
Abstract
Blending of poly(l-lactide) and poly(d-lactide) (PLLA, PDLA) results in the formation of poly(lactide) stereocomplex crystallites (SC). For the preparation method, numerous reports employed solution mixing and stirring to receive a product with uniform performance. However, the effect of stirring time during solution mixing on the subsequent crystallization behavior did not clear yet, and which is important for solution crystallization, casting and spinning. In this paper, linear PLLA and PDLA with various molecular weights were synthesized. The PLLA and PDLA solutions were mixed together and stirred for different times before casting. Results revealed that the structures of PLA SC and homochiral crystallites (HC) did not vary with change in the stirring time. More amount of content of SC with uniform structure and less amount of content of HC were produced in the specimens stirred for longer time. When the specimens crystallized either from the melt or from the melt-quenching, less amount of content of crystallites (total crystallinity of SC and HC) was produced and the melting temperature of SC and HC depressed in the specimens that were stirred for longer time. With the increase in molecular weights, the effect of stirring time on the crystallization behaviors declined. This investigation provides a route to tailor the crystallization behavior of PLLA/PDLA specimens.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Poly(lactide) (PLA) is a biodegradable macromolecule which has been investigated intensively because it is producible from renewable resources and its product is non-toxic to human body and the natural environment [1]. As the chiral carbon atom exists in the lactyl repeat unit, three isomeric forms of PLA would be produced, i.e., poly(l-lactide) (PLLA), poly(d-lactide) (PDLA) and poly(racemic lactide) (PLDA). The PLLA and PDLA could develop crystallites, and their crystallization capacities and melting temperatures reduce rapidly as optical purity decreases [2,3,4]. The poly(L, d-lactide) (PLDA), which is an amorphous material, exhibits a glass transition temperature (Tg), similar to PLLA and PDLA, ~ 60 °C.
Stereocomplexation could occur among the PLLA and PDLA chains or segments either in solution [5], in the molten [6] or in the rubbery state [7], and it results in the formation of stereocomplex crystallites (SC) that are different from the homochiral crystallites (HC) developed in neat PLLA or PDLA [5]. The SC have a higher melting temperature (~ 250 °C) [8, 9] and exhibit more compact structure than HC [10]; thus, the PLA SC endow materials with higher mechanical performance [11], higher heat deflection temperature [12] and better hydrolysis resistance [13] than that of specimens only HC develop. However, both the SC and HC would form in the PLLA/PDLA blends with higher molecular weights [6], and the formation of HC reduces the thermal and mechanical performances of the blends. To make the SC efficiently form in the PLLA/PDLA blends with higher molecular weights, numerous methods, such as varying chemical structure [14, 15]; blending at a proper temperature [16,17,18]; stereocomplexing in supercritical fluid [19]; low-temperature sintering [20,21,22]; reactive melting blend [23]; applying shearing in the PLLA/PDLA melts [24, 25]; and incorporating polyethylene glycol [26], polymethyl methacrylate [27, 28], block copolymers [29, 30] or nucleate agents [31,32,33,34] as a third component, etc., have been described. During the preparation of PLLA/PDLA casted films [35,36,37,38,39] and nanofibers [40, 41], numerous reports employed solution blending, and the stirring is essential to receive a product with uniform performance. As a matter of fact, the stirring in solution or in the precipitant could supply a flow field, which could facilitate the formation of fibrous crystal or shish-kebabs during solution crystallization [42,43,44,45]. However, the effect of stirring the solution on the subsequent crystallization behavior and morphology of PLLA/PDLA casted film has not been described systemically. Tsuji et al. [46] reported that the content of SC reduced apparently when the PLLA/PDLA precipitate was obtained without stirring. During solution mixing, the PLLA and PDLA molecular chains would disperse evenly by stirring, which facilities the formation of SC. Notwithstanding, the influence of stirring time on the crystallization behavior and whether the SC have memory effect after stirring for longer time are still unclear; yet, these issues are important for the application of solution crystallization, casting and spinning of PLLA/PDLA blends. In this paper, the PLLA/PDLA blends were prepared by solution casting, and the effects of stirring time during solution mixing on the subsequent crystallization and melting behaviors of the casted films were investigated.
Experimental
Materials
The ethyl acetate, toluene, ethanol and dichloromethane were domestic agents with analytical grade. The ethyl acetate was refluxed under calcium hydride for 24 h before distillation. The toluene was refluxed under sodium for 24 before distillation. All other agents were used directly as received. The levorotatory-lactide and dextrorotatory-lactide (l-la and D-LA, optical purity ≥ 99.5%) were purchased from Changchun SinoBiomaterials Co., Ltd., China, and were recrystallized in anhydrous ethyl acetate for three times before being used. The tin(II) 2-ethylhexanoate (Sn(Oct)2, > 95%) was bought from Aladdin and used directly as received. The isopropanol (≥ 99.5%) was purchased from Shanghai Aladdin Co., Ltd. (China) and was dehydrated under calcium hydride for 24 h before distillation. All the purified agents and Sn(Oct)2 were preserved in inert atmosphere before being used.
