
Abstract
Pendant carboxyl groups were introduced into donor–acceptor polyfluorene carrying benzothiadiazole as an acceptor moiety. The emission color from its solution was different depending on the solvent. While the methanol solution of the polymer exhibited blue emission, yellow emission was observed from its tetrahydrofuran solution. Influence of solvent polarity, solution concentration, temperature, and addition of acid on photoluminescence properties indicated that the observed change in emission color was due to polymer aggregation coming from inter and/or intramolecular interaction between pendant carboxyl groups. Polyfluorene carrying benzothiadiazole moiety with pendant amino groups also exhibited emission color change and was useful as a fluorimetric sensor for polyacid.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Donor–acceptor (D–A) conjugated polymers (CPs) are currently of interest due to the built-in intramolecular charge transfer which can facilitate ready manipulation of the electronic structure (HOMO/LUMO levels). A number of D–A CPs have been prepared as functional materials, such as printable field-effect transistors [1,2,3], low-cost photovoltaic cells [4,5,6,7,8,9], and near-IR emitting OLED [10]. They also have attracted particular interest as environmentally responsive photoluminescence materials because it is possible to change their emission properties by tuning D–A interaction. For example, Altınok et al. [11] prepared two-dimensional, D–A poly(phenylene ethynylene)s with pendant tetracene moiety as an acceptor unit to observe photoluminescence response upon exposure to oxygen. Nakamura et al. [12] reported fluorescence solvatochromism of polymers containing fluorene and maleimide units in the main chain. Yang et al. [13] prepared D–A polyfluorene carrying naphthoselenadiazole (NSeD) and found that this polyfluorene exhibits different emission spectra in solution and solid film. In solution, the copolymer exhibits two emission peaks at 450 nm and 700 nm, which are corresponding to fluorene (donor) and NSeD (acceptor) segments, respectively. On the other hand, the emission at 450 nm decreased remarkably in the solid state due to the interchain interaction.
We are interested in fluorene-based D–A polymers which exhibit photoluminescence response. Wang and Bazan [14] reported pH-responsive aggregation-mediated optical properties of conjugated polyelectrolyte copolymers with carboxyl functionalities, indicating that the fluorescence property in solution can be tuned if we successfully control the aggregation of D–A polyfluorene molecules. Our idea is to introduce pendant carboxyl functionalities onto D–A polyfluorene with benzothiadiazole (BT). The carboxyl functionalities are expected to induce controlled polymer aggregation arising from reversible interaction between carboxyl groups. Further, existence of carboxyl groups makes it possible to solubilize the resulting D–A polyfluorene in a polar solvent such as methanol which is usually non-solvent for polyfluorene. Figure 1 shows chemical structures of polyfluorenes studied in this work. We report here preparation of D–A polyfluorenes carrying pendant carboxyl or amino functionalities and their photoluminescence properties.

