYG
The ability to rapidly introduce a transgene into differentiated plant organs, combined with strong and balanced expression that does not require antibiotic selection, provides a valuable tool for fundamental and applied plant molecular biology. A simple, single-step transformation system that achieves high transformation frequencies may significantly shorten the gene-to-protein time and make multiple protein evaluations manageable. For example, such a transformation strategy may improve the efficiency of the rhizosecretion-based protein production system (Borisjuk et al. 1999) in terms of speed, multiple target validation, and yield potential. As with many other production systems, including the periplasmic space of Escherichia coli (Veiga et al. 1999), the growth medium of mammalian cell cultures (Fouser et al. 1992), or the mammary glands of transgenic animals (Simons et al. 1987), rhizosecretion relies on extracellular targeting and secretion to achieve the highest possible yields.
To test the applicability of the pRYG-based transformation system to rhizosecretion, we cloned the human secreted alkaline phosphatase (SEAP) gene with its secretion signal peptide into pRYG under the control of the same genetic elements as employed for GUS, thereby creating a pRYG(SEAP) transformation vector (Fig. 1D). We used this vector to inoculate aseptic tobacco leaf sections in vitro; no exogenous auxins were added throughout the time course of the experiment. As expected, transgenic roots developed from the inoculation area within 9–12 days, and expression of the SEAP gene was observed at the time of root development. When the emerging root tips were excised from the origin points, heat treated to eliminate the endogenous phosphatase activity, and incubated in the presence of an alkaline phosphatase substrate, the transformed root tissue developed a characteristic blue stain resulting from a substrate breakdown by heat-stable recombinant SEAP (Fig. 5B). The control roots transformed with the pRYG vector alone showed no SEAP activity (Fig. 5A). To further confirm the expression of SEAP, several leaf sections carrying the pRYG(SEAP) roots were transferred to liquid medium and cultivated for 7 days. A portion of the fast-growing root was cut and placed on top of a PVDF membrane with slight pressure applied to ensure complete contact of the root surface with the membrane in a process similar to tissue printing. After the printed root tissue was removed, the membrane was heat-treated and stained for SEAP activity. As expected, SEAP activity was easily detected on the outside surface of the root (Fig. 5C).

SEAP production and secretion from the pRYG(SEAP)-transformed hairy roots. A, B Light microscopy (×100) of the emerging hairy roots induced by pRYG (A) and pRYG(SEAP) (B). SEAP activity was visualized as a blue staining of the root tissues producing the recombinant protein. C SEAP activity detected on the surface of the 10-day-old pRYG(SEAP) root clone cultivated in the liquid medium (×10); root tissue was printed onto the PVDF membrane with slight pressure applied and removed prior to staining. D, E Note the visual difference in SEAP secretion from slow- (D) and fast-growing (E) populations of hairy roots cultivated on the surface of hormone-free MSR medium in a vertical orientation. All stained samples were pre-incubated at 65°C for 30 min to remove background phosphatase activity and then incubated overnight at 37°C in a thin layer of BCIP-phosphatase substrate
Hairy roots generated on the tobacco leaf sections exhibited different rates of growth when kept on the surface of the MSR medium in a vertical orientation. Since root growth-promoting rol genes and the SEAP gene were located on the same transformation vector, we wanted to determine whether the rate of root growth could be used for positive visual selection of those root clones with higher levels of transgene expression. Hairy roots from the same transformation experiment were divided into slow- and fast- growing populations based on their elongation rates (Table 1). Slow-growing roots elongated on average 2.4-fold slower than their fast-growing counterparts, and produced four- to five-fold less SEAP.
Both numbers were significant (t-test, P<0.05). Some roots developing after the inoculation may have had partial or complete silencing of the entire transgene locus due to chromatin confirmation and/or co-suppression and thus did not grow efficiently on the hormone-free medium. Therefore, the slow-growing roots produced much less transgenic protein, and thus can be effectively eliminated after simple visual selection. To further confirm this hypothesis through the visual observations, we excised the tip portions of the roots together with the supporting block of agar medium and stained them for the secreted SEAP. As expected, the fast-growing roots developed a stronger blue-colored zone typical of the greater SEAP enzymatic activity released into the medium (Fig. 5D, E).
The mature plants from independent transgenic lines regenerated from different roots selected within the fast-growing population still showed some variations in SEAP production. Some shoots regenerated spontaneously from the green regions of hairy roots grown on the surface of the MSR medium; other shoots were regenerated by transferring segments of the fast-growing roots onto the regeneration medium after 4 weeks in culture. Fully transgenic regenerants retained the ability to secrete SEAP into hydroponic culture medium. Eighty-seven percent (26/30) of the selected regenerants secreted per day 6–28 μg/g root DW of biologically active SEAP from their roots (Fig. 6).

