
Abstract
We assessed the detection rate of antineutrophil cytoplasmic antibody (ANCA) and investigated the clinical significance of ANCA positivity at diagnosis in patients with IgA vasculitis (Henoch-Schönlein purpura). We retrospectively reviewed their medical records of 86 IgA vasculitis patients. We divided IgA vasculitis patients based on ANCA positivity and compared variables at diagnosis and poor outcomes and medication during follow-up between the two groups. All-cause mortality, relapse, chronic kidney disease (CKD) (stage 3–5) and end-stage renal disease (ESRD) were defined as poor outcomes. We assessed the renal histological features based on the International Study of Kidney Disease in Children (ISKDC) classification and Oxford classification. Comparison of cumulative survivals was analysed by the Kaplan–Meier survival analysis. Five of 86 IgA vasculitis patients (5.8%) had ANCA and all ANCA-positive patients had myeloperoxidase (MPO)-ANCA. IgA vasculitis patients with ANCA exhibited pulmonary and nervous involvement of IgA vasculitis more frequently than those without. There was no significant difference in renal involvement between the two groups. There were no significant differences in renal histological features and poor outcomes related to renal function between IgA vasculitis patients with and without ANCA. In addition, 5 IgA vasculitis patients did not meet the classification criteria for ANCA-associated vasculitis (AAV). Particularly, there were no significant differences in CKD and ESRD-free survival rates between IgA vasculitis patients with and without ANCA. 5.8% of IgA vasculitis patients had MPO-ANCA and poor outcomes of IgA vasculitis were not affected by the presence of ANCA.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
IgA vasculitis, that is identically called Henoch-Schönlein purpura, is a systemic autoimmune vasculitis which primarily affects small-sized vessels including capillaries, arterioles and venules [1]. IgA vasculitis is characterised by various clinical manifestations: arthralgia/arthritis, abdominal pain, glomerulonephritis, lung lesions and neuropathies may occur based on skin purpura [1, 2]. IgA vasculitis commonly occurs in patients of age under 20 years and the annual incidence in children ranges from 10 to 20 cases of 100,000 patients per year, while that in adults is as lower as 1.3 cases of 100,000 patients per year [3, 4]. Early kidney involvement of IgA vasculitis may provoke seriously poor outcomes during follow-up such as relapse, chronic kidney disease (CKD) and end-stage renal disease (ESRD) [5, 6].
According to the 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides (the 2012 CHCC definitions), small vessel vasculitis can be divided into two categories such as antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV) and immune complex vasculitis characterised by immune complex deposits [1, 7]. IgA vasculitis is one of immune complex vasculitis and described as vasculitis with IgA1 and complement 3 (C3)-dominant immune deposits. Thus, ANCA may theoretically be rarely detected in patients with IgA vasculitis. For this reason, so far, there have been only a few case reports on ANCA-positive IgA vasculitis patients to date [8]. However, there was no report on the clinical implication of ANCA in a considerable number of adult patients with IgA vasculitis yet. Hence, in this study, we assessed the detection rate and the subtype of ANCA and investigated the clinical significance of ANCA positivity at diagnosis in patients with IgA vasculitis. Furthermore, we reclassified IgA vasculitis patients with ANCA based on the classification criteria for both IgA vasculitis and AAV.
Patients and methods
Patients
We collected the list of 97 patients with IgA vasculitis using the Clinical Data Repository System and retrospectively reviewed their medical records. The inclusion and exclusion criteria were as follows: (1) patients who had been first classified as IgA vasculitis at the Department of Internal Medicine, Yonsei University College of Medicine, Severance Hospital, from March 2005 to January 2019; (2) patients who fulfilled the European League against Rheumatism/Paediatric Rheumatology International Trials Organisation/Paediatric Rheumatology European Society (EULAR/PRINTO/PRES) criteria for IgA vasculitis. Since the American College of Rheumatology criteria for IgA vasculitis has an item of age ≤ 20 years at disease onset and our study included only adult patients, we utilised the EULAR/PRINTO/PRES criteria. The EULAR/PRINTO/PRES criteria consist of mandatory palpable purpura, not thrombocytopenia/petechiae and more than one of the four conditions such as diffuse abdominal pain, typical leukocytoclastic vasculitis with predominant IgA deposits or proliferative glomerulonephritis with predominant IgA deposit on histology, arthritis or arthralgia and renal involvement (proteinuria: > 0.3 g/24 h or > 30 mmol/mg of urine albumin to creatinine ratio on a spot morning sample; and/or haematuria, red blood cell (RBC) casts: > 5 RBC per high power field or ≥ 2 + on dipstick or RBC casts in the urinary sediment) [9]; (3) patients who had well-documented medical records to collect clinical and laboratory data at diagnosis of IgA vasculitis and to confirm the fulfilment of the classification criteria; (4) patients who had the results of both indirect immunofluorescence assay and antigen-specific assay for ANCA such as perinuclear (P)-ANCA, C-ANCA, MPO-ANCA and proteinase 3 (PR3)-ANCA at diagnosis. In patients who tested positive by indirect fluorescence assay but negative by antigen-specific assays, P-ANCA positive was considered as MPO-ANCA positive and C-ANCA positive as PR3-ANCA; (5) patients who had been followed up for at least ≥ 12 weeks; (6) patients who had never been classified as other vasculitis other than IgA vasculitis or medical history to affect either the classification of IgA vasculitis or ANCA positivity prior to or at diagnosis, particularly, chronic liver diseases, coexisting malignancies, serious comorbidities and serious infection, which were identified by the 10th revised International Classification Diseases (ICD-10); (7) patients who had never received immunosuppressive drugs for IgA vasculitis prior to diagnosis, which might be related to ANCA positivity and were verified by the Korean Drug Utilisation Review (DUR) system.
Of 97 patients screened, we excluded five IgA vasculitis patients due to the insufficient medical records, three IgA vasculitis patients due to no results of testing for ANCA at diagnosis and one IgA vasculitis patient due to a concomitant malignancy. Moreover, we further excluded two patients who fulfilled the EULAR/PRINTO/PRES criteria for IgA vasculitis and furthermore met the 2007 European Medicines Agency algorithm (the 2007 EMA algorithm) for AAV and classic polyarteritis nodosa (cPAN), in which authors added the modified contents of the 2012 CHCC definitions [7, 10]. Finally, we included 86 IgA vasculitis patients in this study.
Clinical and laboratory data at diagnosis and poor outcomes and medications during follow-up
As was described in Table 1, we obtained demographic, clinical and laboratory data at the time of diagnosis. Diabetes mellitus and hypertension at diagnosis of IgA vasculitis were counted as comorbidities. In this study, we reviewed the frequency of all-cause mortality, relapse, CKD (stage 3–5) and ESRD as poor outcomes of IgA vasculitis. All-cause mortality was defined as death for any reason during follow-up. Relapse was defined as a new development of symptoms of IgA vasculitis in a previously asymptomatic patient (at least for 1 month) [11]. CKD (stage 3–5) was defined as an estimated glomerular filtration rate < 60 mL/min/1.73 m2, which was calculated by the equation proposed by Chronic Kidney Disease Epidemiology Collaboration (eGFR (CKD-EPI)) [12] and ESRD was defined as an impairment of renal function which requires dialysis. Thus, CKD (stage 3–5) included ESRD in this study. The follow-up duration was determined as the gap time from the diagnosis of AAV to the last visit. In patients with poor outcomes, the follow-up duration was defined as the gap time from the diagnosis of IgA vasculitis to death for the dead patients, to relapse for the patients with relapse, to the first notice of eGFR < 60 mL/min/1.73 m2 to CKD (stage 3–5) patients and to the initiation of dialysis for the ESRD patients. Also, we counted the number of patients who received glucocorticoid, cyclophosphamide, azathioprine, calcineurin inhibitors and mycophenolate mofetil for the treatment of IgA vasculitis.
Histological assessment
We assessed the renal histological features based on the International Study of Kidney Disease in Children (ISKDC) classification and Oxford classification [13, 14]. We simplified the ISKDC classification into six grades as follows: minimal alterations (grade I), mesangial proliferation (grade II), mesangial proliferation or sclerosis with < 50% crescents (grade III), mesangial proliferation or sclerosis with 50–75% crescents (grade IV), mesangial proliferation or sclerosis > 75% crescents (grade V) and membranoproliferative-like glomerulonephritis (grade VI). We also used the Oxford classification as five simplified categories without subclasses such as mesangial hypercellularity, endocapillary hypercellularity, segmental glomerulosclerosis/adhesion, tubular atrophy/interstitial fibrosis and cellular/fibro-cellular crescents.
Statistical analyses
All statistical analyses were conducted using SPSS software (version 23 for Windows; IBM Corp., Armonk, NY, USA). Continuous variables were expressed as a median and interquartile range (IQR)/a mean ± standard deviation (SD) and categorical variables were expressed as number and the percentage. Significant differences in categorical variables between the two groups were analysed using the Chi square and Fisher’s exact tests. Significant differences in continuous variables between the two groups were compared using the Mann–Whitney test. Comparison of cumulative survivals between the two groups was analysed by the Kaplan–Meier survival analysis. P values less than 0.05 were considered statistically significant.
Results
Characteristics of 86 patients with IgA vasculitis
The detection rate of ANCA in IgA vasculitis patients was 5.8% (5 of 86 patients) and all ANCAs were MPO-ANCA. Based on the EULAR/PRINTO/PRES criteria, cutaneous manifestation (purpura) was found in all patients and renal manifestation was the second common manifestation (58.1%), followed by gastrointestinal manifestation (44.2%) and arthritis/arthralgia (33.7%). The median serum IgA and C3 were 308.0 mg/dL and 112.2 mg/dL. Proteinuria and haematuria were detected in 58.1% and 53.5% of patients, respectively. Seven patients (8.1%) had diabetes mellitus and 19 (22.1%) hypertension at diagnosis. The median follow-up duration was 38.5 months. One patient died and 12 experienced relapse. Of 13 patients with CKD (stage 3–6), 5 patients had ESRD. The most commonly administered drug was glucocorticoid (51.2%), followed by azathioprine (7.0%) (Table 1).
Comparison of clinical and laboratory data
IgA vasculitis patients with ANCA were older than those without. Among clinical manifestations at diagnosis, patients with ANCA exhibited pulmonary (20.0% vs. 1.2%, P = 0.007) and nervous systemic manifestations (40.0% vs. 2.5%, P < 0.001) more frequently than those without. Among laboratory results, patients with ANCA exhibited the significantly higher median ESR (72.0 vs. 31.0 mm/h, P = 0.007) and CRP (8.8 vs. 2.6 mg/L, P = 0.035) than those without. However, there were no significant differences in comorbidities at diagnosis and poor outcomes and medications during follow-up between the two groups (Table 2).
Comparison of renal histological features
Fifty of the 86 patients exhibited renal manifestation, which was defined by the EULAR/PRINTO/PRES criteria, and 30 of the 50 patients with renal manifestation underwent a renal biopsy. For this reason, we could analyse the renal histological features only in these 30 patients. Two of 30 patients with renal histology had ANCA (6.7%). There were no significant differences in renal histological features based on both the ISKDC and Oxford criteria between IgA vasculitis patients with and without ANCA. Moreover, ANCA positivity did not affect CKD (stage 3–5) or ESRD occurrence (Table 3).
Comparison of the cumulative survival rates
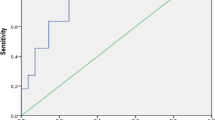
The cumulative patients’ survival rate and relapse-free survival rate did not seem to be affected by ANCA positivity at diagnosis in IgA vasculitis patients. Also, there were no significant differences in CKD (stage 3–5) and ESRD-free survival rates between IgA vasculitis patients with and without ANCA (Fig. 1).

Comparison of the cumulative survival rates of poor outcomes between IgA vasculitis patients with and without ANCA. There were no significant differences in all-cause mortality, relapse, CKD (stage 3–5) and ESRD-free survival rates between IgA vasculitis patients with and without ANCA. ANCA antineutrophil cytoplasmic antibody, CKD chronic kidney disease, ESRD end-stage renal disease
Application of the classification criteria for AAV to IgA vasculitis patients with ANCA
Although at the entry of this study, we already excluded two patients with both IgA vasculitis and AAV, to reconfirm the classification of IgA vasculitis in patients with ANCA, we applied the 2007 EMA algorithm to five IgA vasculitis patients with ANCA. All patients with ANCA did not meet the 2007 EMA algorithm modified by the revised 2012 CHCC definitions for MPA, granulomatosis with polyangiitis (GPA) and eosinophilic GPA (EGPA) at all [7, 10]. Two IgA vasculitis patients with ANCA underwent renal biopsy which revealed histological features compatible with IgA vasculitis nephritis. Thus, they could be distinguished from MPA, whereas three IgA vasculitis patients with ANCA did not undergo renal biopsy but two of them exhibited proteinuria without haematuria, which was compatible with the definition of renal involvement of IgA vasculitis based on the EULAR/PRINTO/PRES criteria. However, the 2007 EMA algorithm defines renal vasculitis as haematuria (RBC casts or > 10% RBC dysmorphism) or haematuria 2 + along with proteinuria 2 + on urine stick. These two patients showed no MPA-specific renal vasculitis and, thus, they could not be reclassified as MPA. The rest one patient without renal histology exhibited neither GPA surrogate marker nor evidence of renal vasculitis and, thus, could not be classified as MPA (Table 4).
Discussion
In this study, we evaluated the frequency of ANCA detected at diagnosis and its clinical significance in 86 patients with IgA vasculitis during follow-up and found that 5.8% of IgA vasculitis patients had MPO-ANCA and poor outcomes of IgA vasculitis were not affected by the presence of ANCA. There have been several studies investigating the clinical implication of ANCA positivity in patients with IgA nephropathy, which is indistinguishable from glomerulonephritis of IgA vasculitis [7]. They reported that ANCA-positive IgA nephropathy exhibited more severe clinical and histological features but they suggested that not all ANCA play pathogenic roles in the pathogenesis of IgA nephropathy [15,16,17], whereas, in Korea, there have been only two case reports to date: one is regarding positive cytoplasmic (C)-ANCA in one of 30 Korean children patients with IgA vasculitis [18] and the other is a case of elderly onset IgA vasculitis with myeloperoxidase (MPO)-ANCA [19]. We have reviewed the literature and could find two hypotheses on the production and detection of ANCA positivity in IgA vasculitis patients. Kim and Shin cited previous studies reporting an increase in TNF-alpha levels and a decrease in regulatory T cell population in IgA vasculitis patients. And they insisted that elevated serum TNF-alpha and deteriorated regulatory T cells might provoke pro-inflammatory cytokine storm, resulting in producing C-ANCA [18], whereas Ronda and colleagues demonstrated ANCA of IgA isotype in IgA vasculitis patients and suggest that the IgA ANCA target might be a membrane-associated protein rather than cytoplasmic protein and it might be different from the targets commonly recognised by IgG ANCA [20, 21]. Thus, we conclude that ANCA can be detected as a result of an inflammatory reaction of IgA vasculitis and, furthermore, it may be a pathogenic cause of IgA-related diseases.