Chemical structures of D–A polyfluorenes studied in this work
Experimental
Materials
4,7-Dibromo-2,1,3-benzothiadiazole (1) [15] and 2,7-dibromo-9,9-bis(6ʹ-butoxycarbonylaminohexyl)fluorene (2) [16] were prepared according to the reported methods. 2,7-Dibromofluorene (3), 9,9-dihexylfluorene-2,7-bis(trimethyleneborate) (4) and trioctylmethylammonium chloride (aliquat® 336) were purchased from Aldrich. Tris{tris[3,5-bis(trifluoromethyl)phenyl] phosphine} palladium was purchased from Wako Pure Chemical Industries, Ltd. All other reagents were purchased from commercial sources and used as received.
9,9-Bis(5-ethoxycarbonylpentyl)-2,7-dibromofluorene (5)
The mixture of 3 (2.6 g, 8.0 mmol) and potassium tert-butoxide (3.4 g, 30 mmol) in 50 mL of THF was stirred at room temperature for 1 h. Then, ethyl 6-bromohexanoate (5.4 g, 24 mmol) was added and the mixture was refluxed for 16 h. The reaction mixture was poured into water and extracted with dichloromethane. The organic layer was dried over anhydrous magnesium sulfate and placed under reduced pressure to remove the solvents. The residue was purified by silica gel column chromatography eluting with a mixture of ethyl acetate and hexane (1:9 by v/v) to obtain 5 as a pale yellow powder (2.9 g, 59%). mp 85 °C; 1H NMR (500 MHz, CDCl3, δ): 7.58 (t, J = 6.8 Hz, 2H, Ar–H), 7.48 (m, 4H, Ar–H), 4.06 (q, J = 7.0 Hz, 4H, CH2), 2.11 (t, J = 7.5 Hz, 4H, CH2), 1.93 (t, J = 3.8 Hz, 4H, CH2), 1.38 (m, 4H, CH2), 1.20 (t, J = 7.0 Hz, 6H, CH3), 1.10 (m, 4H, CH2), 0.59 (m, 4H, CH2); 13C NMR (125 MHz, CDCl3, δ): 173.6, 152.1, 139.2, 130.3, 126.0, 121.5, 121.1, 60.1, 55.5, 40.0, 34.1, 29.3, 24.5, 23.3, 14.2; IR (KBr) 1724 (C=O); Anal. calcd for C29H36Br2O4: C, 57.25; H, 5.96. found: C, 57.46; H, 5.85.
Polyfluorene carrying benzothiadiazole with pendant ethoxycarbonyl groups (PFBT-CO2Et)
To a solution of 4 (452 mg, 0.90 mmol), 5 (274 mg, 0.45 mmol), and 1 (132 mg, 0.45 mmol) in 30 mL of toluene was added cesium fluoride (273 mg, 1.8 mmol) and tris{tris[3,5-bis(trifluoromethyl)phenyl] phosphine} palladium (36 mg). The reaction mixture was refluxed under a nitrogen atmosphere for 3 days and poured into methanol to precipitate the polymer. The crude polymer was washed with methanol using Soxhlet apparatus for 24 h to remove oligomers and catalyst residue to obtain PFBT-CO2Et as a yellow solid. Yield: 160 mg (31%). GPC: Mw = 6820, PDI = 2.8.
Polyfluorene carrying benzothiadiazole with pendant carboxyl groups (PFBT-CO2H)
The mixture of PFBT-CO2Et (60 mg), THF (4 mL), and 1.5 M KOH solution (8 mL) was refluxed for 40 h. After cooling, 1 M HCl solution was added until the pH = 4. The precipitate was recovered by centrifugation and washed with water to obtain PFBT-CO2H as a yellow solid. Yield 50 mg (83%).
Polyfluorene carrying benzothiadiazole with pendant butoxycarbonylamino groups (PFBT-NHBoc)
To a mixture of 4 (402 mg, 0.80 mmol), 2 (289 mg, 0.40 mmol), 1 (118 mg, 0.45 mmol), tris{tris[3,5-bis(trifluoromethyl)phenyl] phosphine} palladium (36 mg), and aliquat 336 (3 drops) in 30 mL of toluene was added aqueous sodium carbonate (2 M, 3 mL). The reaction mixture was refluxed under a nitrogen atmosphere for 2 days and poured into methanol to precipitate the polymer. The crude polymer was washed with acetone using Soxhlet apparatus for 24 h to remove oligomers and catalyst residue to obtain PFBT-NHBoc as a yellow solid. Yield: 330 mg (60%). GPC: Mw = 5270, PDI = 1.7.
Polyfluorene carrying benzothiadiazole with pendant amino groups (PFBT-NH2)
To a solution of PFBT-NHBoc (60 mg) in 30 mL of 1,4-dioxane was added 9 mL of 37% hydrochloric acid. The reaction mixture was stirred for 2 days at room temperature. After removal of the solvent under reduced pressure, 50 mL of acetone was added to obtain a precipitate. To the precipitate were added THF (6 mL) and 50%NaOH (4 mL) and the reaction mixture was stirred at room temperature for 1 h. The separated organic layer was washed with water and dried over anhydrous MgSO4. The organic layer was placed under reduced pressure to remove the solvent to obtain PFBT-NH2 as a yellow solid. Yield: 55 mg (90%).
Measurements
1H and 13C NMR spectra were recorded at room temperature on a JEOL A-500 nuclear magnetic resonance spectrometer. Samples were dissolved in CDCl3, and tetramethylsilane (TMS) was used as the internal standard. Gel permeation chromatography (GPC) was carried out on a Tosoh HLC-8020 chromatograph equipped with a set of polystyrene gel columns (Tosoh TSK gel G2500H + G3000H) and refractive/ultraviolet dual-mode detectors. Tetrahydrofuran (THF) was used as the eluent at a flow rate of 1.0 mL/min. The calibration curves for GPC analysis were obtained using polystyrene standards. Photoluminescence spectra were recorded on a HAMAMATSU Multi Channel Analyzer PMA-11 with exciting wavelength of 365 nm. Infrared and UV–Vis spectra were recorded on a JASCO FT/IR-4100 and SHIMADZU UV-2550, respectively.
Results and discussion
Preparation of PFBT-CO2H
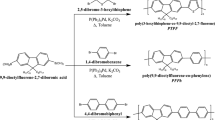
In order to introduce pendant carboxyl functionalities onto D–A polyfluorene, we prepared 9,9-bis(5-ethoxycarbonylpentyl)-2,7-dibromofluorene (5) that carries carboxyl functionality masked as the ethyl ester as a comonomer (Fig. 2).