The synthesis of PLLA and PDLA
The PLLA and PDLA were synthesized by ring-opening polymerization of L-LA and D-LA in anhydrous toluene (the weight of LA/volume of toluene is 1/10) at 120 °C for 24 h, respectively, in the presence of Sn(Oct)2 (0.5 wt%, as the initiator) and isopropanol (as co-initiator). The molecular weights of PLLA and PDLA were regulated by the molar ratio of LA/isopropanol [8, 14]. The synthesized PLLA and PDLA were purified by repeated dissolution and precipitation; dichloromethane was used as solvent and ethanol as the precipitant. The purified solid products were dried at 60 °C in vacuum to constant weight before used. The molecular weights and melting temperatures of PLLA and PDLA are listed in Table 1. The PLLA and PDLA were abbreviated as L and D separately. The numbers after the L and D were their number-average molecular weights (Mn).
Preparation the PLLA/PDLA cast films
The PLLA and PDLA were separately dissolved in dichloromethane at concentrations of 10.0 g/l. After the specimens were dissolved completely, the solutions were mixed together and stirred at room temperature. After stirring for a given time (14, 50 and 100 h), 25 ml mixed solution was taken out and poured into a flat Petri dish. The specimens were obtained by solvent evaporation at room temperature and further vacuum-dried at 40 °C to constant weight. The weight ratio of PLLA: PDLA was fixed at 1:1 for all the specimens. The PLLA/PDLA blends with different molecular weights were separately coded as L32/D31, L58/D67 and L134/D102, where the numbers after L or D were their Mn.
Characterizations
The Mn and molecular weight distributions (PDI) of PLLA and PDLA were evaluated using a gel permeation chromatography (GPC, Waters, USA), which equipped with two Styragel HR gel columns (HR2 and HR4, Waters). The measurements were carried out at 35 °C by using chloroform as an elution solvent, and the flow rate of elution was 1 ml/min. Monodispersed polystyrene standards, with molecular weights ranging from 1790 to 6.0 × 105 g/mol, were adopted to generate the calibration curve. The crystalline structures were characterized by wide-angle X-ray diffraction (WAXD). WAXD profiles were recorded on D8 Advance X-ray diffractometer (Bruker Co., Germany), and the reflection mode was employed, the Cu Kα radiation source (λ = 0.154 nm) was adopted, and the operating voltage and current were 40 kV and 25 mA, respectively. The scanning ranged from 10 to 30° at a scanning speed of 1°/min. Fourier transform infrared (FTIR) spectra were recorded using a Nicolet 6700 spectrophotometer in KBr pellets. All spectra were recorded by averaging 32 scans at a resolution of 2 cm−1 and were baseline corrected. The thermal properties were analyzed by differential scanning calorimetry (DSC) with a DSC 200F3 (NETZSCH, Germany) under a 50 ml/min nitrogen gas flow. For the neat PLLA and PDLA, ~ 5 mg samples were heated from the room temperature to 200 °C at a fixed rate of 10 °C/min. After cooling to room temperature, the second heating curves were recorded. The PLLA/PDLA cast films (5 ± 0.5 mg) were first heated from room temperature to 255 °C at a rate of 10 °C/min, and then: (1) the specimens were kept at 255 °C for 1 min, cooled to room temperature at 5 °C/min to investigate crystallization behavior from the melt, and heated to 250 °C at a rate of 10 °C/min; or (2) kept at 255 °C for 1 min, quenched in liquid nitrogen for 5 min, then heated to 250 °C at 10 °C/min to observe the crystallization and melting behaviors from the melt-quenched state; or (3) annealed at 255 °C for 1, 5 and 10 min, respectively, cooled to room temperature at 5 °C/min, and heated to 250 °C at a rate of 10 °C/min subsequently. The morphologies of the specimens were characterized by using scanning electron microscopes (S-3400N SEM from Hitachi), and the specimens were coated with gold (Au) under vacuum before observation.
Results and discussion
Original data of the specimens
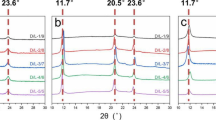
The WAXD profile of the original cast films are shown in Fig. 1. Where the diffraction peaks locate at 16.9° and 19.1° are attributed to the (200/110) and (203) planes of HC, respectively, the diffractions at 12.1°, 21.0° and 24.0° are assigned to (110), (300/030) and (220) planes of SC, respectively [5, 14]. In the L32/D31 specimen, only diffraction peaks assigned to SC were observed, and the intensity of these diffraction peaks enhanced as the specimen was stirred for longer times. In the L58/D67 specimen stirred for 14 h, the diffraction peaks at 12.0°, 16.9°, 19.1° and 21.0° appeared, indicating that both SC and HC developed. Stirred for longer times, the diffraction peaks assigned to SC intensified, while the strength of diffractions assigned to HC did not change obviously, and the ratio of peak intensity at 12.0° and 16.9° (I12.0°/I16.9) and the crystallinity of SC (Xsc) increased with stirring time (Figure S1 and Table S1). In the case of L134/D102, both diffraction peaks assigned to SC and HC enhanced, and the I12.0°/I16.9° and Xsc also increased with stirring time (Figure S1 and Table S1). The WAXD results revealed that the stirring time did not vary the structures of PLLA/PDLA specimens, only SC formed in the specimen with low molecular weight, and both SC and HC produced in the specimens with higher molecular weights. The diffraction peaks assigned to SC intensified as the specimens were stirred for longer times.

The WAXD of PLLA/PDLA blends with stirred for various times
The FT-IR spectra of all the PLLA/PDLA samples are listed in Fig. 2. The absorbances at 921 cm−1 and 909 cm−1 are the characteristic absorption bands of PLA α (α′) crystallites (103 helix) and stereocomplex crystallites (31 helix), respectively [37, 47]. In the L32/D31 specimen with different stirring times, only the absorbance at 909 cm−1 were found, which indicated that only SC developed. For the L58/D67 specimen stirred for 14 h, both the absorbances at 921 cm−1 and 909 cm−1 were observed. After the specimen was stirred for longer times, the absorbance at 921 cm−1 reduced gradually, while the absorbance at 909 cm−1 became more obvious. In the case of L134/D102 specimen, the variations in vibrational bands were similar to that of L58/D67 blend.

The FT-IR spectra of PLLA/PDLA specimens with stirred for various times
DSC scans of the as-cast specimens are presented in Fig. 3, and the melting points and enthalpies calculated from DSC are listed in Table S2. For the L32/D31 specimen stirred for 14 h, two endothermic peaks were found, and the magnitude of the peak at 227 °C was smaller than that at 244 °C. Combined with the WAXD and FTIR results above mentioned, the two endothermic peaks should be assigned to the melting of SC [9, 48]. Stirring for longer times, the temperatures of these two peaks decreased slightly, and the peak intensity at the lower temperature enhanced, while the peak at higher temperature reduced. Notwithstanding, the total melting enthalpy (ΔHsc) of these two peaks did not vary obviously (Table S2). In the L58/D67 specimen stirred for 14 h, two separate melting peaks appeared at 175 °C and 220 °C. The signal at 175 °C should be assigned to the melting of HC (Thc) and the peak at 220 °C was the melting of SC (Tsc) [3, 8, 14]; prolonging the stirring time, the intensity of cold crystallization and the Thc reduced, while the Tsc became narrower although ΔHsc did not vary obviously. These results indicated that SC with more uniform structure developed as the specimen was stirred for longer times, and fewer homochiral PLA molecules or segments were incorporated into the HC lattice. The variation of the L134/D102 specimen was similar to the L58/D67 specimen. From the WAXD and FTIR, the signals assigned to SC enhanced, while the DSC results indicated that the ΔHsc did not vary obviously with stirring time, which should be due to the part of SC developed during heating (from the cold crystallization or after the melting of HC).

DSC of PLLA/PDLA casted films at various molecular weights
In order to observe the structure developed in the specimens, ~ 3-mg casted films were soaked in 6 ml dichloromethane for 24 h. After that, the photographs of these solutions were listed in Figure S2. For the L32/D31 specimen with different stirring times, the film shape was preserved in all the specimens. For the L58/D67 specimen stirred for 14 h, the film disintegrated into lots of small pieces. After stirring for longer time, the disintegrated pieces became larger. The variation of the L134/D102 specimen was similar to that of L58/D67. The unsolved pieces of these specimens were observed by SEM, and their pictures are presented in Fig. 4. For the L32/D31 specimen stirred for 14 h, pores with a size of 1–3 μm distributed evenly in the specimen. Stirred for longer time, the number of pores reduced, and only a few pores were detected after the specimens were stirred for 100 h. For the L58/D67 specimen stirred for 14 h, number of pores with different sizes was observed, and the differences of the pore size were great, parts of the pores were not intact. The pore size reduced after stirred for 50 h. Stirred for 100 h, lots of spherical particles with a size of 1–2 μm were fulfilled the pores (Figure S3). In the case of L134/D102 specimen, the species with uniform size of pores were observed when stirred 14 h. Stirred for 50 h, the size of pore reduced obviously, and a network was observed. Stirred for 100 h, lots of spherical particles were covered on the network (Figure S3).

The SEM of PLLA/PDLA specimens with stirred for various times (the unsolved pieces extracted from the dichloromethane)
Since the formation of SC originates from hydrogen bond interaction between different isomers, the SC would preferentially develop in the specimens during crystallization from the melt [49,50,51] and solution [52, 53], irrespective of the composition and molecular weights. For the formation of HC, it relays on the Van der Waals force among homochiral chains and segments. When the stirring was exerted on the PLLA/PDLA solutions, it could facilitate interaction between isomers, and the SC would produce during the stirring. In the solution, the SC with stable structure developed at the first stage of stereomcoplexation, which presented as network structure (Scheme 1) [54]. For the blends with low molecular weights, the SC network developed sufficiently at a fast rate even stirring for a short time. In addition, the network structure could restrict the movement of PLLA and PDLA polymers, which resulted in the formation of SC with microsphere at the second stage, and the spherical SC had lower melting temperature than that of network. Stirring for longer time, part of network structure would be destroyed due to the low density of chain entanglement in the flow field, and more content of microsphere developed in the specimen with lower molecular weights.

The speculated stereocomplex formation model of the PLLA/PDLA blends
For the PLLA/PDLA blends with higher molecular weights, the network developed at a lower rate, and the network with a larger pore size developed at the first stage. The size of pores reduced with stirring time. After the network developed sufficiently, the SC microsphere also developed at the second stage. As the chain entanglement became obvious and microphase separation occurred between PLLA and PDLA molecular chains with higher molecular weights, only parts of segments participated in the formation of network and microsphere. Thus, the melting temperatures of SC with different structures did not exhibit such obvious differences as specimens with lower molecular weights. Stirred for longer time, the SC network and microsphere distributed evenly in the specimen, and these SC melted in a narrower temperature range. Furthermore, the preformed SC had two effects to the formation of HC, i.e., nucleation and restriction [36, 55]. A small amount of SC would accelerate the formation of HC. As more content of SC developed in the specimens, the restriction effect became more obvious, and less content of HC developed during solution evaporation.
The crystallization from the melt
After melted at 255 °C for 1 min, the non-isothermal crystallization DSC curves are displayed in Fig. 5, and the data are listed in Table S3. In the L32/D31 specimen stirred for 14 h, a clear exothermic peak (Tc) appeared at 119.1 °C. The Tc and enthalpy of this exothermic peak (ΔHc) decreased dramatically after the specimen was stirred for longer times. In the L58/D67 specimen, both the ΔHc and Tc decreased gradually with stirring for longer times. In the case of the L134/D102 blend, the Tc and ΔHc increased at first and then decreased slightly with the increase the stirring time, and this could be ascribed to the reason that both SC and HC formed in the L134/D102 blend when it was stirred for 14 h (the formation of HC and SC were revealed by the DSC in Fig. 6 and the WAXD in Figure S4), and the formation of HC and SC would restrict with each other, resulting in the lower Tc and ΔHc. Stirred for longer time (50 h), only SC formed which could develop at a higher temperature (Fig. 6). Further increasing the stirring time, the Tc and ΔHc reduced.

The non-isothermal DSC profile of the specimens

The second heating DSC scans after the cooling shown in Fig. 3
The second heating of DSC curves (Fig. 6) indicated that the Tsc was 201.7 °C in the L32/D31 specimen stirred for 14 h, and this value was 169.1 °C for L32/D31 blend which was stirred 100 h. (The structure developed in the L32/D31 blend with different stirring times was attested by the WAXD presented in Figure S5.) The reduction of Tsc was 32.5 °C, and the decrement of ΔHsc was 35 J/g. For the L58/D67 specimen, that depression of Tsc was 22.0 °C and the decrement of ΔHsc was 21.3 J/g. And these values were 11.0 °C and 9.3 J/g, respectively, for the L134/D102 specimen. The cooling and second heating DSC curves implied that the crystallization capacity reduced in the specimens that were stirred for longer times during solution mixing, and the depression of Tsc and ΔHsc became smaller with increasing molecular weights of PLA.
When the SC were melted and kept at a relative low temperature, the PLLA and PDLA molecular chain couples would preserve their interactions which resulting in a heterogeneous melt, and this phenomenon was called the “memory effect.” This heterogeneous melt produced crystallites easily [56]. However, the heterogeneous melt transformed to homogeneous melt gradually at higher temperature or melted for longer time, and the interactions between PLLA and PDLA segments are randomly distributed in a homogeneous melt, which could prevent the subsequent crystallization. In the specimens with relative low molecular weights, the “heterogeneous” to “homogeneous” transformation conducted with a higher rate due to this lower viscosity and chain entanglements. For the blends with higher molecular weight, this transformation proceeded at a lower rate, which thanks to the higher viscosity and entanglement density. Consequently, the crystallization capacity depressed at a lower rate in the specimens with higher molecular weights. In the PLLA/PDLA blends stirred for longer times, the SC developed evenly in the specimens and they were melted at a narrower temperature range than that of specimens stirred for short time. After melting at the same temperature, more amount of content of homogeneous melt produced and less “memory effect” preserved in the specimens stirred for longer time. Thus, the crystallization capacity declined with stirring time.
The crystallization from the melt-quenched specimens
In order to attest the assumption above, the DSC curves of the melt-quenched specimens are presented in Fig. 7, and their data are listed in Table S4. A cold crystallization peak appeared at 75 °C (Tcc) in the L32/D31 specimen irrespective of stirring time, but the enthalpy of cold crystallization (ΔHcc), the Tsc and ΔHsc decreased with increasing the stirring time. For the L58/D67 specimen, a broad cold crystallization peak appeared at 93.7 °C, and two separated endothermic peaks were observed at 170.2 and 220.5 °C when the specimen was stirred for 14 h. Based on the WAXD result exhibited in the Figure S6, the signals at 220 °C was assigned to the melting of PLA SC, and the peak at 170 °C was assigned to the melting of HC. Stirring for longer times (50 and 100 h), a narrower cold crystallization peak appeared at ~ 77 °C, and only one endothermic peak at 215 °C was detected, which indicated that only SC formed. The crystallization of L134/D102 specimen was similar to that of variation of the L58/D67. In addition, both the SC and HC appeared in the L134/D102 specimen regardless of stirring time, and the Thc and ΔHhc also reduced with the increase in the stirring time. Most of the melt-quenched specimens exhibited similar regularity, i.e., the Thc, ΔHhc and Tsc, ΔHsc decreased as the specimens were stirred for longer times. The Tcc and ΔHcc decreased gradually in the L58/D67 and L134/D102 specimens stirred for longer times, which could be interpreted that both the HC and SC formed during heating in the specimens stirred for a short time, and the formation of HC and SC would compete with and impeded each other during crystallization. Thus, the cold crystallization appeared at a higher temperature. In the specimens stirred for longer time, mainly SC produced during heating, and it crystallized at relative lower temperature. The cold crystallization temperature increased as the molecular weight of PLLA and PDLA increased, which would be due to the reason that the mobility of chains and segments declined with enlarging molecular weights.

The heating DSC curves of the specimens after melt-quenching
The crystallization and melting behaviors of the quenched PLLA/PDLA specimens with different stirring times were similar to that of melt-crystallization specimens exhibited in Fig. 6. However, the depression of Tsc in the quenched specimens was 2–6 °C, and the decrement of ΔHsc was 0–16 J/g in all the specimens, which were lower than counterparts of melt-crystallization specimens. Further, more content of PLA HC developed in the quenched L58/D67 and L134/D102 specimens. These results implied that the content of crystallites produced from the melted specimens were lower than that of quenched specimens; this diversity should be due to the specimens experienced different annealing history. For the melting crystallization, the PLLA and PDLA molecular chain couples kept the melt state for longer time. Thus, more content of homogenous melt produced and less “memory effect” preserved, which resulted in lower crystallization capacity.
The crystallization from the melt after annealing at various times
For the L32/D31 specimen which stirred for 50 h, the specimen was melted at 255 °C at first, after annealing at this temperature for different times, the cooling and second heating DSC curves are displayed in Fig. 8. After melting at 255 °C for longer time, both the Tc and ΔHc declined rapidly during cooling, the Tcc increased, while the Tsc and ΔHsc depressed gradually in the second heating. For the L58/D67 specimen that annealed at the same history (Figure S7), it presented similar regularity to L32/D31. Figures 6, 7 and 8 indicated that, the more memory effect was preserved, and higher crystallization ability was received when the PLLA/PDLA specimens suffered from shorter melt history.

The cooling and second heating of the L32/D31 specimen stirred for 50 h
Conclusion
In this study, the effect of stirring time during solution mixing on the crystallization behaviors of PLLA/PDLA casted films was examined. Results revealed that the crystallization behaviors of PLLA/PDLA were strongly depended on the stirring time. For the original casting films, the content of SC increased, while both the melting temperature (Thc) and enthalpy (ΔHhc) of poly(lactide) homochiral crystallites (HC) depressed gradually as the specimens were stirred for longer time. During crystallization from the melt or melt-quenching, less content of SC and HC were produced, and the Thc and Tsc depressed in the specimens stirred for longer times. The crystallization capacity was suppressed after the specimens were stirred for longer times during solution mixing. Higher crystallization ability could be preserved when the PLLA/PDLA specimens suffer from shorter melting history. More crystallization capacity preserved with enlarging the molecular weights of PLLA/PDA specimens. This investigation could provide more bases for the solution crystallization, casting and spinning.
References
Jem K J, van der Pol J, de Vos S (2010) Microbial lactic acid, its polymer poly(lactic acid), and their industrial applications. In: Chen GG-Q (ed) Plastics from bacteria, vol 14, 1st edn. Springer, Berlin, pp 323–346. https://doi.org/10.1007/978-3-642-03287-5_13
Saeidlou S, Huneault MA, Li H, Park CB (2012) Poly(lactic acid) crystallization. Prog Polym Sci 37:1657–1677
Jun S, Yan-Long L, Sheng X, Xin-Chao B, Jing-Ru S, Gao L, Chen XS, Hou HQ (2015) The stereocomplex formation and phase separation of PLLA/PDLA blends with different optical purities and molecular weights. Chin J Polym Sci 33:1–8. https://doi.org/10.1007/s10118-015-1715-y
Ikada Y, Jamshidi K, Tsuji H, Hyon SH (1987) Stereocomplex formation between enantiomeric poly(lactides). Macromolecules 20:904–906
Tsuji H, Ikada Y (1996) Crystallization from the melt of poly(lactide)s with different optical purities and their blends. Macromol Chem Phys 197:3483–3499
Tsuji H, Ikada Y (1993) Stereocomplex formation between enantiomeric poly(lactic acid)s 9. Stereocomplexation from the melt. Macromolecules 26:6918–6926
Wang L, Feng C, Zhou D, Shao J, Hou H, Li G (2018) The crystallization and phase transition behaviors of asymmetric PLLA/PDLA blends: from the amorphous state. Polym Cryst 1:e10006
Shao J, Xiang S, Bian X, Sun J, Li G, Chen X (2015) Remarkable melting behavior of PLA stereocomplex in linear PLLA/PDLA blends. Ind Eng Chem Res 54:2246–2253
Hu X, Shao J, Zhou D, Li G, Ding J, Chen X (2017) Microstructure and melting behavior of a solution-cast polylactide stereocomplex: effect of annealing. J Appl Polym Sci. https://doi.org/10.1002/app.44626
Okihara T, Tsuji M, Kawaguchi A, Katayama KL, Tsuji H, Hyon SH, Ikada Y (1991) Crystal structure of stereocomplex of poly(L-lactide) and poly(D-lactide). J Macromol Sci Part B Phys 30:119–140. https://doi.org/10.1080/00222349108245788
Tsuji H, Ikada Y (1999) Stereocomplex formation between enantiomeric poly(lactic acid)s. XI. Mechanical properties and morphology of solution-cast films. Polymer 40:6699–6708
Hirata M, Kimura Y (2008) Thermo mechanical properties of stereoblock poly(lactic acid)s with different PLLA/PDLA block compositions. Polymer 49:2656–2661
Karst D, Yang Y (2008) Effect of arrangement of L-lactide and D-lactide in poly[(L-lactide)-co-(D-lactide)] on its resistance to hydrolysis studied by molecular modeling. Macromol Chem Phys 209:168–174
Shao J, Sun J, Bian X, Cui Y, Li G, Chen X (2012) Investigation of poly(lactide) stereocomplexes: 3-armed poly(L-lactide) blended with linear and 3-armed enantiomers. J Phys Chem B 116:9983–9991
Biela T, Duda A, Penczek S (2006) Enhanced melt stability of star-shaped stereocomplexes as compared with linear stereocomplexes. Macromolecules 39:3710–3713
Liu Y, Sun J, Bian X, Feng L, Xiang S, Sun B, Chen Z, Li G, Chen X (2013) Melt stereocomplexation from poly(l-lactic acid) and poly(d-lactic acid) with different optical purity. Polym Degrad Stab 98:844–852
Bao RY, Yang W, Jiang WR, Liu ZY, Xie BH, Yang MB, Fu Q (2012) Stereocomplex formation of high-molecular-weight polylactide: a low temperature approach. Polymer 53:5449–5454
Liu Z, Fu M, Ling F, Sui G, Bai H, Zhang Q, Fu Q (2019) Stereocomplex-type polylactide with bimodal melting temperature distribution: toward desirable melt-processability and thermomechanical performance. Polymer 169:21–28
Purnama P, Kim SH (2009) Stereocomplex formation of high molecular weight polylactide using supercritical fluid. Macromolecules 43:1137–1142
Bai D, Liu H, Bai H, Zhang Q, Fu Q (2017) Low-temperature sintering of stereocomplex-type polylactide nascent powder: effect of crystallinity. Macromolecules 50:7611–7619
Bai D, Liu H, Bai H, Zhang Q, Fu Q (2016) Powder metallurgy inspired low-temperature fabrication of high-performance stereocomplexed polylactide products with good optical transparency. Sci Rep 6:20260
Fu M, Liu Z, Bai D, Ling F, Bai H, Zhang Q, Fu Q (2018) Low-temperature sintering of stereocomplex-type polylactide nascent powder: from compression molding to injection molding. Macromol Mater Eng 303:1800178
Deng S, Bai H, Liu Z, Zhang Q, Fu Q (2019) Toward supertough and heat-resistant stereocomplex-type polylactide/elastomer blends with impressive melt stability via in situ formation of graft copolymer during one-pot reactive melt blending. Macromolecules 52:1718–1730
Song Y, Zhang X, Yin Y, Zhang C, de Vos S, Wang R, Joziasse CAP, Liu G, Wang D (2016) Crystallization of equimolar poly(l-lactide)/poly(d-lactide) blend below the melting point of α crystals under shear. Eur Polym J 75:93–103
Song Y, Zhang XQ, Yin YA, de Vos S, Wang RY, Joziasse CAP, Liu GM, Wang DJ (2015) Enhancement of stereocomplex formation in poly(L-lactide)/poly(D-lactide) mixture by shear. Polymer 72:185–192
Bao RY, Yang W, Wei XF, Xie BH, Yang MB (2014) Enhanced formation of stereocomplex crystallites of high molecular weight poly(l-lactide)/poly(d-lactide) blends from melt by using poly(ethylene glycol). ACS Sustain Chem Eng 2:2301–2309
Bao RY, Yang W, Liu ZY, Xie BH, Yang MB (2015) Polymorphism of a high-molecular-weight racemic poly(l-lactide)/poly(d-lactide) blend: effect of melt blending with poly(methyl methacrylate). RSC Adv 5:19058–19066
Samuel C, Cayuela J, Barakat I, Muller AJ, Raquez JM, Dubois P (2013) Stereocomplexation of polylactide enhanced by poly(methyl methacrylate): improved processability and thermomechanical properties of stereocomplexable polylactide-based materials. ACS Appl Mater Interfaces 5:11797–11807
Fukushima K, Furuhashi Y, Sogo K, Miura S, Kimura Y (2005) Stereoblock poly(lactic acid): synthesis via solid-state polycondensation of a stereocomplexed mixture of poly(L-lactic acid) and poly(D-lactic acid). Macromol Biosci 5:21–29
Cao ZQ, Sun XR, Bao RY, Yang W, Xie BH, Yang MB (2016) Role of carbon nanotube grafted poly(l-lactide)-block-poly(d-lactide) in the crystallization of poly(l-lactic acid)/poly(d-lactic acid) blends: suppressed homocrystallization and enhanced stereocomplex crystallization. Eur Polym J 83:42–52
Xiong ZJ, Zhang XQ, Wang R, de Vos S, Wang RY, Joziasse CAP, Wang DJ (2015) Favorable formation of stereocomplex crystals in poly(l-lactide)/poly(d-lactide) blends by selective nucleation. Polymer 76:98–104
Han L, Pan P, Shan G, Bao Y (2015) Stereocomplex crystallization of high-molecular-weight poly(l-lactic acid)/poly(d-lactic acid) racemic blends promoted by a selective nucleator. Polymer 63:144–153
He S, Bai H, Bai D, Ju Y, Zhang Q, Fu Q (2019) A promising strategy for fabricating high-performance stereocomplex-type polylactide products via carbon nanotubes-assisted low-temperature sintering. Polymer 162:50–57
Chen Y, Hua WQ, Zhang ZC, Xu JZ, Bian FG, Zhong GJ, Xu L, Li ZM (2019) An efficient, food contact accelerator for stereocomplexation of high-molecular-weight poly(l-lactide)/poly(d-lactide) blend under nonisothermal crystallization. Polymer 170:54–64
Shao J, Guo Y, Xiang S, Zhou D, Bian X, Sun J, Li G, Hou H (2016) The morphology and spherulite growth of PLA stereocomplex in linear and branched PLLA/PDLA blends: effects of molecular weight and structure. CrystEngComm 18:274–282
Shao J, Sun J, Bian X, Cui Y, Zhou Y, Li G, Chen X (2013) Modified PLA homochiral crystallites facilitated by the confinement of PLA stereocomplexes. Macromolecules 46:6963–6971
Shao J, Sun J, Bian X, Zhou Y, Li G, Chen X (2013) The formation and transition behaviors of the mesophase in poly(D-lactide)/poly(L-lactide) blends with low molecular weights. CrystEngComm 15:6469–6476
Tsuji H, Ikada Y, Hyon SH, Kimura Y, Kitao T (1994) Stereocomplex formation between enantiomeric poly(lactic acid). VIII. Complex fibers spun from mixed solution of poly(D-lactic acid) and poly(L-lactic acid). J Appl Polym Sci 51:337–344
Tsuji H (2005) Poly(lactide) stereocomplexes: formation, structure, properties, degradation, and applications. Macromol Biosci 5:569–597
Tsuji H, Nakano M, Hashimoto M, Takashima K, Katsura S, Mizuno A (2006) Electrospinning of poly(lactic acid) stereocomplex nanofibers. Biomacromol 7:3316–3320
Lv R, Tian R, Na B, Zhang P, Liu Q (2015) Strong confinement effects on homocrystallization by stereocomplex crystals in electrospun polylactide fibers. J Phys Chem B 119:15530–15535
Kyotani M (1982) Stirring-induced crystallization of nylon 6. J Macromol Sci B 21:275–285
Pennings AJ, Kiel AM (1965) Fractionation of polymers by crystallization from solution, III. On the morphology of fibrillar polyethylene crystals grown in solution. Kolloid Z Z Polym 205:160–162
Pennings AJ (1967) Fractionation of polymers by crystallization from solutions. II. J Polym Sci Polym Symp 16:1799–1812
Keller A, Willmouth FM (1972) Lattice orientation within stirring-induced crystals of polyethylene. J Macromol Sci B 6:539–544
Tsuji H, Hyon SH, Ikada Y (1991) Stereocomplex formation between enantiomeric poly(lactic acid)s. 4. Differential scanning calorimetric studies on precipitates from mixed solutions of poly(D-lactic acid) and poly(L-lactic acid). Macromolecules 24:5657–5662
Zhang JM, Tashiro K, Tsuji H, Domb AJ (2007) Investigation of phase transitional behavior of poly(L-lactide)/poly(D-lactide) blend used to prepare the highly-oriented stereocomplex. Macromolecules 40:1049–1054
Brzeziński M, Bogusławska M, Ilčíková M, Mosnáček J, Biela T (2012) Unusual thermal properties of polylactides and polylactide stereocomplexes containing polylactide-functionalized multi-walled carbon nanotubes. Macromolecules 45:8714–8721
Sun JR, Yu HY, Zhuang XL, Chen XS, Jing XB (2011) Crystallization behavior of asymmetric PLLA/PDLA blends. J Phys Chem B 115:2864–2869
Bouapao L, Tsuji H (2009) Stereocomplex crystallization and spherulite growth of low molecular weight poly(L-lactide) and poly(D-lactide) from the melt. Macromol Chem Phys 210:993–1002
Tsuji H, Bouapao L (2012) Stereocomplex formation between poly(L-lactic acid) and poly(D-lactic acid) with disproportionately low and high molecular weights from the melt. Polym Int 61:442–450
Tsuji H, Horii F, Hyon SH, Ikada Y (1991) Stereocomplex formation between enantiomeric poly(lactic acid)s. 2. Stereocomplex formation in concentrated solutions. Macromolecules 24:2719–2724
Tsuji H, Hyon SH, Ikada Y (1991) Stereocomplex formation between enantiomeric poly(lactic acid)s. 3. Calorimetric studies on blend films cast from dilute solution. Macromolecules 24:5651–5656
Saeidlou S, Huneault MA, Li HB, Sammut P, Park CB (2012) Evidence of a dual network/spherulitic crystalline morphology in PLA stereocomplexes. Polymer 53:5816–5824
Wei XF, Bao RY, Cao ZQ, Yang W, Xie BH, Yang MB (2014) Stereocomplex crystallite network in asymmetric PLLA/PDLA blends: formation, structure, and confining effect on the crystallization rate of homocrystallites. Macromolecules 47:1439–1448
He Y, Xu Y, Wei J, Fan ZY, Li SM (2008) Unique crystallization behavior of poly(L-lactide)/poly(D-lactide) stereocomplex depending on initial melt states. Polymer 49:5670–5675
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grants Nos. 51403089 and 21574060), the Major Special Projects of Jiangxi Provincial Department of Science and Technology (Grant No. 20114ABF05100), the Project of Jiangxi Provincial Department of Education (GJJ170229), the Technology Plan Landing Project of Jiangxi Provincial Department of Education (GCJ2011-243), the Outstanding Youth Foundation of Jiangxi Normal University, the China Postdoctoral Science Foundation (2019M652282), and Postdoctoral Science Foundation of Jiangxi Province (2018KY37).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Shao, J., Xu, L., Pu, S. et al. The crystallization behavior of poly(l-lactide)/poly(d-lactide) blends: effect of stirring time during solution mixing. Polym. Bull. 78, 147–163 (2021). https://doi.org/10.1007/s00289-020-03103-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00289-020-03103-9