In this study, ANCA subtype which was observed in five patients with IgA vasculitis was MPO-ANCA but not PR3-ANCA. In terms of renal vasculitis, MPO-ANCA is primarily associated with necrotising crescentic glomerulonephritis of MPO-ANCA vasculitis but is rarely detected in IgA nephropathy. Ogawa and colleagues described the phenomenon of MPO-ANCA positivity in patients with IgA vasculitis (or IgA nephropathy) as the consequence of IgA-MPO-ANCA rather than the independent generation of MPO-ANCA [22]. Valenzuela and colleagues proposed a concept of microscopic polyangiitis and IgA vasculitis overlapping syndrome which is characterised by the synergistic effect of MPO-ANCA and IgA-containing immune complex [23]. Similarly, in our study, the frequencies of lung and neurological manifestations were significantly higher in IgA vasculitis patients with ANCA than those without (20.0% vs. 1.2%, P = 0.007 and 40.0% vs. 2.5%, P < 0.001). Given that central and peripheral nervous system dysfunction or pulmonary manifestation are known as very uncommon in IgA vasculitis patients [24, 25], we assume that ANCA might be involved in systemic symptoms other than those common in IgA vasculitis patients in the form of MPO-ANCA vasculitis rather than IgA-containing immune complex-induced vasculitis.
We have mentioned the possibility of MPA and IgA vasculitis overlapping syndrome in the process of explaining the association of MPO-ANCA (or MPO-ANCA IgA) with systemic manifestations. However, we applied the 2007 EMA algorithm for AAV and the 2012 CHCC definitions to all patients with IgA vasculitis and found that none fulfilled those criteria. In addition, we applied the ACR/EULAR 2017 provisional classification criteria for GPA to five patients with ANCA, which consist of nine items with different points. The total score of 5 or greater can classify a patient as GPA [26]. None of them could be classified as GPA by these criteria either. Thus, we conclude that the overlapping syndrome cannot explain ANCA positivity in IgA vasculitis in this study.
We intended to investigate poor outcomes, particularly renal failure, in IgA vasculitis patients; however, there are few previous studies on the effect of ANCA on renal function during the disease course of IgA vasculitis. Instead, given that IgA vasculitis is indistinguishable from IgA nephropathy as long as there are no extrarenal manifestations [7], we reviewed previous studies regarding the effect of ANCA on the deterioration in renal function in patients with IgA nephropathy. Huang and colleagues suggested that identifying and discerning between pathogenic and non-pathogenic ANCA should be required in patients with IgA nephropathy [15]. Yang and colleagues provide information on the renal pathogenic role of ANCA in IgA nephropathy by demonstrating that ANCA-positive patients with IgA nephropathy exhibited more severe and progressive renal and histological feature than ANCA-negative patients with IgA nephropathy. Furthermore, they suggested that immune complex containing IgA in ANCA-positive patients with IgA nephropathy might not be directly associated with ANCA production [16]. Meanwhile, Xie and colleagues focused on the presence of systemic (extrarenal) manifestations in ANCA-positive patients with IgA nephropathy [17]. Therefore, we conclude that ANCA could be considered a risk factor for the worsening of IgA nephropathy including IgA vasculitis nephritis despite the uncertain mechanism.
In this study, we assessed the detection rate and the subtype of ANCA and investigated the clinical significance of ANCA positivity at diagnosis in a considerable number of Korean adult patients with IgA vasculitis for the first time. Particularly, we unveiled that the effect of ANCA on the outcomes of IgA vasculitis was not clinically significant unlike previous studies in other countries [15,16,17]. However, our study has several issues. First, since our study is a monocentric retrospective study, the number of IgA vasculitis patients was not large enough to represent the clinical significance of ANCA in Korean patients with IgA vasculitis. For this reason, the simple comparison analysis of variables between IgA vasculitis patients with and without ANCA may have several limitations. And furthermore, in particular, the number of patients with biopsy results was too small to give forth the firmly more reliable data. However, as you know, so far, previous studies have reported the detection of ANCA in IgA vasculitis patients at a too low rate to conduct statistical analysis. Therefore, we believe that this study is meaningful as a pilot study to report the detection rate of ACA in a considerable number of patients with IgA vasculitis. Second, the follow-up duration was not long enough to reflect the outcomes of IgA vasculitis. Third, not all patients had undergone a renal biopsy. Future studies with a larger number of IgA vasculitis patients and a considerable follow-up period will provide clear information on the clinical significance of ANCA in Korean patients with IgA vasculitis.
In conclusion, 5 out of 86 patients with IgA vasculitis had ANCA but they did not fulfil the classification criteria for AAV at all. In addition, ANCA positivity tended to be clinically frequent in lung and neurological symptoms but did not seem to affect renal histology or overall prognosis in patient with IgA vasculitis. These results might be due to the results of excluding patients who met the classification criteria for AAV. Therefore, we suggest that in real clinical practice, physicians should first apply the classification criteria for AAV to patients with ANCA positivity and then, if not for AAV, apply the classification criteria for IgA vasculitis.
References
Yang YH, Yu HH, Chiang BL (2014) The diagnosis and classification of Henoch-Schönlein purpura: an updated review. Autoimmun Rev 13(4–5):355–358. https://doi.org/10.1016/j.autrev.2014.01.031
Blanco R, Martínez-Taboada VM, Rodríguez-Valverde V, García-Fuentes M, González-Gay MA (1997) Henoch-Schönlein purpura in adulthood and childhood: two different expressions of the same syndrome. Arthritis Rheum 40(5):859–864. https://doi.org/10.1002/1529-0131(199705)40:5%3c859:AID-ART12%3e3.0.CO;2-J
Nielsen HE (1988) Epidemiology of Schönlein-Henoch purpura. Acta Paediatr Scand 77(1):125–131
Fervenza FC (2003) Henoch-Schönlein purpura nephritis. Int J Dermatol 42(3):170–177
Sano H, Izumida M, Shimizu H, Ogawa Y (2002) Risk factors of renal involvement and significant proteinuria in Henoch-Schönlein purpura. Eur J Pediatr 161(4):196–201
Kang Y, Park JS, Ha YJ, Kang MI, Park HJ, Lee SW, Lee SK, Park YB (2014) Differences in clinical manifestations and outcomes between adult and child patients with Henoch-Schönlein purpura. J Korean Med Sci 29(2):198–203. https://doi.org/10.3346/jkms.2014.29.2.198
Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, Flores-Suarez LF, Gross WL, Guillevin L, Hagen EC, Hoffman GS, Jayne DR, Kallenberg CG, Lamprecht P, Langford CA, Luqmani RA, Mahr AD, Matteson EL, Merkel PA, Ozen S, Pusey CD, Rasmussen N, Rees AJ, Scott DG, Specks U, Stone JH, Takahashi K, Watts RA (2013) 2012 Revised international Chapel Hill consensus conference nomenclature of vasculitides. Arthritis Rheum 65(1):1–11. https://doi.org/10.1002/art.37715
Torraca PF, Castro BC, Filho Hans G (2016) Henoch-Schönlein purpura with c-ANCA antibody in an adult. An Bras Dermatol 91(5):667–669. https://doi.org/10.1590/abd1806-4841.20164181
Ozen S, Pistorio A, Iusan SM, Bakkaloglu A, Herlin T, Brik R, Buoncompagni A, Lazar C, Bilge I, Uziel Y, Rigante D, Cantarini L, Hilario MO, Silva CA, Alegria M, Norambuena X, Belot A, Berkun Y, Estrella AI, Olivieri AN, Alpigiani MG, Rumba I, Sztajnbok F, Tambic-Bukovac L, Breda L, Al-Mayouf S, Mihaylova D, Chasnyk V, Sengler C, Klein-Gitelman M, Djeddi D, Nuno L, Pruunsild C, Brunner J, Kondi A, Pagava K, Pederzoli S, Martini A, Ruperto N, Paediatric Rheumatology International Trials Organisation (PRINTO) (2010) EULAR/PRINTO/PRES criteria for Henoch-Schönlein purpura, childhood polyarteritis nodosa, childhood Wegener granulomatosis and childhood Takayasu arteritis: Ankara, 2008. Part II: final classification criteria. Ann Rheum Dis 69(5):798–806. https://doi.org/10.1136/ard.2009.116657
Watts R, Lane S, Hanslik T, Hauser T, Hellmich B, Koldingsnes W, Mahr A, Segelmark M, Cohen-Tervaert JW, Scott D (2007) Development and validation of a consensus methodology for the classification of the ANCA-associated vasculitides and polyarteritis nodosa for epidemiological studies. Ann Rheum Dis 66(2):222–227. https://doi.org/10.1136/ard.2006.054593
Calvo-Río V, Hernández JL, Ortiz-Sanjuán F, Loricera J, Palmou-Fontana N, González-Vela MC, González-Lamuño D, González-López MA, Armesto S, Blanco R, González-Gay MA (2016) Relapses in patients with Henoch-Schönlein purpura: analysis of 417 patients from a single center. Medicine (Baltimore) 95(28):e4217. https://doi.org/10.1097/MD.0000000000004217
Levey AS, Stevens LA, Schmid CH, Zhang YL, Castro AF 3rd, Feldman HI, Kusek JW, Eggers P, Van Lente F, Greene T, Coresh J, CKD-EPI (Chronic Kidney Disease Epidemiology Collaboration) (2009) A new equation to estimate glomerular filtration rate. Ann Intern Med 150(9):604–612. https://doi.org/10.7326/0003-4819-150-9-200905050-00006
Haas M (1997) Histologic subclassification of IgA nephropathy: a clinicopathologic study of 244 cases. Am J Kidney Dis 29(6):829–842
Trimarchi H, Barratt J, Cattran DC, Cook HT, Coppo R, Haas M, Liu ZH, Roberts IS, Yuzawa Y, Zhang H, Feehally J, IgAN Classification Working Group of the International IgA Nephropathy Network and the Renal Pathology Society; Conference Participants (2017) Oxford Classification of IgA nephropathy 2016: an update from the IgA Nephropathy Classification Working Group. Kidney Int 91(5):1014–1021. https://doi.org/10.1016/j.kint.2017.02.003
Huang X, Wang Y, Xie L, Zhang Y, Tang S, Yin S, Gao X, Cai J, Wang W, Zhang J, Zhao J, Huang Y, Li Y, Zhang J (2015) IgA nephropathy with anti-neutrophil cytoplasmic antibody seropositivity. Clin Nephrol 84(3):156–164. https://doi.org/10.5414/CN108571
Yang YZ, Shi SF, Chen YQ, Chen M, Yang YH, Xie XF, Zou R, Lv JC, Liu LJ, Zhang H (2015) Clinical features of IgA nephropathy with serum ANCA positivity: a retrospective case-control study. Clin Kidney J 8(5):482–488. https://doi.org/10.1093/ckj/sfv078
Xie L, He J, Liu X, Tang S, Wang W, Li F, Zhang Y, Zhang J, Huang Y, Zhao J, Li Y, Zhang J (2018) Clinical value of systemic symptoms in IgA nephropathy with ANCA positivity. Clin Rheumatol 37(7):1953–1961. https://doi.org/10.1007/s10067-017-3931-z