Preparation of 5
Figure 3 shows synthetic pathway for PFBT-CO2H. Since dibromide 5 carries ester functionalities which are subject to hydrolysis under conventional Suzuki coupling polymerization using basic reagents, we employed base-free Suzuki coupling polymerization developed by Brookins et al. [17]. The obtained ester-functionalized polyfluorene (PFBT-CO2Et) was finally converted to its diacid form (PFBT-CO2H) by alkaline hydrolysis followed by acidifying.

Preparation of PFBT-CO2H
The 1H NMR spectrum of PFBT-CO2Et is shown in Fig. 4a. The peaks at 4.0 and 1.3 ppm were ascribed to CH2 and CH3 of ethoxy groups, respectively. The composition of the resulting copolymer was determined by 1H NMR through the peak area ratio between the signals coming from the CH3 protons of hexyl groups (at 0.8 ppm) and the ones belonging to the OCH3 protons (at 1.4 ppm) to be m:n = 19:81. The structural composition of the copolymer matched that expected on the basis of the feed ratio (m:n = 1:4). The hydrolysis reaction of PFBT-CO2Et was carried out in basic conditions. The complete conversion of ethoxycarbonyl to carboxyl functionalities was confirmed by disappearance of protons due to OC2H5 groups (Fig. 4b).

1H NMR spectra of a PFBT-CO2Et and b PFBT-CO2H in CDCl3
Table 1 summarizes solubilities of PFBT-CO2Et and PFBT-CO2H. It is noteworthy that PFBT-CO2H became soluble in both THF and methanol which is a non-solvent for PFBT-CO2Et. The observed enhancement of solubility in polar solvent is due to the incorporation of polar carboxyl groups in PFBT-CO2H.
Optical properties of PFBT-CO2H
Figure 5 shows photoluminescence spectra of PFBT-CO2H in methanol (dotted line), THF (solid line), and acetonitrile (dashed line) at the polymer concentration of 0.2 mg/mL. The spectrum from methanol solution exhibited emission peaks around 440 nm. On the other hand, a strong emission at 580 nm emerged from THF and acetonitrile solutions instead of the emission around 440 nm. Therefore, the emission colors were quite different to our naked eye. While the emission from methanol solution was blue, THF and acetonitrile solutions gave yellow emission. Although both methanol and acetonitrile are polar solvents, the emissions from these solvents were quite different, suggesting that hydrogen bonding played an important role in determining emission properties. Since PFBT-CO2H exhibited a bimodal emission, the band at 440 is assigned to a unimer emission and the band at 580 is an excimer emission. The emission at longer wavelength is indicative of inter and/or intramolecular aggregation through π–π stacking interactions.

Photoluminescence spectra of PFBT-CO2H in methanol (a, dotted line), THF (b, solid line), and acetonitrile (c, dashed line) (λex = 365 nm) at the concentration of 0.2 mg/mL
Figure 6 shows UV–Vis spectra of PFBT-CO2H in methanol and THF. The observed wavelengths of maximum absorbance in methanol and THF were 473 and 435 nm, respectively. A blue shift in absorption was observed in THF solution. The optical bandgaps were determined from the lowest energy onset in methanol and THF solutions to be 1.86 and 2.42 eV, respectively. Probably, the decreased bandgap in THF solution is because of distortion of π-system arising from interactions among carboxyl groups.

UV–Vis spectra of PFBT-CO2H in methanol (a) and THF (b)
Figure 7 shows the effect of polymer concentration of PFBT-CO2H in methanol on the emission properties. In dilution conditions, where aggregation is negligible, the emission of PFBT-CO2H was mainly in the blue region (a). On the other hand, the emission at longer wavelength was more pronounced in increased polymer concentration (b and c). This shift can be ascribed to aggregation due to increased interchain contacts.

Photoluminescence spectra of PFBT-CO2H in methanol at 0.2 mg/mL (a, dotted line), 0.4 mg/mL (b, solid line), and 1.0 mg/mL (c, dashed line) (λex = 365 nm) at the temperature of 20 °C
Therefore, the observed change in photoluminescence spectra in Fig. 6 can be explained by considering polymer aggregation arising from inter and/or intramolecular interaction between pendant carboxyl groups. In THF and acetonitrile, the PFBT-CO2H chains aggregated due to the hydrogen-bonding interaction between carboxyl groups as shown in Fig. 8. On the other hand, the PFBT-CO2H chain assumes a fully extended conformation in methanol because hydrogen bonds can be easily disrupted in methanol.

Schematic illustration of PFBT-CO2H chains through hydrogen bonding between pendant carboxyl groups
Another evidence of polymer aggregation due to hydrogen bonding is fluorescence change of PFBT-CO2H in THF solution at two different temperatures (Fig. 9). As the temperature increased from 20 to 60 °C, the emission at 420 nm emerged, indicating that inter and/or intramolecular interaction was weakened by the partial cleavage of hydrogen bonding at higher temperature.

Photoluminescence spectra of PFBT-CO2H in THF at 20 °C (a) and 60 °C (b) (λex = 365 nm) at the concentration of 0.2 mg/mL
Effect of non-solvent addition into PFBT-CO2H in methanol solution was examined (Fig. 10). Addition of water resulted in decrease in blue emission and increase in yellow emission. This emission shift is reasonably explained by considering the polymer aggregation.

Changes in fluorescence spectrum of PFBT-CO2H (0.5 mg of PFBT-CO2H in 2 mL of methanol) upon addition of water (a, 0 mL; b, 1 mL; c, 2 mL) (λex = 365 nm) at the temperature of 20 °C
Finally, we examined the effect of hydrochloric acid. Since hydrogen bonding can be cleaved by strong acid such as hydrochloric acid, the photoluminescence spectrum of PFBT-CO2H in THF solution was measured at different concentrations of hydrochloric acid. The results are shown in Fig. 11. As expected, increase in the acid concentration led to a decrease in emission at 580 nm which comes from aggregated PFBT-CO2H. However, the change in emission spectrum was not remarkable. Since hydrochloric acid is a non-solvent for PFBT-CO2H, the effect of cleaving hydrogen bonds was compensated for polymer aggregation due to addition of non-solvent.

Changes in fluorescence spectrum of PFBT-CO2H (0.5 mg of PFBT-CO2H in 2 mL of THF) upon addition of hydrochloric acid (a, 0 mL; b, 1 mL; c, 1.5 mL) (λex = 365 nm) at the temperature of 20 °C
Preparation of PFBT-NH2
D–A polyfluorene carrying BT moiety with pendant amino groups (PFBT-NH2) was then examined as a material for photoluminescence sensor for polyacid, because polyacid can induce polymer aggregation due to formation of polymer complex through electrostatic interaction between amino and acidic functionalities.
Polyfluorene carrying BT moiety with pendant amino groups was prepared as shown in Fig. 12. Suzuki coupling terpolymerization between 1, 2, and 4 was carried out to obtain PFBT-NHBoc which was converted to PFBT-NH2 after the deprotection of Boc-group under acidic conditions.

Preparation of PFBT-NH2
Figure 13 shows 1H NMR spectra of PFBT-NHBoc and PFBT-NH2. The peaks at 4.4 and 1.4 ppm were ascribed to amide NH protons and methyl protons of Boc groups, respectively. After deprotection reaction, these two peaks disappeared completely.

1H NMR spectra of a PFBT-NHBoc and b PFBT-NH2 in CDCl3
Optical properties of PFBT-NH2
Figure 14 shows fluorescence spectra of PFBT-NH2 in methanol and THF. The emission from its methanol solution gave blue emission. On the other hand, the emission color from its THF solution was yellow. This behavior is almost same as PFBT-CO2H. Therefore, the observed change in photoluminescence spectrum of PFBT-NH2 can be explained by aggregation of amino groups because primary amines are known to form hydrogen bonds with each other.

Photoluminescence spectra of PFBT-NH2 in methanol (a) and THF (b) (λex = 365 nm) at the concentration of 0.2 mg/mL
In order to obtain an evidence of polymer–polymer interaction which accounts for the yellow emission at 580 nm observed in THF solution, photoluminescence spectrum was examined at different polymer concentration in methanol solution (Fig. 15). Apparently, the emission at 580 nm increased with increase in PFBT-NH2 concentration, indicating that the emission at 580 nm was coming from interaction between emitting polymers.

Photoluminescence spectra of PFBT-NH2 in methanol at 0.3 mg/mL (a, blue line), 0.7 mg/mL (b, green line), and 1.7 mg/mL (c, red line) (λex = 365 nm) at the temperature of 20 °C
Fluorometric sensor for polyacid
Fluorescent-based sensors appear as one of promising methodology for chemical sensing with high sensitivity and easy operation [18]. Since polyacid can induce aggregation of polyamine through acid–base interaction, PFBT-NH2 is expected to act as a sensor for polyacid. We added polyvinylsulfonic acid (PVSA) as a target molecule to the solution of PFBT-NH2 in methanol. Figure 16 shows fluorescence spectra of PFBT-NH2 in methanol containing different amount of PVSA. The emission at shorter wavelength around 410 nm decreased with increase in PVSA. On the other hand, the emission at longer wavelength around 550 nm increased with increase in PVSA. To our naked eye, the emission color changed from blue to yellow upon addition of PVSA. This change in emission color is thought to be coming from aggregation of PFBT-NH2, indicating that PFBT-NH2 can be useful as fluorometric sensor for polyacid.

Changes in photoluminescence spectrum of PFBT-NH2 (1 mg of PFBT-NH2 in 3 mL of methanol) at different amount of PVSA (a, 0 mg; b, 5 mg; c, 10 mg) (λex = 365 nm) at the temperature of 20 °C
Conclusions
We designed and synthesized novel D–A type polyfluorenes with pendant carboxyl or amino groups which are soluble in both THF and methanol. The emission color of the polyfluorene with BT acceptor carrying pendant carboxyl groups changed depending on solvent polarity, acidity, temperature, and polymer concentration. Aggregation of the polymer chain due to the hydrogen bonding between pendant carboxyl functionalities is considered to be the reason for the observed photoluminescence properties. The polyfluorenes with BT acceptor carrying pendant amino groups also exhibited emission color change depending on the solvent. The shift in emission color of PFBT-NH2 was useful to detect polyacid due to the polyacid-induced aggregation. These polyfluorenes are expected to be new candidates for sensors and molecular imaging materials.
References
Arias AC, MacKenzie JD, McCulloch I, Rivnay J, Salleo A (2010) Materials and applications for large area electronics: solution-based approaches. Chem Rev 110:3–24
Coropceanu V, Cornil J, da Silva Filho DA, Olivier Y, Silbey R, Brédas J-L (2007) Charge transport in organic semiconductors. Chem Rev 107:926–952
Zaumseil J, Sirringhaus H (2007) Electron and ambipolar transport in organic field-effect transistors. Chem Rev 107:1296–1323
Cheng Y-J, Yang S-H, Hsu C-S (2009) Synthesis of conjugated polymers for organic solar cell applications. Chem Rev 109:5868–5923
Günes S, Neugebauer H, Sariciftci NS (2007) Conjugated polymer-based organic solar cells. Chem Rev 107:1324–1338
Clarke TM, Durrant JR (2010) Charge photogeneration in organic solar cells. Chem Rev 110:6736–6767
Lee JF, Hsu SLC, Lee PI, Chuang HY, Yang ML, Chen JS, Chou WY (2010) A new intramolecular donor–acceptor polyfluorene copolymer for bulk heterojunction solar cells. Sol Energy Mater Sol Cells 94:1166–1172
Smith KA, Lin Y-H, Mok JW, Yager KG, Strzalka J, Nie W, Mohite AD, Verduzco R (2015) Molecular origin of photovoltaic performance in donor-block-acceptor all-conjugated block copolymers. Macromolecules 48:8346–8353
Cimrová V, Morávková Z, Pokorná V, Výprachický D (2017) Investigation of donor–acceptor copolymer films and their blends with fullerene in the active layers of bulk heterojunction solar cells by Raman microspectroscopy. