
Abstract
Discovering the genes underlying fundamental processes that enable cells to live and reproduce is a technical challenge, because loss of gene function in mutants results in organisms that cannot survive. This study describes a forward genetics method to identify essential genes in fungi, based on the propensity for Agrobacterium tumefaciens to insert T-DNA molecules into the promoters or 5′ untranslated regions of genes and by placing a conditional promoter within the T-DNA. Insertions of the promoter of the GAL7 gene were made in the human pathogen Cryptococcus neoformans. Nine strains of 960 T-DNA insertional mutants screened grew on media containing galactose, but had impaired growth on media containing glucose, which suppresses expression from GAL7. T-DNA insertions were found in the homologs of IDI1, MRPL37, NOC3, NOP56, PRE3 and RPL17, all of which are essential in ascomycete yeasts Saccharomyces cerevisiae or Schizosaccharomyces pombe. Altering the carbon source in the medium provided a system to identify phenotypes in response to stress agents. The pre3 proteasome subunit mutant was further characterized. The T-DNA insertion and phenotype co-segregate in progeny from a cross, and the growth defect is complemented by the reintroduction of the wild type gene into the insertional mutant. A deletion allele was generated in a diploid strain, this heterozygous strain was sporulated, and analysis of the progeny provided additional genetic evidence that PRE3 is essential. The experimental design is applicable to other fungi and has other forward genetic applications such as to isolate over-expression suppressors or enhance the production of traits of interest.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
All antifungal drugs used in medical mycology target molecules that are required for fungal viability; however, the identification and characterization of genes that encode essential functions is difficult. The following study provides a new insertional mutagenesis method to identify essential genes in the human pathogen Cryptococcus neoformans. While the focus of this research is on potential targets for drug development in human pathogens, the same insertional mutagenesis approach would also be applicable to investigations of other aspects of fungal biology.
The limited numbers of antimicrobial agents and narrowing pipeline to develop new ones are well-recognized problems across the fields of microbial pathogenesis. These are challenges faced in medical mycology, where antifungal drug resistance is also a major global problem (Calderone et al. 2014; Denning and Bromley 2015). The underlying reasons for this are multifactorial. However, antifungal agents are rarely sought in programs aiming to develop antimicrobials and just one new class of antifungal, the echinocandins, has been released in decades. Current interests therefore have focused on agents already approved for other uses now tested for their antifungal properties (Krysan 2015; Roemer and Krysan 2014). Nevertheless, suitable drug targets are needed, that is proteins or metabolites that are crucial for microbe viability.
The Cryptococcus neoformans species complex is a monophyletic group of related fungal species, currently with seven proposed species (Hagen et al. 2015), all of which are capable of causing diseases in the human population, in native wild life, and in domestic pets. An initiate estimate made by the United States Centers for Disease Control and Prevention was ~600,000 deaths occurred each year from this disease (Park et al. 2009). A revised calculation was presented at the 9th International Conference on Cryptococcus and Cryptococcosis in 2014, of about half that number (Idnurm and Lin 2015; Park et al. 2014). Thus, cryptococcosis continues to rank as a major causes of mortality attributed to infectious diseases.
Beyond the interest as a major global pathogen, the Cryptococcus genus is a member of the Basidiomycota phylum of fungi. The species complex has become one of the few models for molecular biology research in the basidiomycetes. This phylum diverged from the Ascomycota, a group carrying most of the known human pathogenic fungi, somewhere ~800–1600 million years ago (Berbee and Taylor 2010; Taylor and Berbee 2006). Hence, analysis of genes that may be suitable as broad-spectrum targets for new antimicrobial drugs should take into consideration Cryptococcus species and other basidiomycetes.
We recently described a forward genetics method to identify essential genes in C. neoformans (Ianiri and Idnurm 2015). Transfer-DNA (T-DNA) molecules from Agrobacterium tumefaciens were inserted randomly into the genome of a diploid strain of C. neoformans. These heterozygous strains were induced to undergo meiosis and sporulation, and the spores analyzed to infer insertions in essential genes by the absence of viable progeny carrying the drug resistance marker within the T-DNA. However, there were several limitations and drawbacks with the method. These included T-DNA insertions that triggered chromosomal translocations and the tendency for T-DNAs to insert between genes and into regions that may be the promoters or 5′ untranslated regions (UTR) of genes, thereby making it necessary to use other methods to test which of the genes on either side of the translocation or insertion is essential for fungal viability. In addition, primary screening required a substantial amount of labor and media. Thus in the following sections, we describe a new method to identify genes that are essential for viability in C. neoformans.
Methods
Construction of shuttle vectors for transformation of multiple species
Two plasmids were created to transform multiple species: the bacteria Escherichia coli and Agrobacterium tumefaciens and the fungi Saccharomyces cerevisiae and Cryptococcus neoformans (Fig. 1). The oligonucleotide primer sequences used in amplification during plasmid construction, and strain characterization, are provided in Table 1.

Construction of two plasmids for transformation of multiple microbes. a Plasmid or DNA maps and the construction process. In step 1, the S. cerevisiae 2 micron circle and URA3 gene were amplified from pRS426 with primers ALID2018-ALID2019 and cloned into the ScaI site of pPZP-201BK, to form plasmid pGI3. In step 2, the NAT cassette was amplified from pPZP-NATcc with primers ai290-ALID2147 and the GAL7 promoter from C. neoformans genomic DNA with primers GI114-GI115, and cloned into pGI3 to form pGI7. AmpR and KanR refer to genes conferring resistance to ampicillin and kanamycin in bacteria. For simplicity, replication components in E. coli and A. tumefaciens are omitted. Tick marks indicate 100 bp intervals. b Sequence of the GI114-GI115 amplicon, including the 1020 bp immediately upstream of the GAL7 open reading frame. Sequences corresponding to primers GI114 and GI115 are underlined. The right border sequence in the T-DNA is in bold, with the three nucleotides that usually integrate into the fungal genome in the box with the grey highlight
Plasmid pGI3 was generated by digesting the A. tumefaciens binary vector pPZP-201BK (Covert et al. 2001) with ScaI restriction enzyme, whose site is outside but close to the right border, and by recombining into it the S. cerevisiae 2 µ circle and URA3 gene amplified from plasmid pRS426 (Sikorski and Hieter 1989) using primers ALID2018-ALID2019 (these two primers have chimerical regions with the pPZP-201BK to allow recombination in S. cerevisiae). Transformation was carried out with the lithium acetate and polyethylene glycol method (Ito et al. 1983), using equimolar amounts of the PCR product and the linearized plasmid pPZP-201BK, in S. cerevisiae strain FY834 [MATα; his3; ura3; leu2; lys2; trp1 (Winston et al. 1995)]. Selection of S. cerevisiae transformants was on yeast nitrogen base (YNB) minimal medium without uracil, supplemented with histidine, leucine, lysine and tryptophan to compensate for these other auxotrophies in strain FY834. DNA from one transformant of S. cerevisiae was extracted and transformed by heat shock into E. coli, with selection on kanamycin (50 µg/ml). Plasmids were prepared from E. coli and inserts sequenced. The resulting plasmid pGI3 contains pPZP-201BK and regions for replication and selection in S. cerevisiae (Fig. 1a).
The promoter of the C. neoformans GAL7 gene, which encodes galactose-1-phosphate uridyltransferase, and the NAT cassette, which confers resistance to nourseothricin when transformed into C. neoformans, were recombined into plasmid pGI3 to generate the plasmid pGI7, as follows. The GAL7 promoter was amplified from C. neoformans strain KN99α with primers GI114-GI115 (Fig. 1b), which are based on primers used previously to amplify this region (Ruff et al. 2009); primer GI114 has a chimerical region for recombination into plasmid pGI3 and GI115 a chimerical region for recombination with the ACT1 promoter of the NAT cassette. Similarly, the NAT cassette (pACT1::NAT::tTRP1) was amplified from plasmid pPZP-NATcc (Walton et al. 2005) with primers ai290 and ALID2147, of which ALID2147 has a chimerical region for recombination into plasmid pGI3. PCRs were performed using a High Fidelity Taq polymerase. S. cerevisiae strain FY834 was transformed with equimolar amounts of the PCR products (GAL7 promoter and NAT) and the plasmid pGI3 linearized with BamHI and KpnI. Transformants were subject to colony PCR to identify positive clones. DNA was extracted from one strain (Pitkin et al. 1996) and the DNA used to electroporate strain EHA105 of A. tumefaciens; transformants were selected on lysogeny broth medium containing 50 µg/ml kanamycin. Plasmids were isolated from A. tumefaciens transformants, and the inserts in one plasmid (pGI7) were sequenced to confirm the correct assembly of the PCR fragments within the T-DNA.
Agrobacterium tumefaciens-mediated transformation
Agrobacterium tumefaciens-mediated transformation (AtMT) was performed using a standard protocol (Bundock et al. 1995; Idnurm et al. 2004), except that galactose replaced glucose in all media types. That is, the induction medium contained galactose rather than glucose (at 0.9 g/L), and growth of C. neoformans transformants after co-cultivation with the Agrobacterium was on yeast extract + peptone + 2% galactose (YPG), supplemented with nourseothricin (100 µg/ml) to select for fungal transformants and cefotaxime (100 µg/ml) or tetracycline (15 µg/ml) to counter select the Agrobacterium.
Strain KN99α (Nielsen et al. 2003) of C. neoformans sensu stricto, i.e., C. neoformans var. grubii or serotype A, was used as the recipient for transformation of T-DNAs from A. tumefaciens strain EHA105 with the pGI7 plasmid.
Identification of mutants with impaired growth and T-DNA insertions in putative essential genes
960 transformants were arranged into ten 96-well plates containing 100 µl of liquid YPG and cultured overnight. 2.5 µl from each culture were spotted onto four agar medium plates: YPD, YPG and YNB + glucose and YNB + galactose. Strains selected from this primary screen as unable to grow on repressive media containing glucose were tested using tenfold serial dilutions.
Genomic DNA was extracted from fungal strains using a CTAB extraction buffer (Pitkin et al. 1996). Inverse PCR was performed to identify the T-DNA insertion sites. Briefly, genomic DNA was digested with BglII or KpnI restriction enzymes, re-ligated with T4 DNA ligase, and the ligation used as the template for PCR using primers GI120-ai077 with ExTaq polymerase (Takara Bio, Japan). Inverse PCR products were sequenced, and the sequences used to search with BLAST the C. neoformans strain H99 genome at the Broad Institute (Janbon et al. 2014). C. neoformans genes were used to search the Saccharomyces Genome Database for the S. cerevisiae orthologs.
Analysis of strain 3A11 and the PRE3 gene
Strain 3A11 was crossed with strain KN99a, which is congenic with the MAT a mating type (Nielsen et al. 2003), on Murashige and Skoog 4% agar medium in the dark at 22 °C (Xue et al. 2007). Basidiospores were isolated using an Olympus microscope with an attached micromanipulation stage and plated onto YPG medium.
A wild type copy of PRE3 was amplified with high fidelity Q5 DNA polymerase (New England BioLabs, Ipswich, MA) with primers MAI0170-MAI0171 and cloned into plasmid pPZP-NEO11, linearized with EcoRV-HindIII, using Gibson assembly (New England BioLabs). A clone without errors was identified by sequencing, and the plasmid was electroporated into A. tumefaciens strain EHA105 and selected on lysogeny broth + kanamycin media. The A. tumefaciens strain was used to transform strain 3A11 as described above. Transformed strains were selected on YPG + G418 (100 µg/ml) + cefotaxime (200 µg/ml).
One copy of PRE3 was deleted by homologous recombination in the diploid strain AI187 [MAT a/MATα ade2/ADE2 ura5/URA5; (Idnurm 2010)]. The 5′ side of the gene was amplified with primers MAI0172-MAI0173 and 3′ side amplified with primers MAI0174-MAI0175. The NAT cassette was amplified with primers ai006-ai290. The three fragments were joined together into the EcoRV-HindIII site of plasmid pPZP-201BK using Gibson assembly. The gene replacement construct was amplified using primers MAI00172-MAI0175. The DNA was precipitated onto gold beads and transformed into strain AI187 using a PDS/He-1000 biolistic apparatus [Bio-Rad, Hercules, CA (Toffaletti et al. 1993)] and plated onto YPD with nourseothricin (100 µg/ml). Mutant strains were identified by PCR, and then inoculated on Murashige and Skoog medium and incubated in the dark at 22 °C to induce meiosis and the production of basidiospores. Basidiospores were micromanipulated onto YPD medium, and allowed to germinate at 30 °C. Colonies were phenotyped for genetic markers (ADE2, MAT, URA5) on YNB + adenine and YNB + uracil media, and by crossing to MAT a and MATα parents on V8 juice agar and/or by PCRs specific for each mating type.
Phenotype characterization of T-DNA insertional mutants
Strains were cultured overnight in YPG medium, tenfold serially diluted, and plated onto yeast extract peptone with 2% galactose or 0.5% galactose + 2% raffinose as carbon sources. One set of plates was incubated at 37 °C and another exposed to UV light (120 Jm−2). Seven plates contained stress agents: 0.04% methyl methanesulfonate (MMS) (Sigma Aldrich, 129925), 0.06% ethidium bromide solution (EtBr) (Sigma Aldrich, E1510), 0.25 mM menadione (Sigma Aldrich, M5625), 0.25 mM paraquat dichloride hydrate (paraquat) (Sigma Aldrich, 36541), 0.4 mM Luperox TBH70X, 0.4 mM tert-butyl hydroperoxide solution (tBOOH) (Sigma Aldrich, 458139), 5 mM hydrogen peroxide solution (H2O2) (Sigma Aldrich, 216763) and 0.01 mM cadmium sulfate (CdSO4) (Sigma Aldrich, 481882). Plates were incubated at 28 °C for 2 days.
Results
Development of shuttle plasmids
A method for altering the transcription of genes adjacent to the insertion of T-DNA molecules was sought for C. neoformans to identify genes that are essential for viability. The strategy was to insert the GAL7 promoter as close as possible to one border of the T-DNA. The right border almost always inserts three bases, so the GAL7 promoter was fused adjacent to this border. Plasmid pGI7 was generated that can replicate in E. coli, A. tumefaciens, S. cerevisiae and transform C. neoformans (Fig. 1).
This new plasmid pGI7 was used to transform T-DNA molecules into C. neoformans, with the transformants cultured on media containing galactose to allow expression of genes adjacent to the insertion event. In the primary screen using transformants arrayed in ten 96-well plates, 17 strains grew poorly on YPD. The same result was obtained on YNB with glucose as the sole carbon source, although the results on this medium were not as clear. Growth of the strains was examined using more accurate serial dilutions, confirmed that nine strains from the screen (0.9%) have reduced growth on YPD compared to YPG (Fig. 2).

Insertional mutants of C. neoformans with impaired growth in the presence of glucose. Nine strains were identified from 960 screened transformed with T-DNAs. Tenfold serial dilutions were plated onto yeast extract peptone (YP) galactose and YP glucose and cultured for 2 days
Identification of genes mutated in T-DNA insertional mutants
Inverse PCR was performed to amplify the regions flanking the insertion sites of the T-DNA in the C. neoformans genome. Sequences were compared to the H99 genome database that features transcriptome information on the genes (Janbon et al. 2014). The positions of the insertions and associated sequences are in Fig. 3. The genes responsible for the inviable phenotype in two strains 5E12 and 5H11 could not be identified, due to obtaining PCR reactions only with the native GAL7 gene. Alternative methods to inverse PCR, or potentially genome sequencing that can be used to identify T-DNA junctions (Chambers et al. 2014; Esher et al. 2015), could potentially identify these insertion events and genes affected. One strain (4E3) inserted into a section of the genome with a complex prediction of genes, including a non-coding RNA molecule. In five strains the GAL7 promoter inserted at the start of genes whose orthologs are essential in S. cerevisiae (Dowell et al. 2010; Giaever et al. 2002). The functions of the genes, IDI1, NOC3, NOP56, PRE3 and RPL17, as characterized in S. cerevisiae are provided in Table 2. The sixth insertion was into the MRPL37 homolog, whose mutation blocks respiration in S. cerevisiae; C. neoformans requires respiration for survival. The homologs of five genes are also essential for viability in a second ascomycete yeast, Schizosaccharomyces pombe (Houchens et al. 2008; Kim et al. 2010). One gene, RPL17, is not essential in S. pombe. However, it is duplicated in both S. cerevisiae and S. pombe; in S. cerevisiae RPL17A is essential while RPL17B is not and in S. pombe both genes can be deleted individually, suggesting redundancy in function in fission yeast (Kim et al. 2010). The RPL17 homolog was identified as essential in A. fumigatus (Firon et al. 2003). In addition to the model yeasts, the NOC3 homolog is has been reported as essential in Candida albicans and Aspergillus fumigatus (Becker et al. 2010; Hu et al. 2007).

Insertion sites of the T-DNA from pGI7 in the C. neoformans strains. a Agrobacterium DNA sequences of the wild type (WT) region and around the T-DNA (boxed). Nucleotides deleted during the insertion are in bold in the wild type sequence. The left and right border sequences are in bold in the mutants. b Diagram of the insertion sites relative to essential genes. The T-DNA insertion sites are black lines perpendicular to and above the sequence. White boxes indicate 5′ UTRs, dark indicates coding regions, and grey indicates introns
The T-DNA inserted before or close to the start of the C. neoformans gene. Two strains are associated with chromosomal rearrangements (3F5 and 8A11), because the sequences from each side of the T-DNA after inverse PCR map to different chromosomes. 8A11 has the insertion within the second exon of the RPL17 gene, which is predicted to remove the first 31 amino acids of the Rpl17 protein. Two strains have insertions in promoter regions and three within the 5′ UTRs (Table 2; Fig. 3). The close proximity to the start of the genes is consistent with these insertions affecting their normal transcriptional regulation.
Additional evidence that PRE3 is essential in C. neoformans
One strain and its mutated gene were examined in more detail. Three experiments provide additional evidence that the T-DNA inserted into the regulatory region of an essential gene. Strain 3A11 has the T-DNA inserted in the PRE3 gene. Pre3 is an essential subunit of the 20S proteasome. The protein is a peptidyl-glutamyl peptide hydrolyzing enzyme, although this activity is dispensable for cell survival (Enenkel et al. 1994; Heinemeyer et al. 1997).
First, strain 3A11 was crossed to strain KN99a, which is congenic to the wild type KN99α but of the opposite MAT a mating type. Basidiospores were micromanipulated and germinated on YP galactose. 24 colonies were obtained, and their growth tested on media with glucose and nourseothricin. Perfect co-segregation between nourseothricin resistance and the reduced growth on glucose was observed. As an independent genetic marker, the mating type of each progeny was determined; the mating type a or α properties encoded by this locus segregate independently of the growth and drug resistance properties, indicating that the progeny from the cross reflect recombination (Fig. 4).

The T-DNA insertion segregates with reduced growth on glucose in progeny from a genetic cross between strain 3A11 (MATα) and wild type KN99a. The three strains at the top are wild type KN99α, 3A11 and KN99a. They and the progeny were plated on three media and cultured 2 days. The diagram indicates mating types of the progeny obtained from the cross: grey circles MAT a and white circles MATα
Second, the wild type copy of PRE3 was amplified, cloned adjacent to a marker for G418 resistance, and strain 3A11 transformed by AtMT with this construct. After co-culture, C. neoformans transformants were selected on YP galactose + G418. The transformants were tested for growth on YPG and YPD. Growth was restored to wild type in the complemented strain on YPD (Fig. 5).

The wild type copy of PRE3 complements the strain 3A11 (pre3 mutant) growth defect on glucose. Tenfold serial dilutions of strains were grown on YP galactose and YP glucose (wild type KN99α, 3A11 pre3::T-DNA and pre3::T-DNA + PRE3)
Third, a large deletion of PRE3 was made in the diploid strain AI187 by replacing the open reading frame via homologous recombination, with a cassette conferring nourseothricin resistance (Fig. 6a, b). The heterozygote strain pre3::NAT/PRE3 was plated on Murashige and Skoog medium to induce meiosis and sporulation. Germination of the basidiospores to form colonies was low (10 from 52 basidiospores plated). However, a similar number (13/52) of basidiospores germinated and produced 2–4 cells before arresting growth (Fig. 6c). Of the ten colonies obtained, only one was nourseothricin resistant, and subsequent PCR analysis indicated it was heterozygous pre3::NAT/PRE3. Phenotypes controlled by three markers that are on separate chromosomes in strain AI187 (MAT a/MATα; ade2/ADE2; ura5/URA5) were tested, showing independent segregation of the mating type (MAT), ADE2 and URA5 loci (Fig. 6d). The lack of nourseothricin-resistant progeny is indicative that the deletion allele of PRE3 is deleterious for cell survival.

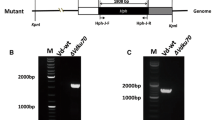
Independent confirmation that PRE3 is essential in C. neoformans by targeted gene replacement into the diploid AI187 strain, sporulation and analysis of progeny. a Diagram of the wild type and gene replacement alleles, with the positions of primers marked with arrows. b PCR confirmation of correct replacement of PRE3. Faint bands in the wild type are non-specific amplification. The sizes are the 1 kb + ladder (Invitrogen). c Microcolony arrested at a four-cell stage after basidiospore germination on YP glucose agar. d Segregation data of colonies after sporulation of the heterozygous pre3/PRE3 strain; the pre3/PRE3 strain is in the bottom right corner. Mating types are grey for MAT a and white for MATα
In vitro stress sensitivity phenotypes are associated with mutations of essential genes
One disadvantage of a complete gene deletion is that this results in an inviable strain. The inducible promoter system has the potential to be manipulated to provide some level of growth, and strains can then be tested for phenotypes that can provide additional information about gene function. Hence, the strains were cultured on plates containing 2% galactose or 0.5% galactose + 2% raffinose and a selection of stress agents or conditions (Fig. 7). On 2% galactose, all strains except 4C9 showed a minor decrease in growth at 37 °C and this phenotype was augmented when grown on 0.5% galactose 2% raffinose medium. Decreasing the amount of galactose had little to no effect on growth without stress; however, greatly enhanced growth phenotypes under stress conditions. This suggests that specific genes also play roles in protecting against specific stresses.

Growth defect phenotypes are associated with mutations in specific essential genes. Overnight cultures of strains grown in the presence of galactose were tenfold serially diluted and dotted onto plates containing stresses and different carbon sources (G = 2% galactose and GR = 0.5% galactose + 2% raffinose). After 2 days growth the plates were photographed. Concentrations or treatments are: no stress (28 °C), 37 °C, UV (120 J m−2), 5 mM hydrogen peroxide solution (H2O2), 0.25 mM paraquat dichloride hydrate (paraquat), 0.25 mM menadione, 0.4 mM tert-butyl hydroperoxide solution (tBOOH), 0.04% methyl methanesulfonate (MMS), 0.06% ethidium bromide (EtBr) and 0.01 mM cadmium sulfate (CdSO4)
Discussion
Essential genes in Cryptococcus neoformans have been identified by a number of approaches. One approach employed a diploid strain, with both targeted and insertional mutagenesis into the strain to mutate one copy of the gene, and those strains were then sporulated and the essentiality of the inserted DNA inferred by the inability to recover those haploid progeny (Ianiri and Idnurm 2015). Targeted mutagenesis is achieved by biolistic transformation; the disadvantage of targeted mutagenesis is that it requires knowledge of the genes to be chosen and since limited information is available for essential genes in basidiomycetes, the choice has to be made based on essential genes known in ascomycetes. Unless aiming to delete all the genes, which is an ongoing project in a haploid strain background of C. neoformans (Homer et al. 2016; Liu et al. 2008), this is a restriction in the discovery of novel genes as it effectively focuses on C. neoformans genes conserved across fungi. To overcome this issue and to be able to identify on a large-scale essential genes of C. neoformans, random insertional mutagenesis can be performed using either Agrobacterium-mediated transformation (AtMT) or biolistic delivery of the DNA (Ianiri and Idnurm 2015). However, the use of random insertional mutagenesis in the diploid strain has a number of drawbacks, and hence was a motivation to develop new approaches.
Insertional mutagenesis using AtMT or biolistic delivery of exogenous DNA is effective for gene function discovery in haploid isolates of C. neoformans. To use a haploid strain of C. neoformans for mutant generation and to overcome issues related with the use of a diploid strain, for essential genes identification we generated a vector for AtMT with the inducible GAL7 promoter. The approach was to generate AtMT strains in a haploid background of C. neoformans, arrange the strains in 96-well plates, and spot them on inducible (+galactose) and repressive (+glucose) media. Strains only able to grow under inducible conditions should have an insertion in the promoter or 5′ UTR regions of genes essential for growth. Further evidence for essentiality can be obtained by Mendelian genetic analysis, by showing co-segregation of the T-DNA with the growth defect and absence of the T-DNA with wild type growth, as illustrated for strain 3A11 (Fig. 6). The Murashige and Skoog medium used for crossing C. neoformans supports the growth of the strains without the need to add galactose.
The GAL7 genes and its promoters have been in use in Cryptococcus species for over two decades (Wickes and Edman 1995). The promoter is able to drive expression of adjacent genes, being repressed by glucose and induced by galactose (Baker and Lodge 2012; Ruff et al. 2009). Placing copies of the IPC1 and TOP1 genes, encoding inositol-phosphoryl ceramide synthase and topoisomerase, under control of the GAL7 promoter was used to infer that these are essential for viability (Del Poeta et al. 1999; Luberto et al. 2001). An alternative promoter could be the CTR4 promoter, which is subject to regulation by the presence or absence of copper ions in the media (Ory et al. 2004).
This approach of construct design and subsequent use as an insertional mutagenesis tool is applicable to other fungal species. The requirement is to produce two DNA fragments; one with a promoter element that enables gene regulation and the other a selectable marker. These can be joined using in vivo recombination in S. cerevisiae into plasmid pGI3. The regulated promoter could be from one of the genes for galactose utilization, as these are present in many fungi and over long evolutionary time (Okuda et al. 2014), or another source, such as the Tet-Off system (Lai et al. 2016). S. cerevisiae transformants are then screened by PCR, and positive clones are subject to plasmid extraction that are electroporated in an A. tumefaciens strain that can be used for AtMT. This avoids cloning procedures that rely on restriction enzyme sites and can also eliminate steps involving replication in E. coli.
A number of advantages are worth highlighting, listed as follows and with the focus on C. neoformans. (i) T-DNA insertions that are unlinked to the mutant phenotype are highly unlikely. In C. neoformans unlinked mutations occur in about a third to one half of mutants isolated after Agrobacterium-mediated transformation (Walton et al. 2005). (ii) The method ameliorates the problem of chromosomal rearrangements that hamper genetic analysis. T-DNA mutagenesis can cause chromosomal rearrangements, yielding genetic segregation results that suggest insertions are into essential genes but are not, plus add substantial challenges in interpreting all the rearrangements to the genome (Ianiri and Idnurm 2015). In addition, only one side of T-DNA insertion, i.e., the region next to the GAL7 promoter, needs to be sought to identify the gene of interest. (iii) In C. neoformans, unlike in other fungi, Agrobacterium cannot be used to target constructs to their native locus. Hence, there is no skew within the transformant population for insertions into the native GAL7 locus. (iv) Agrobacterium has a tendency to insert the T-DNA into intergenic regions (Walton et al. 2005), that would include promoters or 5′ UTRs and the locations in which placement of a new promoter would work. (v) There is generally precise integration of the T-DNA at the right border of three nucleotides from that border sequence, in contrast to the more variable left border (Michielse et al. 2005). (vi) It is possible to find phenotypes associated with impairing these genes beyond an inviable state, by manipulating the carbon sources.
While there are many advantages to this type of screen, of the seven insertion sites identified, six were in genes known to be essential in other fungi, while the seventh (strain 4E3) was in an area of gene complexity making it not possible to establish clearly which gene was affected. At least three caveats are worth raising about the ability to identify all essential genes in C. neoformans using this approach. (i) Overexpression of certain proteins or misregulated expression could have a deleterious effect on the cell, e.g., by the production of inadvertent artificial “toxic” products or altering the cell cycle. Hence, some essential genes may not be possible to be detected. However, these genes may still be sought, e.g., an alternative screen would be to plate transformants onto YPD and then screen for strains with growth defects on YPG. (ii) Insertions into genes influencing galactose utilization may appear as essential. These genes can be eliminated as essential by creating an independent deletion allele, such as was performed for PRE3. (iii) The T-DNA insertions into regulatory regions cannot distinguish between “very sick” and “essential” mutants, because there is some growth of the strains on YPG. In C. neoformans the question of essentiality can be further addressed by making a gene deletion allele in a diploid strain and sporulation.
In summary, we report here a method that can be used to find essential genes by the insertion of a conditional promoter into the regulatory regions of those genes. This same type of approach could be extended to other fungi or to other phenotypes whereby the differential regulation of gene expression can provide new information about gene functions.
References
Baker LG, Lodge JK (2012) Galactose-inducible promoters in Cryptococcus neoformans var. grubii. Methods Mol Biol 845:211–226
Becker JM, Kauffman SJ, Hauser M, Huang L, Lin M, Sillaots S, Jiang B, Xu D, Roemer T (2010) Pathway analysis of Candida albicans survival and virulence determinants in a murine infection model. Proc Natl Acad Sci USA 107:22044–22049
Berbee ML, Taylor JW (2010) Dating the molecular clock in fungi—how close are we? Fungal Biol Rev 24:1–16
Bundock P, den Dulk-Ras A, Beijersbergen A, Hooykaas PJJ (1995) Trans-kingdom T-DNA transfer from Agrobacterium tumefaciens to Saccharomyces cerevisiae. EMBO J 14:3206–3214
Calderone R, Sun N, Gay-Andrieu F, Groutas W, Weerawarna P, Prasad S, Alex D, Li D (2014) Antifungal drug discovery: the process and outcomes. Future Microbiol 9:791–805
Chambers K, Lowe RGT, Howlett BJ, Zander M, Batley J, Van de Wouw AP, Elliott CE (2014) Next-generation genome sequencing can be used to rapidly characterise sequences flanking T-DNA insertions in random insertional mutants of Leptosphaeria maculans. Fungal Biol Biotechnol 1:10
Covert SF, Kapoor P, Lee M-H, Briley A, Nairn CJ (2001) Agrobacterium tumefaciens-mediated transformation of Fusarium circinatum. Mycol Res 105:259–264
Del Poeta M, Toffaletti DL, Rude TH, Dykstra CC, Heitman J, Perfect JR (1999) Topoisomerase I is essential in Cryptococcus neoformans: role in pathobiology and as an antifungal target. Genetics 152:167–178
Denning DW, Bromley MJ (2015) How to bolster the antifungal pipeline. Science 347:1414–1416
Dowell RD, Ryan O, Jansen A, Cheung D, Agarwala S, Danford T, Bernstein DA, Rolfe PA, Heisler LE, Chin B, Nislow C, Giaever G, Phillips PC, Fink GR, Gifford DK, Boone C (2010) Genotype to phenotype: a complex problem. Science 328:469
Enenkel C, Lehmann H, Kipper J, Gückel R, Hilt W, Wolf DH (1994) PRE3, highly homologous to the human major histocompatibility complex-linked LMP2 (RING12) gene, codes for a yeast proteasome subunit necessary for the peptidylglutamyl-peptide hydrolyzing activity. FEBS Lett 341:193–196
Esher SK, Granek JA, Alspaugh JA (2015) Rapid mapping of insertional mutations to probe cell wall regulation in Cryptococcus neoformans. Fungal Genet Biol 82:9–21
Firon A, Villalba F, Beffa R, d’Enfert C (2003) Identification of essential genes in the human fungal pathogen Aspergillus fumigatus by transposon mutagenesis. Eukaryot Cell 2:247–255
Gamalinda M, Jakovljevic J, Babiano R, Talkish J, de la Cruz J, Woolford JL Jr (2013) Yeast polypeptide exit tunnel ribosomal proteins L17, L35 and L37 are necessary to recruit late-assembling factors required for 27SB pre-rRNA processing. Nucleic Acids Res 41:1965–1983
Gautier T, Bergès T, Tollervey D, Hurt E (1997) Nucleolar KKE/D repeat proteins Nop56p and Nop58p interact with Nop1p and are required for ribosome biogenesis. Mol Cell Biol 17:7088–7098
Giaever G, Chu AM, Ni L, Connelly C, Riles L, Véronneau S, Dow S, Lucau-Danila A, Anderson K, André B, Arkin AP, Astromoff A, El-Bakkoury M, Bangham R, Benito R, Brachat S, Campanaro S, Curtiss M, Davis K, Deutschbauer A, Entian K-D, Flaherty P, Foury F, Garfinkel DJ, Gerstein M, Gotte D, Güldener U, Hegemann JH, Hempel S, Herman Z, Jaramillo DF, Kelly DE, Kelly SL, Kötter P, LaBonte D, Lamb DC, Lan N, Liang H, Liao H, Liu L, Luo C, Lussier M, Mao R, Menard P, Ooi SL, Revuelta JL, Roberts CJ, Rose M, Ross-Macdonald P, Scherens B, Schimmack G, Shafer B, Shoemaker DD, Sookhai-Mahadeo S, Storms RK, Strathern JN, Valle G, Voet M, Volckaert G, Wang C-Y, Ward TR, Wilhelmy J, Winzeler EA, Yang Y, Yen G, Youngman E, Yu K, Bussey H, Boeke JD, Snyder M, Philippsen P, Davis RW, Johnston M (2002) Functional profiling of the Saccharomyces cerevisiae genome. Nature 418:387–391
Hagen F, Khayhan K, Theelen B, Kolecka A, Polacheck I, Sionov E, Falk R, Parnmen S, Lumbsch HT, Boekhout T (2015) Recognition of seven species in the Cryptococcus gattii/Cryptococcus neoformans species complex. Fungal Genet Biol 78:16–48
Hanlon SE, Xu Z, Norris DN, Vershon AK (2004) Analysis of the meiotic role of the mitochondrial ribosomal proteins Mrps17 and Mrpl37 in Saccharomyces cerevisiae. Yeast 21:1241–1252
Heinemeyer W, Fischer M, Krimmer T, Stachon U, Wolf DH (1997) The active sites of the eukaryotic 20 S proteasome and their involvement in subunit precursor processing. J Biol Chem 272:25200–25209
Homer CM, Summers DK, Goranov AI, Clarke SC, Wiesner DL, Diedrich JK, Moresco JJ, Toffaletti D, Upadhya R, Caradonna I, Petnic S, Pessino V, Cuomo CA, Lodge JK, Perfect J, Yates JR 3rd, Nielsen K, Craik CS, Madhani HD (2016) Intracellular action of a secreted peptide required for fungal virulence. Cell Host Microbe 19:849–864
Houchens CR, Perreault A, Bachand F, Kelly TJ (2008) Schizosaccharomyces pombe Noc3 is essential for ribosome biogenesis and cell division but not DNA replication. Eukaryot Cell 7:1433–1440
Hu W, Sillaots S, Lemieux S, Davison J, Kauffman S, Breton A, Linteau A, Xin C, Bowman J, Becker J, Jiang B, Roemer T (2007) Essential gene identification and drug target prioritization in Aspergillus fumigatus. Plos Pathog 3:e24
Ianiri G, Idnurm A (2015) Essential gene discovery in the basidiomycete Cryptococcus neoformans for antifungal drug target prioritization. mBio 6:e02334
Idnurm A (2010) A tetrad analysis of the basidiomycete fungus Cryptococcus neoformans. Genetics 185:153–163
Idnurm A, Lin X (2015) Rising to the challenge of multiple Cryptococcus species and the diseases they cause. Fungal Genet Biol 78:1–6
Idnurm A, Reedy JL, Nussbaum JC, Heitman J (2004) Cryptococcus neoformans virulence gene discovery through insertional mutagenesis. Eukaryot Cell 3:420–429
Ito H, Fukuda Y, Murata K, Kimura A (1983) Transformation of intact yeast cells treated with alkali cations. J Bacteriol 153:163–168
Janbon G, Ormerod KL, Paulet D, Byrnes EJ 3rd, Yadav V, Chatterjee G, Mullapudi N, Hon C-C, Billmyre RB, Brunel F, Bahn Y-S, Chen W, Chen Y, Chow EWL, Coppée J-Y, Floyd-Averette A, Gaillardin C, Gerik KJ, Goldberg J, Gonzalez-Hilarion S, Gujja S, Hamlin JL, Hsueh Y-P, Ianiri G, Jones S, Kodira CD, Kozubowski L, Lam W, Marra M, Mesner LD, Mieczkowski PA, Moyrand F, Nielsen K, Proux C, Rossignol T, Schein JE, Sun S, Wollschlaeger C, Wood IA, Zeng Q, Neuvéglise C, Newlon CS, Perfect JR, Lodge JK, Idnurm A, Stajich JE, Kronstad JW, Sanyal K, Heitman J, Fraser JA, Cuomo CA, Dietrich FS (2014) Analysis of the genome and transcriptome of Cryptococcus neoformans var. grubii reveals complex RNA expression and microevolution leading to virulence attenuation. Plos Genet 10:e1004261
Kim D-U, Hayles J, Kim D, Wood V, Park H-O, Won M, Yoo H-S, Duhig T, Nam M, Palmer G, Han S, Jeffery L, Baek S-T, Lee H, Shim YS, Lee M, Kim L, Heo K-S, Noh EJ, Lee A-R, Jang Y-J, Chung K-S, Choi S-J, Park J-Y, Park Y, Kim HM, Park S-K, Park H-J, Kang E-J, Kim HB, Kang H-S, Park H-M, Kim K, Song K, Song KB, Nurse P, Hoe K-L (2010) Analysis of a genome-wide set of gene deletions in the fission yeast Schizosaccharomyces pombe. Nat Biotechnol 28:617–623
Krysan DJ (2015) Toward improved anti-cryptococcal drugs: novel molecules and repurposed drugs. Fungal Genet Biol. 78:93–98
Lai W-C, Sun H-FS, Lin P-H, Lin H, Shieh J-C (2016) A new rapid and efficient system with dominant selection developed to inactivate and conditionally express genes in Candida albicans. Curr Genet 62:213–235
Liu OW, Chun CD, Chow ED, Chen C, Madhani HD, Noble SM (2008) Systematic genetic analysis of virulence in the human fungal pathogen Cryptococcus neoformans. Cell 135:174–188
Luberto C, Toffaletti DL, Wills EA, Tucker SC, Casadevall A, Perfect JR, Hannun YA, Del Poeta MM (2001) Roles for inositol-phosphoryl ceramide synthase 1 (IPC1) in pathogenesis of C. neoformans. Genes Dev 15:201–212
Mayer MP, Hahn FM, Stillman DJ, Poulter CD (1992) Disruption and mapping of IDI1, the gene for isopentenyl diphosphate isomerase in Saccharomyces cerevisiae. Yeast 8:743–748
Merz S, Westermann B (2009) Genome-wide deletion mutant analysis reveals genes required for respiratory growth, mitochondrial genome maintenance and mitochondrial protein synthesis in Saccharomyces cerevisiae. Genome Biol 10:R95
Michielse CB, Hooykaas PJJ, van den Hondel CAMJJ, Ram AFJ (2005) Agrobacterium-mediated transformation as a tool for functional genomics in fungi. Curr Genet 48:1–17
Milkereit P, Gadal O, Podtelejnikov A, Trumtel S, Gas N, Petfalski E, Tollervey D, Mann M, Hurt E, Tschochner H (2001) Maturation and intranuclear transport of pre-ribosomes requires Noc proteins. Cell 105:499–509
Nielsen K, Cox GM, Wang P, Toffaletti DL, Perfect JR, Heitman J (2003) Sexual cycle of Cryptococcus neoformans var. grubii and virulence of congenic a and α isolates. Infect Immun 71:4831–4841
Okuda T, Ando A, Sakuradani E, Kikukawa H, Kamada N, Ochiai M, Shima J, Ogawa J (2014) Characterization of galactose-dependent promoters from an oleaginous fungus Mortierella alpina 1S-4. Curr Genet 60:175–182
Ory JJ, Griffith CL, Doering TL (2004) An efficiently regulated promoter system for Cryptococcus neoformans utilizing the CTR4 promoter. Yeast 21:919–926
Park BJ, Wannemuehler KA, Marston BJ, Govender N, Pappas PG, Chiller TM (2009) Estimation of the current global burden of cryptococcal meningitis among persons living with HIV/AIDS. AIDS 23:525–530
Park J, Rajasingham R, Smith RM, Boulware DR (2014) Update on the global burden of cryptococcosis. Mycoses 57S1:6
Pitkin JW, Panaccione DG, Walton JD (1996) A putative cyclic peptide efflux pump encoded by the TOXA gene of the plant-pathogenic fungus Cochliobolus carbonum. Microbiology 142:1557–1565
Roemer T, Krysan DJ (2014) Antifungal drug development: challenges, unmet clinical needs, and new approaches. Cold Spring Harb Perspect Med 4:a019703
Ruff JA, Lodge JK, Baker LG (2009) Three galactose inducible promoters for use in C. neoformans var. grubii. Fungal Genet Biol 46:9–16
Sikorski RS, Hieter P (1989) A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122:19–27
Taylor JW, Berbee ML (2006) Dating divergences in the fungal tree of life: review and new analyses. Mycologia 98:838–849
Toffaletti DL, Rude TH, Johnston SA, Durack DT, Perfect JR (1993) Gene transfer in Cryptococcus neoformans by use of biolistic delivery of DNA. J Bacteriol 175:1405–1411
Walton FJ, Idnurm A, Heitman J (2005) Novel gene functions required for melanization of the human pathogen Cryptococcus neoformans. Mol Microbiol 57:1381–1396
Wickes BL, Edman JC (1995) The Cryptococcus neoformans GAL7 gene and its use as an inducible promoter. Mol Microbiol 16:1099–1109
Winston F, Dollard C, Ricupero-Hovasse SL (1995) Construction of a set of convenient Saccharomyces cerevisiae strains that are isogenic to S288C. Yeast 11:53–55
Xue C, Tada Y, Dong X, Heitman J (2007) The human fungal pathogen Cryptococcus can complete its sexual cycle during a pathogenic association with plants. Cell Host Microbe 1:263–273
Zhang Y, Yu Z, Fu X, Liang C (2002) Noc3p, a bHLH protein, plays an integral role in the initiation of DNA replication in budding yeast. Cell 109:849–860
Acknowledgements
This research was initiated at the University of Missouri-Kansas City: from there we thank Colleen Mayhue, Chris Naumann and Nghi Chau for their technical help. The work was supported by grants from the United States National Institutes of Health (AI094364) and Australian Research Council (FT130100146).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by M. Kupiec.
Rights and permissions
About this article

Cite this article
Ianiri, G., Boyce, K.J. & Idnurm, A. Isolation of conditional mutations in genes essential for viability of Cryptococcus neoformans . Curr Genet 63, 519–530 (2017). https://doi.org/10.1007/s00294-016-0659-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00294-016-0659-2