Quantification of SEAP secretion from the roots of 30 individual transgenic lines (gray columns) regenerated from a fast-growing population of pRYG(SEAP) root clones compared with a control exudate from the wild-type plant (white column). Recombinant SEAP secretion is quantified as micrograms of secreted protein per gram root dry weight per day. Each bar represents the mean values of three replicates. The experiment was repeated with similar results
Marker-free transformation has been successfully demonstrated by using several plant and non-plant genes that are capable of promoting organ differentiation or explant regeneration (Zuo et al. 2002). However, a number of morphological aberrations resulting from the activity of some of these genes (i.e., rol cluster) partially limits the use of this strategy as a plant transformation tool aiming at expressing a target protein throughout an entire plant (Cui et al. 2001). On the contrary, the pRYG-based system presented here has not been developed as a merely transformation strategy. Using rol genes as a phenotypic selection marker provides an additional advantage of generating a higher ratio of young, metabolically active root tissue with multiple adventitious root growth (Baiza et al. 1998), resulting in higher rates of protein expression. Since a large number of transformed roots can be induced rapidly and every hairy root emerges as the result of an independent transformation event (Tepfer 1984), the described system may provide new means for simultaneous analysis of multiple protein targets and/or cDNA libraries on a large scale. This system also enables the simultaneous subcloning and expression of more than one target gene—for example, a heavy and a light chain of an antibody (Komarnytsky et al. unpublished).
Conclusions
The utilization of the pRYG vector harboring three rol genes from A. rhizogenes is a fast and effective method for the generation of transgenic hairy roots without the introduction of antibiotic resistance. The system utilizes phenotypic selection in the absence of exogenously supplied hormones and can be visually evaluated for the relative strength of transgene expression. The pRYG-based transformation system can also be used for the rhizosecretion of heterologous proteins, as has been shown for SEAP. Root clones with the highest secretion of the heterologous protein can be readily regenerated into transgenic lines that retain the ability to secrete the target protein over their lifetime. In addition, this approach allows the production of large numbers of independently transformed roots within a small in vitro system or on intact plants. It is likely that future careful selection of genetic elements that regulate target protein expression and secretion will further increase the speed and efficiency of the pRYG-based transformation system. Root clones expressing individual transgenes may be easily propagated as hairy root cultures or regenerated into mature plants, adding to the versatility of the system.
Abbreviations
- GUS::
-
β-Glucuronidase
- SEAP::
-
Secreted alkaline phosphatase
- rolABC::
-
Cluster of rolA, rolB, and rolC genes
References
Baiza AM, Quiroz A, Ruiz JA, Maldonado-Mendoza I, Loyola-Vargas VM (1998) Growth patterns and alkaloid accumulation in hairy root and untransformed root cultures of Datura stramonium. Plant Cell Tissue Organ Cult 54:123–130
Borisjuk NV, Borisjuk LG, Logendra S, Petersen F, Gleba Y, Raskin I (1999) Production of recombinant proteins in plant root exudates. Nat Biotechnol 17:466–469
Borisjuk N, Borisjuk L, Komarnytsky S, Timeva S, Hemleben V, Gleba Y, Raskin I (2000) Tobacco ribosomal DNA spacer element stimulates amplification and expression of heterologous genes. Nat Biotechnol 18:1303–1306
Bradford MM (1976) A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Cardarelli M, Mariotti D, Pomponi M, Spano L, Capone I, Costantino P (1987) Agrobacterium rhizogenes T-DNA genes capable of inducing hairy root phenotype. Mol Gen Genet 209:475–480
Chilton MD, Tepfer DA, Petit A, David C, Casse-Delbart F, Empe J (1982) Agrobacterium rhizogenes inserts T-DNA into the genomes of the host plant root cells. Nature 295:432–434
Constantino P, Capone I, Cardarelli M, De Poalis A, Mauro ML, Trovato M (1994) Bacterial plant oncogenes: the rol genes saga. Genetica 94:203–211
Cui M, Takayanagi K, Kamada H, Nishimura S, Handa T (2001) Efficient shoot regeneration from hairy roots of Antirrhinum majus L. transformed by the rol-type MAT vector system. Plant Cell Rep 20:55–59
Damiani F, Paolocci F, Consonni G, Crea F, Tonelli C, Arcioni S (1998) A maize anthocyanin transactivator induces pigmentation in hairy roots of dicotyledonous species. Plant Cell Rep 17:339–344
Ebinuma H, Sugita K, Matsunaga E, Yamakado M, Komamine A (1997) Principle of MAT vector. Plant Biotechnol 14:133–139
Ebinuma H, Sugita K, Matsunaga E, Endo S, Yamada K, Komamine A (2001) Systems for the removal of a selection marker and their combination with a positive marker. Plant Cell Rep 20:383–392
Endo S, Sugita K, Sakai M, Tanaka H, Ebinuma H (2002) Single-step transformation for generating marker-free transgenic rice using the ipt-type MAT vector system. Plant J 30:115–122
Fouser LA, Swanberg SL, Lin BY, Benedict M, Kelleher K, Cumming DA, Riedel GE (1992) High level expression on a chimeric anti-ganglioside GD2 antibody: genomic kappa sequences improve expression in COS and CHO cells. Biotechnology 10:1121–1127
Gaume A, Komarnytsky S, Borisjuk N, Raskin I (2003) Rhizosecretion of recombinant proteins from plant hairy roots. Plant Cell Rep 21:1188–1193
Hellens RP, Edwards EA, Leyland NR, Bean S, Mullineaux PM (2000) pGreen: a versatile and flexible binary Ti vector for Agrobacterium-mediated plant transformation. Plant Mol Biol 42:819–832
Jefferson RA, Kavanagh TA, Bevan MW (1987) GUS fusions: β-glucuronidase as a sensitive and versatile gene fusion marker in higher plants. EMBO J 6:3901–3907
Kay E, Vogel TM, Bertolla F, Nalin R, Simonet P (2002) In situ transfer of antibiotic resistance genes from transgenic (transplastomic) tobacco plants to bacteria. Appl Environ Microbiol 68:3345–3351
Kunkel T, Niu QW, Chan YS, Chua NH (1999) Inducible isopentenyl transferase as a high-efficiency marker for plant transformation. Nat Biotechnol 17:916–919
Langridge W, Fitzgerald K, Koncz C, Shell J, Szalay A (1989) Dual promoter of Agrobacterium tumefaciens mannopine synthase genes is regulated by plant growth hormones. Proc Natl Acad Sci USA 86:3219–3223
Leung J, Fukuda H, Wing D, Schell J, Masterson R (1991) Functional analysis of cis-elements, auxin response and early developmental profiles of the mannopine synthase bi-directional promoter. Mol Gen Genet 230:463–474
Mauro ML, Trovato M, De Paolis A, Gallelli A, Costantino P, Altamura MM (1996) The plant oncogene rolD stimulates flowering in transgenic tobacco plants. Dev Biol 180:693–700
Murashige T, Skoog F (1962) A revised medium for rapid growth and bioassays with tobacco tissue culture. Physiol Plant 15:493–497
Nicoll SM, Brigham LA, Wen F, Hawes MC (1995) Expression of transferred genes during hairy root development in pea. Plant Cell Tissue Organ Cult 42:57–66
Schmulling T, Schell J, Spena A (1988) Single genes from Agrobacterium rhizogenes influence plant development. EMBO J 7:2621–2629
Simons JP, McClenaghan M, Clark AJ (1987) Alteration of the quality of milk by expression of sheep β-lactoglobulin in transgenic mice. Nature 328: 530–532
Simpson RB, Spielmann A, Margossian L, McKnight TD (1986) A disarmed binary vector from Agrobacterium tumefaciens functions in Agrobacterium rhizogenes. Plant Mol Biol 6:403–415
Slightom JL, Durand-Tardif M, Jouanin L, Tepfer D (1986) Nucleotide sequence analysis of TL-DNA of Agrobacterium rhizogenes agropine-type plasmid. Identification of open reading frames. J Biol Chem 261:108–121
Stougaard J, Abildsten D, Marcker KA (1987) The Agrobacterium rhizogenes pRi TL-DNA segment as a gene vector system for transformation of plants. Mol Gen Genet 207:251–255
Teeri T, Levaslaiho H, Franck M, Uotila J, Heino P, Palva E, Van Montagu M, Herrera-Estrella L (1989) Gene fusions to lacZ reveal new expression pattern of chimeric genes in transgenic plants. EMBO J 8:343–350
Tepfer D (1984) Transformation of several species of higher plants by Agrobacterium rhizogenes: sexual transmission of the transformed genotype and phenotype. Cell 37:959–967
Torregroza L, Bouquet A (1997) Agrobacterium rhizogenes and Agrobacterium tumefaciens co-transformation to obtain grapevine hairy roots producing the coat protein of grapevine chrome mosaic nepovirus. Plant Cell Tissue Organ Cult 49:53–62
Veiga E, de Lorenzo V, Fernandez LA (1999) Probing secretion and translocation of a β-autotransporter using a reporter single-chain Fv as a cognate passenger domain. Mol Microbiol 33:1232–1243
Vilaine F, Casse-Delbart F (1987) Independent induction of transformed roots by the TL and TR regions of the Ri plasmid of agropine type Agrobacterium rhizogenes. Mol Gen Genet 206:17–23
Zuo J, Niu QW, Ikeda Y, Chua NH (2002) Marker-free transformation: increasing transformation frequency by the use of regeneration-promoting genes. Curr Opin Biotechnol 13:173–180
Acknowledgements
The authors thank Dr. Stanton Gelvin (Purdue University) for the mas2′ promoter, and Dr. Thomas Schmulling (Freie Universitat Berlin) for the cluster of rol genes. We are grateful to Ivan Jenkins for his technical assistance in the greenhouse, and to Nir Yakoby for helpful discussions and for reviewing the manuscript. A. Gaume is a recipient of a Swiss Science Foundation postdoctoral fellowship.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by A. Altman
Rights and permissions
About this article
Cite this article
Komarnytsky, S., Gaume, A., Garvey, A. et al. A quick and efficient system for antibiotic-free expression of heterologous genes in tobacco roots. Plant Cell Rep 22, 765–773 (2004). https://doi.org/10.1007/s00299-004-0761-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00299-004-0761-7