
Abstract
Manganese peroxidase (MnP) is a major extracellular component of the lignin-degrading system of the white-rot fungus, Phanerochaete chrysosporium. Homologous expression of recombinant MnP isozyme 1 (rMnP1) in P. chrysosporium was achieved using a novel transformation system for this fungus, which utilizes the Streptomyces hygroscopicus bialaphos-resistant gene, bar, as the selectable marker. The transformation frequency for this system is approximately 100 bialaphos-resistant transformants per microgram of plasmid DNA. Transformed strains all contain plasmid DNA, ectopically integrated into the fungal genome. Using this transformation system, the promoter region of the P. chrysosporium translation elongation factor gene was used to drive expression of mnp1, encoding MnP1, in primary metabolic cultures of P. chrysosporium, where endogenous MnP was not expressed. Approximately 2–3 mg of active recombinant MnP1 per liter of extracellular medium was produced in agitated cultures of transformants.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The white-rot basidiomycete, Phanerochaete chrysosporium, has been the focus of numerous studies on lignin (Gold et al. 1989; Kirk and Farrell 1987) and aromatic pollutant degradation (Bumpus and Aust 1987; Hammel 1989; Reddy et al. 1998). The major extracellular components of the lignin degradation system in this fungus are two families of peroxidases—lignin peroxidase (LiP) and manganese peroxidase (MnP)—along with an extracellular H2O2-generating system (Gold and Alic 1993; Gold et al. 1989; Kirk and Farrell 1987). Both MnP and LiP have been characterized by a variety of biochemical and kinetic techniques (Gold and Alic 1993; Gold et al. 1989, 2000). Crystal structures of both peroxidases have been reported (Piontek et al. 1993; Poulos et al. 1993; Sundaramoorthy et al. 1994). The genes encoding various LiPs and MnPs from P. chrysosporium have been isolated and sequenced over the past 15 years (Cullen 1997; Gold and Alic 1993; Gold et al. 2000). The regulation of MnP gene expression has also been studied, using promoter–reporter systems (Gettemy et al. 1997; Godfrey et al. 1994; Ma et al. 2001).
To study structure–function relationships in MnP and LiP, efficient expression systems for the recombinant form of these enzymes are essential. Owing to complex post-translational modification of these extracellular proteins, homologous expression in P. chrysosporium is advantageous, because the resulting recombinant enzymes closely resemble the wild-type enzymes (Gold et al. 2000; Mayfield et al. 1994; Sollewijn Gelpke et al. 1999). However, productive homologous expression requires an efficient DNA transformation system for P. chrysosporium. Two strategies are employed for the selection of transformed P. chrysosporium strains. The first method, pioneered in our laboratory, involves selection of prototrophic transformants upon complementation of auxotrophic P. chrysosporium strains with wild-type genes. We reported integrative transformation systems using both heterologous and homologous ade (Alic et al. 1989, 1990) and ura (Akileswaran et al. 1993) genes as the selectable markers. The ade and ura transformation systems were employed for homologous production of recombinant P. chrysosporium MnP (Mayfield et al. 1994), LiP (Sollewijn Gelpke et al. 1999), and cellobiose dehydrogenase (Li et al. 2000). Nonetheless, this selection method has disadvantages. First, it requires isolating P. chrysosporium auxotrophic strains and characterizing genes that complement the mutations in these strains. Second, many P. chrysosporium auxotrophic strains grow less robustly than the wild-type strain and reversion of auxotrophic mutations is a theoretical possibility. A second method for selection of P. chrysosporium transformants uses dominant selectable markers, which are genes introduced into wild-type strains to confer drug resistance. A bacterial kanamycin resistance determinant of Tn903 was reported to confer G418 resistance in transformed P. chrysosporium (Randall and Reddy 1991; Randall and Reddy 1992; Randall et al. 1989, 1991). Recently, transformation systems were reported which utilize the phleomycin (Ph) resistance gene from Streptoallotiechus hindustanus as the selectable marker (Birch et al. 1998; Gessner and Raeder 1994). In each case, the transforming plasmids were, apparently, maintained extrachromosomally. However, the transformation frequency in both systems was too low (6–10 transformants/μg of plasmid DNA) for efficient protein expression studies or other applications. Hence, the establishment of a transformation system using a dominant selectable marker with high transformation frequency would be advantageous for homologous expression studies in P. chrysosporium; and such a system could also aid in gene disruption experiments in this organism.
In this report, we describe a new integrative transformation system for P. chrysosporium with relatively high transformation frequency, using the bialaphos resistance gene (bar) from the bacterium Streptomyces hygroscopicus as a selectable marker. Bialaphos is a potent, nonselective herbicide produced by S. hygroscopicus which is composed of two l-alanine residues and an analogue of glutamic acid, phosphinothricin (PPT) (Thompson et al. 1987). PPT, the active component of bialaphos, is a potent inhibitor of glutamine synthetase. The bar gene isolated from S. hygroscopicus encodes an acetyl-transferase that acetylates PPT (Thompson et al. 1987). This gene has been introduced into plants, including tobacco, tomato, and potato (De Block et al. 1987), and into fungi, including Neurospora crassa (Avalos et al. 1989), Erysiphe graminis (Chaure et al. 2000), Cercospora kikuchii (Upchurch et al. 1994), and Pleurotus ostreatus (Yanai et al. 1996), resulting in high-level resistance to bialaphos.
Using a transformation system based on the bar gene, we constructed a relatively efficient homologous expression system for recombinant Phanerochaete chrysosporium MnP isozyme 1 (rMnP1). The promoter region of the P. chrysosporium translation elongation factor (TEF) gene was used to drive expression of mnp1, encoding MnP1, in primary metabolic cultures of P. chrysosporium, where endogenous MnP was not expressed. Using this system, approximately 2–3 mg of active rMnP1 per liter of extracellular fluid was produced.
Materials and methods
Chemicals
PPT (glufosinate) was obtained from Sigma–Aldrich Laborchemikalien, Germany.
Organisms
Homokaryon 8, a homokaryon derivative of P. chrysosporium strain OGC101, was isolated by fruiting and selecting a single basidiospore-derived colony as described by Alic et al. (1987) and Gold and Cheng (1978, 1979). Escherichia coli strain DH5α was used for plasmid construction and preparation.
Construction of pGBar and pGBar(i)
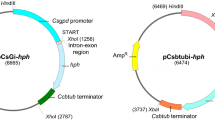
pCB1635, containing bar, was obtained from the Fungal Genetics Stock Center (University of Kansas Medical Center, Kansas City, Kan.). pGBar contained a bar expression cassette subcloned into pUC18 (New England Biolabs), which included, from 5′ to 3′, 1.1 kb of the P. chrysosporium gpd promoter (Mayfield et al. 1994), the bar coding sequence, and 250 bp of the P. chrysosporium mnp1 3′ untranslated region (UTR; Fig. 1A). The gpd promoter was fused with the bar coding sequence at the translation start codon, using the megaprimer polymerase chain reaction (PCR; Barik 1997). In the first PCR for constructing the megaprimer, the template was pGPDstu1.8, which contained 1.8 kb of gpd DNA (including the 1.1-kb gpd promoter and the 0.8-kb gpd coding sequence) subcloned as a XbaI-EcoRI fragment into pUC18 (Mayfield et al. 1994). The forward primer, gpd-HindIII, corresponded to the gpd promoter sequence 170 bp upstream of the start codon and contained a HindIII site (Table 1). The reverse primer, bar-gpd, was composed of 17 nt of the 5′ end of the bar coding sequence followed by 17 nt of the 3′ end of the gpd promoter sequence (Table 1). The amplified megaprimer was purified by agarose gel electrophoresis and extracted using a gel extraction kit (Qiagen). The second PCR mixture included the forward primer used in the first PCR, the megaprimer, and pCB1635 as the template. The reverse primer used in this PCR, bar-kpnI, corresponded to the 3′ end of the bar coding sequence and included a KpnI site at its 5′ end (Table 1). The final PCR product was a 0.7-kb gpd-bar fragment, containing the 170-bp gpd promoter joined with the entire bar coding sequence. This fragment was subsequently digested with HindIII and KpnI. The 250-bp mnp1 3′UTR with an introduced KpnI site at its 5′ end was amplified by PCR using mnp1 genomic DNA as the template, and m1-3′UTR-K and m1-3′UTR-RI as the forward and reverse primers, respectively (Table 1). The amplified fragment was digested with KpnI and EcoRI. Finally, the HindIII-KpnI gpd-bar fragment and the KpnI-EcoRI mnp1 3′UTR fragment were ligated to HindIII-EcoRI-digested pGPDstu1.8, yielding pGBar. pGBar(i) was similar to pGBar, except that the bar expression cassette in pGBar(i) contained the 1.1-kb gpd promoter together with the gpd exon 1 (6 bp), intron 1 (55 bp) and the 5′ end of exon 2 (9 bp) joined with the ATG-less bar coding sequence (Fig. 1B). The same materials and megaprimer PCR procedure for constructing pGBar were used to construct pGBar(i), with the exception of the reverse primer used in the first PCR. For constructing pGBar(i), this primer was gpd-bar(i) containing 17 nt of the 5′ end of the bar coding sequence (excluding the start codon), followed by 9 nt of the 5′ end of the gpd exon 2 and 8 nt of the 3′ end of the gpd intron 1 (Table 1).