Kim JE, Shin JI (2014) Positive c-ANCA in Henoch-Schonlein purpura: what is the mechanism? Comment on: Adult-onset Henoch-Schonlein purpura with positive c-ANCA (anti-proteinase 3): case report and review of literature (Rheumatol Int. 2013 Feb; 33(2):493-496). Rheumatol Int 34(7):1017. https://doi.org/10.1007/s00296-013-2832-y
Yu JH, Lee KB, Lee JE, Kim H, Kim K, Jang KS, Park MH (2012) A case of elderly-onset crescentic Henoch-Schönlein purpura nephritis with hypocomplementemia and positive MPO-ANCA. J Korean Med Sci 27(8):957–960. https://doi.org/10.3346/jkms.2012.27.8.957
Ronda N, Esnault VL, Layward L, Sepe V, Allen A, Feehally J, Lockwood CM (1994) Antineutrophil cytoplasm antibodies (ANCA) of IgA isotype in adult Henoch-Schönlein purpura. Clin Exp Immunol 95(1):49–55. https://doi.org/10.1111/j.1365-2249.1994.tb06013.x
Shaw G, Ronda N, Bevan JS, Esnault V, Griffiths DF, Rees A (1992) Antineutrophil cytoplasmic antibodies (ANCA) of IgA class correlate with disease activity in adult Henoch-Schönlein purpura. Nephrol Dial Transplant 7(12):1238–1241. https://doi.org/10.1093/ndt/7.12.1238
Ogawa N, Yano S, Yamane Y, Nishiki M, Yamaguchi T, Tsukamoto T, Muso E, Sugimoto T (2007) MPO-ANCA-positive IgA nephropathy successfully treated with tonsillectomy. Clin Exp Nephrol 11(4):326–331. https://doi.org/10.1007/s10157-007-0506-3
Nagasaka T, Miyamoto J, Ishibashi M, Chen KR (2009) MPO-ANCA- and IgA-positive systemic vasculitis: a possibly overlapping syndrome of microscopic polyangiitis and Henoch-Schoenlein purpura. J Cutan Pathol 36(8):871–877. https://doi.org/10.1111/j.1600-0560.2008.01145.x
Garzoni L, Vanoni F, Rizzi M, Simonetti GD, Goeggel Simonetti B, Ramelli GP, Bianchetti MG (2009) Nervous system dysfunction in Henoch-Schonlein syndrome: systematic review of the literature. Rheumatology (Oxford) 48(12):1524–1529. https://doi.org/10.1093/rheumatology/kep282
Rajagopala S, Shobha V, Devaraj U, D’Souza G, Garg I (2013) Pulmonary hemorrhage in Henoch-Schönlein purpura: case report and systematic review of the English literature. Semin Arthritis Rheum 42(4):391–400. https://doi.org/10.1016/j.semarthrit.2012.07.004
Yoo J, Kim HJ, Ahn SS, Jung SM, Song JJ, Park YB, Lee SW (2018) The utility of the ACR/EULAR 2017 provisional classification criteria for granulomatosis with polyangiitis in Korean patients with antineutrophil cytoplasmic antibody-associated vasculitis. Clin Exp Rheumatol 36(Suppl 111(2)):85–87
Funding
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2017R1D1A1B03029050) and a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute, funded by the Ministry of Health and Welfare, Republic of Korea (HI14C1324).
Author information
Authors and Affiliations
Contributions
All authors contributed to conception and design, or acquisition of data, or analysis and interpretation of data and participated in drafting the manuscript or revising it critically for important intellectual content. Also, all authors gave final approval of the version to be published and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This study was approved by the Institutional Review Board (IRB) of Severance Hospital (4-2017-0673).
Informed consent
The patient’s written informed consent was waived by the approving IRB, as this was a retrospective study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kim, J.Y., Choi, H., Kim, M.K. et al. Clinical significance of ANCA positivity in patients with IgA vasculitis: a retrospective monocentric study. Rheumatol Int 39, 1927–1936 (2019). https://doi.org/10.1007/s00296-019-04397-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00296-019-04397-3