Restriction maps of plasmid constructs. A pGBar, B pGBar(i), C pBar3.8, D pBarTM1. The locations of the gpd promoter, bar coding sequence (bar CDS), mnp1 3′untranslated region (3′UTR), TEF-1 gene promoter, and mnp1 genomic sequence are indicated. For pGBar(i), the short intron-containing sequence is also indicated. For pBar3.8, the restriction sites in parentheses are not present and this has been confirmed by restriction analysis. The unique restriction sites in the multiple cloning sites in pBar3.8 are in bold
Construction of pBar3.8 and pBarTM1
A shorter version of the bar expression cassette from pGBar, limited to 300 bp of the gpd promoter followed by the bar coding sequence and the mnp1 3′UTR, was amplified by PCR. This fragment was ligated to SspI-digested pUC18, yielding pBar3.8 (Fig. 1C). pBarTM1 was constructed by inserting the mnp1 expression cassette into pBar3.8 between the multiple cloning sites, XbaI and EcoRI. The mnp1 expression cassette contained the 870-bp promoter region of the P. chrysosporium TEF gene, followed by the 2.6-kb P. chrysosporium mnp1 genomic DNA (including 1.5 kb of the coding sequence and 1.0 kb of the 3′UTR; Fig. 1D). The TEF gene promoter was isolated as follows: the deduced amino acid sequence of TEF-1α from Aureobasidium pullulans (Thornewell et al. 1995) was used to search for its homologue in the translated P. chrysosporium genomic nucleotide sequence (http://www.jgi.doe.gov/programs/whiterot), using the BLAST program (Altschul et al. 1997). A putative P. chrysosporium TEF coding sequence was located on scaffold 2 at nt 202,805–204,593. An 870-bp fragment containing the P. chrysosporium TEF gene promoter was amplified by PCR, using the genomic DNA of homokaryon 8 as the template and the forward and reverse primers, TEFpr870-5′ and TEFpr870-3′, respectively (Table 1). The TEF gene promoter was joined with the mnp1 genomic sequence at the translation start codon, using the megaprimer PCR. In the first PCR, a megaprimer was prepared containing 21 bp of the 3′ end of TEF gene promoter linked with 144 bp of the mnp1 coding sequence at the start codon. In this PCR, the mnp1 genomic DNA was used as the template. The forward primer was TEFpr-mnp1, which comprised 21 bp of the 3′ end of the TEF gene promoter followed by 18 bp of the 5′ end of the mnp1 coding sequence (Table 1). The reverse primer was mnp1-NotI, which complemented a segment of the mnp1 coding sequence 144 bp downstream of the start codon and 92 bp downstream of a unique NotI site (Table 1). The second PCR included the forward primer TEFpr870-5′, the megaprimer from the first PCR, the reverse primer mnp1-NotI, and the 870-bp TEF gene promoter DNA as the template. Finally, a 1.0-kb fragment (tef-mnp fragment) containing the 870-bp TEF gene promoter joined with the 144-bp of mnp1 coding sequence was amplified. This fragment was digested at a XbaI site introduced at its 5′ end and the NotI site in mnp1. The XbaI-NotI tef-mnp fragment, a NotI-EcoRI mnp1 fragment (containing a part of the mnp1 coding sequence and 1.0 kb of the 3′UTR), and XbaI-EcoRI-digested pBar3.8 were ligated to yield pBarTM1.
Culture medium
GK medium contained a mineral solution (Kirk et al. 1978), 1% glucose and 12 mM ammonium tartrate in 10 mM potassium phosphate, pH 5.0. Minimal medium contained 2% glucose, 0.2% asparagine, and 3 μM thiamine in 10 mM potassium phosphate, pH 5.0 (Alic et al. 1989). GK agar and GK top agar media were the basic GK medium supplemented, respectively, with 0.5 M MgSO4 and 1.5% agar and with 0.4 M MgSO4 and 1% agar. High-carbon, high-nitrogen (HCHN) medium contained Kirk′s salts, 2% glucose, and 12 mM ammonium tartrate in 20 mM 2,2-dimethyl succinate buffer, pH 4.5.
Fungal transformations
Plasmids containing the bar expression cassette were prepared using a Miniprep Kit (Qiagen). Subsequently, 1 μg of plasmid DNA was linearized at the unique EcoRI site and used to transform protoplasts (2×106) generated from the basidiospores of homokaryon 8, as described by Alic et al. (1991). The following modifications were made in the transformation procedure. The diluted protoplast–DNA mixture, containing approximately 4×104 protoplasts and 25 ng of DNA, was added to 3 ml of melted GK top agar medium and immediately spread onto a plate containing 20 ml of solid GK agar medium. After the plates were incubated at 37 °C for 2 h, PPT was diluted in another 3 ml of melted GK top agar medium and spread on each plate to a final concentration of 30 μg/ml.
Southern blot
Genomic DNA from various transformants was extracted as described by Alic et al. (1989). Genomic DNA (1 μg) from each transformant was digested with a selected restriction enzyme and the resulting fragments were resolved on a 0.7% agarose gel and subsequently transferred to a Nylon membrane. The bar coding sequence DNA was used as the template to synthesize random primed [α-32P]dCTP-labeled probes. Southern hybridization and autoradiography were performed as described by Alic et al. (1989).
Production of rMnP1
Approximately 40 PPT-resistant transformants carrying pBarTM1 were assayed for rMnP activity by the o-anisidine plate assay (Mayfield et al. 1994). The transformant exhibiting the highest rMnP activity in the plate assay was purified further, by fruiting and selecting single basidiospore isolates (Alic et al. 1987; Gold and Cheng 1978, 1979). The purified transformant strain was then grown from a conidial inoculum in 20-ml stationary HCHN cultures (with the addition of 0.2% tryptone) at 37 °C for 3 days. Subsequently, the mycelial mat from two stationary cultures was homogenized for 20 s in a blender and used to inoculate 500 ml of HCHN medium in 1-l flasks. The cultures were grown at 37 °C on a rotary shaker at 150 rpm and MnP activity from the extracellular fluid was measured by monitoring the formation of Mn3+-malonate at 270 nm, using a Shimadzu model UV-260 spectrophotometer, as described by Glenn and Gold (1985) and Wariishi et al. (1992). The enzyme reaction mixture (1 ml) contained 0.1 ml of extracellular fluid, 5 mM MnSO4, and 0.1 mM H2O2 in 50 mM sodium malonate, pH 4.5.
Western blot
Extracellular proteins from 3-day-old HCHN shake-cultures of various transformants were resolved on SDS-PAGE and then transferred to a nitrocellulose membrane. A polyclonal antibody against MnP (Pribnow et al. 1989) and the Western-light protein detection kit (Tropix, Bedford, Mass.) were used to detect the MnP signal, following the manufacturer′s directions.
Results and discussion
Transforming P. chrysosporium to PPT resistance
The sensitivity of P. chrysosporium strain OGC101 to PPT was examined in minimal medium, which was used in transformation studies with ade or ura genes as the selectable markers (Akileswaran et al. 1993; Alic et al. 1989, 1991) and in GK medium. PPT (40 μg/ml) completely inhibited the germination of conidial spores from strain OGC101 on solid GK medium (GK supplemented with 1.5% agar). In contrast, 80 μg/ml of PPT were required to inhibit conidial spore germination on solid minimal medium (minimal medium supplemented with 1.5% agar). Because of its genetic homogeneity, a homokaryon derivative of strain OGC101 was chosen as the host for our transformation studies. PPT (30 μg/ml) completely inhibited the germination of basidiospores from homokaryon 8 on solid GK medium. Thus, GK medium and a final PPT concentration of 30 μg/ml were utilized in transformation experiments.
Two plasmids were constructed, pGBar and pGBar(i), which carry bar-expression cassettes. The bar-expression cassette in pGBar contained the 1.1-kb P. chrysosporium gpd promoter followed by the bar coding sequence and the 250-bp P. chrysosporium mnp1 3′UTR (Fig. 1A). The bar-expression cassette in pGBar(i) was similar to that in pGBar, except that an extra sequence, containing the gpd exon 1 (6 bp), intron 1 (55 bp), and the 5′ end of exon 2 (9 bp), was inserted at the 5′ end of the bar coding sequence (Fig. 1B). Transformation of the homokaryon 8 protoplasts with 1 μg of linearized pGBar or pGBar(i) yielded 80–100 PPT-resistant transformants; and approximately 0.1% of the viable protoplasts were transformed to PPT resistance. In contrast, no transformants were observed in minus DNA controls. The transformants first appeared after incubation at 37 °C for 3 days and continued to emerge until day 7. During this period, the background growth of non-transformants was much weaker than the growth of PPT-resistant transformants; and, thus, the transformants could readily be distinguished. Forty transformants carrying either pGBar or pGBar(i) were stably maintained on slants containing 2 ml of GK solid medium supplemented with 60 μg PPT/ml for six generations, suggesting that they are mitotically stable. Moreover, five transformants carrying either pGBar or pGBar(i) were induced to fruit on nutrient-poor media (Gold and Cheng 1979) and over 80% of the isolated basidiospore progenies of each transformant grew robustly on GK slants supplemented with 60 μg PPT/ml, suggesting the transformants are meiotically stable. Since the DNA-treated P. chrysosporium protoplasts were plated in a high concentration on plates and the protoplasts could undergo fusion during the transformation process, some of the isolated transformants were likely to be heterokaryons, containing both transformed and untransformed nuclei.
Previously, we demonstrated that, in P. chrysosporium, significant expression of the enhanced green fluorescent protein gene (egfp) driven by either the gpd or the mnp1 promoter was observed only when an intron-containing sequence was inserted at the 5′ end of egfp (Ma et al. 2001). In addition, in Schizophyllum commune, inserted introns were required for the efficient expression of several homologous and heterologous genes (Lugones et al. 1999; Scholtmeijer et al. 2001), including the hygromycin B resistance gene from E. coli. However, our present results show that both intron-less and intron-containing bar-expressing plasmids can transform P. chrysosporium to PPT resistance with equal transformation frequency. One possible explanation is that even a low level of bar expression might still be sufficient to confer resistance to PPT at the concentration of 30 µg/ml.
The transformation frequency obtained with both pGBar and pGBar(i) is about 80–100 transformants/μg DNA. This frequency is comparable with those obtained with plasmids containing ade (100–300 transformants/μg DNA) or ura genes (100–600 transformants/μg DNA) from either P. chrysosporium or S. commune as selectable markers (Akileswaran et al. 1993; Alic et al. 1989, 1990, 1991). Furthermore, these frequencies are significantly higher than those reported for plasmid constructs, containing either the bacterial kanamycin resistance gene or the Ph resistance gene as selectable markers (6–10 transformants/μg DNA; Birch et al. 1998; Gessner and Raeder 1994; Randall and Reddy 1991, 1992; Randall et al. 1989, 1991).
Varying lengths of gpd promoters were tested for their ability to drive bar expression in P. chrysosporium. A minimum of 300 bp of the gpd promoter was sufficient for bar expression, as no reduction in transformation frequency was observed with a plasmid containing this short promoter. For subsequent subcloning, the bar expression cassette, containing the 300-bp gpd promoter, the bar coding sequence, and the mnp1 3′UTR, was inserted at a unique SspI site in pUC18, freeing the multiple cloning site for additional inserts. This plasmid was named pBar3.8 (Fig. 1C). Southern blot analysis was performed on DNA extracted from the homokaryon derivatives of three randomly chosen PPT-resistant transformants carrying pBar3.8, using the 32P-labeled bar coding sequence as the probe (Fig. 2). The absence of rapidly migrating bands with undigested transformant DNA (lanes 4–6) indicates that the transforming plasmids integrated into the chromosomes rather than being carried in an autonomously replicating form. This contrasts with previous reports of the transformation systems using the bacterial kanamycin resistance gene or Ph resistance gene as the selectable markers (Birch et al. 1998; Randall and Reddy 1991, 1992; Randall et al. 1989). In those earlier studies, the transforming plasmids contained characterized sequences (Randall et al. 1989, 1991; Rao and Reddy 1984) or some unidentified sequences (Birch et al. 1998; Randall and Reddy 1991), which apparently allowed autonomous and extrachromosomal replication. Such sequences are presumably lacking in our bar-expressing plasmids. The Southern blot also shows that the three transformants carry one or more copies of integrated pBar3.8 (Fig. 2, lanes 8–10, lanes 12–14). Multiple copy integration is frequently observed in fungal transformations (Fincham 1989; Gold and Alic 1993). In conclusion, an efficient and integrative transformation system, utilizing bar as a selectable marker was developed for P. chrysosporium.

Southern blot analysis of DNA from homokaryon 8, a basidiospore derivative of Phanerochaete chrysosporium strain OGC101 and transformants carrying pBar3.8. The blot was hybridized with the 32P-labeled bar coding sequence. Lane 1 100 pg of undigested pBar3.8, lane 2 100 pg of EcoRI-linearized pBar3.8, lanes 3–6 undigested genomic DNA from homokaryon 8 and transformants 2, 3, and 4, respectively, carrying pBar3.8, lanes 7–10 the same genomic DNA as in lanes 3–6 digested with PstI, lanes 11–14 the same genomic DNA digested with HindIII. The positions of molecular mass standards are shown on the left
Homologous expression of rMnP1 in P. chrysosporium using bar as a selectable marker
The plasmid pBarTM1, containing the 870-bp promoter region of the P. chrysosporium TEF gene joined with the 2.6-kb fragment encompassing the P. chrysosporium mnp1 genomic coding sequence and 3′UTR, was constructed using pBar3.8 for homologous expression of rMnP1 in P. chrysosporium (Fig. 1D). In this fungus, endogenous mnp genes are expressed only during the secondary metabolic phase, which is triggered by the depletion of nutrient nitrogen (Gold and Alic 1993; Kirk and Farrell 1987). Coupling the mnp1 genomic coding sequence to the TEF gene promoter enables production of rMnP1 during the primary metabolic phase, when nutrient nitrogen is sufficient and the endogenous mnp genes are not expressed. Using a similar strategy, we previously constructed pAGM1, in which the 1.1-kb gpd promoter was joined with the mnp1 genomic sequence and the ade5 gene was used as the selectable marker (Mayfield et al. 1994). The transformant carrying pAGM1 (transformant T15), produced approximately 2 mg of rMnP1 per liter of extracellular culture fluid. The rMnP1 produced in T15 exhibited physical, kinetic, and spectral characteristics essentially identical to those of the native MnP1.
Forty PPT-resistant transformants carrying pBarTM1 were examined for rMnP activity using our o-anisidine plate assay (Mayfield et al. 1994). Among these transformants, 80% exhibited rMnP activity. In contrast, neither the untransformed homokaryon 8 nor transformants carrying the blank vector pBar3.8 exhibited rMnP activity. This indicates that the rMnP activity in transformants carrying pBarTM1 results from the expression of mnp1 driven by the TEF gene promoter. The level of rMnP activity varies among the transformants carrying pBarTM1, as observed for transformants carrying pAGM1 (Mayfield et al. 1994). Transformed strain T1, carrying pBarTM1, exhibiting the highest rMnP activity in the plate assay, was purified by isolating single basidiospores and analyzed further in liquid shake-cultures. Fig. 3 shows a time-course for rMnP activity in shake-cultures of T1 (carrying pBarTM1), T15 (carrying pAGM1), and T1 (carrying pBar3.8 as a control). While no MnP activity was observed in control cultures, similar rMnP activity profiles were exhibited in T1 (pBarTM1) and T15 (pAGM1). As previously shown for T15 (pAGM1; Mayfield et al. 1994), the rMnP activity in cultures of T1 carrying pBarTM1 was highest when grown in nitrogen-sufficient medium with an initial pH of 6.5, while the endogenous MnP was most abundant when grown in nitrogen-limited medium with an initial pH of 4.5 (data not shown). These data also confirm that the activity measured is, indeed, from recombinant, not endogenous MnP. Small differences exist between the rMnP activity profiles exhibited by T1 (pBarTM1) and T15 (pAGM1). As shown in Fig. 3, rMnP activity at pH 4.5 peaked on day 2 and increased again on day 5 in both the T1 (pBarTM1) and T15 (pAGM1) cultures. On day 6, the rMnP activity of T1 (pBarTM1) was 5-fold higher than that of T15 (pAGM1). At pH 6.5, similar rMnP activity was reached in both the T1 (pBarTM1) and T15 (pAGM1) cultures; however, T1 (pBarTM1) peaked on day 3 and T15 (pAGM1) peaked on day 4. Figure 4 shows a Western blot of purified P. chrysosporium wild-type MnP1 and proteins in the extracellular fluid from 3-day-old shake-cultures of T1 (pBar3.8), T1 (pBarTM1), and T15 (pAGM1). As shown, rMnP1 expressed in either T1 (pBarTM1) or T15 (pAGM1) migrated with the same speed on SDS-PAGE as the purified wild-type MnP1. Approximately 2–3 mg of rMnP1 per liter of extracellular culture fluid was produced in both T1 (pBarTM1) and T15 (pAGM1) cultures. This yield is approximately 30% of the endogenous MnP produced in the wild-type strain OGC101 from multiple mnp genes under nitrogen-limiting conditions.

Manganese peroxidase (MnP) activity in the extracellular fluid in primary metabolic cultures of P. chrysosporium transformants. The culture conditions are described in the Materials and methods. The pH of the cultures was adjusted to 4.5 or 6.5. The data from one of three independent experiments (with similar results) are shown. Squares Transformant 1 (T1) carrying pBar3.8, triangles transformant 15 (T15) carrying pAGM1, circles transformant 1 (T1) carrying pBarTM1

Western blot analysis of MnP expressed in T1 carrying pBarTM1 and in T15 carrying pAGM1. Lane 1 100 ng of purified P. chrysosporium MnP1, lane 2 extracellular media containing 15 μg of total protein from 3-day-old agitated high-carbon, high-nitrogen cultures of T1 (pBar3.8), lane 3 T1 (pBarTM1), lane 4 T15 (pAGM1). All samples were separated on 12% SDS-PAGE and blotted with polyclonal antibody raised against MnP, as described in the Materials and methods. The positions of the following molecular mass standards are indicated on the left. From the top: bovine serum albumin (66,300 kDa), glutamate dehydrogenase (55,440 kDa), lactate dehydrogenase (36,500 kDa), and carbonic anhydrase (31,000 kDa)
The production of rMnP was also examined in other organisms, including E. coli (Whitwam et al. 1995) and Aspergillus niger (Conesa et al. 2000, 2002). In E. coli, rMnP was expressed in the form of inclusion bodies and, after reconstitution, a low yield of active rMnP could be obtained. In A. niger, the active rMnP was secreted into the culture medium and, with hemoglobin supplementation, the production of rMnP could reach 100 mg per liter of extracellular culture fluid. However, the rMnP produced by A. niger exhibited a slightly lower mobility on SDS-PAGE than the native MnP, suggesting different post-translational modification in A. niger compared with that in P. chrysosporium. The advantage of a homologous expression system is that the rMnP produced is essentially identical to native MnP. However, improvement of rMnP yield from the homologous expression system must be addressed. In our present study, both homologous rMnP1 expression systems, employing different transformation systems (with ade5 and bar as the selectable markers) and different promoters (gpd and TEF gene promoter) to drive the mnp1 expression, exhibited similar levels of rMnP1 production. Future experimentation is needed to further improve rMnP production in this homologous expression system.
References
Akileswaran L, Alic M, Clark EK, Hornick JL, Gold MH (1993) Isolation and transformation of uracil auxotrophs of the lignin-degrading basidiomycete Phanerochaete chrysosporium. Curr Genet 23:351–356
Alic M, Letzring C, Gold MH (1987) Mating system and basidiospore formation in the lignin-degrading basidiomycete Phanerochaete chrysosporium. Appl Environ Microbiol 53:1464–1469
Alic M, Kornegay JR, Pribnow D, Gold MH (1989) Transformation by complementation of an adenine auxotroph of the lignin-degrading basidiomycete Phanerochaete chrysosporium. Appl Environ Microbiol 55:406–411
Alic M, Clark EK, Kornegay JR, Gold MH (1990) Transformation of Phanerochaete chrysosporium and Neurospora crassa with adenine biosynthetic genes from Schizophyllum commune. Curr Genet 17:305–311
Alic M, Mayfield MB, Akileswaran L, Gold MH (1991) Homologous transformation of the lignin-degrading basidiomycete Phanerochaete chrysosporium. Curr Genet 19:491–494
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acid Res 25:3389–3402
Avalos J, Geever RF, Case ME (1989) Bialaphos resistance as a dominant selectable marker in Neurospora crassa. Curr Genet 16:369–372
Barik S (1997) Mutagenesis and gene fusion by megaprimer PCR. Methods Mol Biol 67:173–182
Birch PRJ, Sims PFG, Broda P (1998) A reporter system for analysis of regulatable promoter functions in the basidiomycete fungus Phanerochaete chrysosporium. J Appl Microbiol 85:417–424
Bumpus JA, Aust SD (1987) Biodegradation of environmental pollutants by the white rot fungus Phanerochaete chrysosporium: involvement of the lignin-degrading system. Bioessays 6:166–170
Chaure P, Gurr SJ, Spanu P (2000) Stable transformation of Erysiphe graminis, an obligate biotrophic pathogen of barley. Nat Biotechnol 18:205–207
Conesa A, Van Den Hondel C, Punt PJ (2000) Studies on the production of fungal peroxidases in Aspergillus niger. Appl Environ Microbiol 66:3016–3023
Conesa A, Jeenes D, Archer DB, Van Den Hondel C, Punt PJ (2002) Calnexin overexpression increases manganese peroxidase production in Aspergillus niger. Appl Environ Microbiol 68:846–851
Cullen D (1997) Recent advances on the molecular genetics of ligninolytic fungi. J Biotechnol 53:273–289
De Block M, Botterman J, Vandewiele M, Dockx J, Thoen C, Gossele V, Movva NR, Thompson C, Van Montagu M, Leemans J (1987) Engineering herbicide resistance in plants by expression of a detoxifying enzyme. EMBO J 6:2513–2518
Fincham JRS (1989) Transformation in fungi. Microbiol Rev 53:148–170
Gessner M, Raeder U (1994) A histone H4 promoter for expression of a phleomycin-resistance gene in Phanerochaete chrysosporium. Gene 142:237–241
Gettemy JM, Li D, Alic M, Gold MH (1997) Truncated-gene reporter system for studying the regulation of manganese peroxidase expression. Curr Genet 31:519–524
Glenn JK, Gold MH (1985) Purification and characterization of an extracellular Mn(II)-dependent peroxidase from the lignin-degrading basidiomycete, Phanerochaete chrysosporium. Arch Biochem Biophys 242:329–341
Godfrey BJ, Akileswaran L, Gold MH (1994) A reporter gene construct for studying the regulation of manganese peroxidase gene expression. Appl Environ Microbiol 60:1353–1358
Gold MH, Alic M (1993) Molecular biology of the lignin-degrading basidiomycete Phanerochaete chrysosporium. Microbiol Rev 57:605–622
Gold MH, Cheng TM (1978) Induction of colonial growth and replica plating of the white rot basidiomycete Phanerochaete chrysosporium. Appl Environ Microbiol 35:1223–1225
Gold MH, Cheng TM (1979) Conditions for fruit body formation in the wood rotting basidiomycete Phanerochaete chrysosporium. Arch Microbiol 121:37–41
Gold MH, Wariishi H, Valli K (1989) Extracellular peroxidases involved in lignin degradation by the white rot basidiomycete Phanerochaete chrysosporium. ACS Symp Ser 389:127–140
Gold MH, Youngs HL, Sollewijn Gelpke MD (2000) Manganese peroxidase. In: Sigel A, Sigel H (eds) Manganese and its role in biological processes. Dekker, New York, pp 559–586
Hammel KE (1989) Organopollutant degradation by ligninolytic fungi. Enzyme Microb Technol 11:776–777
Kirk TK, Farrell RL (1987) Enzymatic ″combustion″: the microbial degradation of lignin. Annu Rev Microbiol 41:465–505
Kirk TK, Schultz E, Connors WJ, Lorenz LF, Zeikus JG (1978) Influence of culture parameters on lignin metabolism by Phanerochaete chrysosporium. Arch Microbiol 177:277–285
Li B, Rotsaert FAJ, Gold MH, Renganathan V (2000) Homologous expression of recombinant cellobiose dehydrogenase in Phanerochaete chrysosporium. Biochem Biophys Res Commun 270:141–146
Lugones L, Scholtmeijer K, Klootwijk R, Wessels JGH (1999) Introns are necessary for mRNA accumulation in Schizophyllum commune. Mol Microbiol 32:681–689
Ma B, Mayfield MB, Gold MH (2001) The green fluorescent protein gene functions as a reporter of gene expression in Phanerochaete chrysosporium. Appl Environ Microbiol 67:948–955
Mayfield MB, Kishi K, Alic M, Gold MH (1994) Homologous expression of recombinant manganese peroxidase in Phanerochaete chrysosporium. Appl Environ Microbiol 60:4303–4309
Piontek K, Glumoff T, Winterhalter K (1993) Low pH crystal structure of glycosylated lignin peroxidase from Phanerochaete chrysosporium at 2.5 Å resolution. FEBS Lett 315:119–124
Poulos TL, Edwards SL, Wariishi H, Gold MH (1993) Crystallographic refinement of lignin peroxidase at 2 Å. J Biol Chem 268:4429–4440
Pribnow D, Mayfield MB, Nipper VJ, Brown JA, Gold MH (1989) Characterization of a cDNA encoding a manganese peroxidase, from the lignin-degrading basidiomycete Phanerochaete chrysosporium. J Biol Chem 264:5036–5040
Randall T, Reddy CA (1991) An improved transformation vector for the lignin-degrading white-rot basidiomycete Phanerochaete chrysosporium. Gene 103:125–130
Randall T, Reddy CA (1992) The nature of extrachromosomal maintenance of transforming plasmids in the filamentous basidiomycete Phanerochaete chrysosporium. Curr Genet 21:255–260
Randall T, Rao TR, Reddy CA (1989) Use of a shuttle vector for the transformation of the white rot basidiomycete, Phanerochaete chrysosporium. Biochem Biophys Res Commun 161:720–725
Randall T, Reddy CA, Boominathan K (1991) A novel extrachromosomally maintained transformation vector for the lignin-degrading basidiomycete Phanerochaete chrysosporium. J Bacteriol 173:776–782
Rao TR, Reddy CA (1984) DNA sequences from a ligninolytic filamentous fungus Phanerochaete chrysosporium capable of autonomous replication in yeast. Biochem Biophys Res Commun 118:821–827
Reddy GVB, Sollewijn Gelpke MD, Gold MH (1998) Degradation of 2,4,6-trichlorophenol by Phanerochaete chrysosporium: Involvement of reductive dechlorination. J Bacteriol 180:5159–5164
Scholtmeijer K, Wosten HAB, Springer J, Wessels JGH (2001) Effect of introns and AT-rich sequences on expression of the bacterial Hygromycin B resistance gene in the basidiomycete Schizophyllum commune. Appl Environ Microbiol 67:481–483
Sollewijn Gelpke MD, Mayfield M, Cereghino GPL, Gold MH (1999) Homologous expression of recombinant lignin peroxidase in Phanerochaete chrysosporium. Appl Environ Microbiol 65:1670–1674
Sundaramoorthy M, Kishi K, Gold MH, Poulos TL (1994) The crystal structure of manganese peroxidase from Phanerochaete chrysosporium at 2.06-Å resolution. J Biol Chem 269:32759–32767
Thompson CJ, Movva NR, Tizard R, Crameri R, Davies JE, Lauwereys M, Botterman J (1987) Characterization of the herbicide-resistance gene bar from Streptomyces hygroscopicus. EMBO J 6:2519–2523
Thornewell SJ, Peery RB, Skatrud PL (1995) Cloning and characterization of the gene encoding translation elongation factor 1a from Aureobasidium pullulans. Gene 162:105–110
Upchurch RG, Meade MJ, Hightower RC, Thomas RS, Callahan TM (1994) Transformation of the fungal soybean pathogen Cercospora kikuchii with the selectable marker bar. Appl Environ Microbiol 60:4592–4595
Wariishi H, Valli K, Gold MH (1992) Manganese(II) oxidation by manganese peroxidase from the basidiomycete Phanerochaete chrysosporium. Kinetic mechanism and role of chelators. J Biol Chem 267:23688–23695
Whitwam RE, Gazarian IG, Tien M (1995) Expression of fungal Mn peroxidase in E. coli and refolding to yield active enzyme. Biochem Biophys Res Commun 216:1013–1017
Yanai K, Yonekura K, Usami H, Hirayama M, Kajiwara S, Yamazaki T, Shishido K, Adachi T (1996) The integrative transformation of Pleurotus ostreatus using Bialaphos resistance as a dominant selectable marker. Biosci Biotechnol Biochem 60:472–475
Acknowledgements
This research was supported by grant MCB-9723725 from the National Science Foundation and grant DE-FG-03-96ER20235 from the United States Department of Energy to M.H.G.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by U. Kück
Rights and permissions
About this article
Cite this article
Ma, B., Mayfield, M.B. & Gold, M.H. Homologous expression of Phanerochaete chrysosporium manganese peroxidase, using bialaphos resistance as a dominant selectable marker. Curr Genet 43, 407–414 (2003). https://doi.org/10.1007/s00294-003-0418-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00294-003-0418-z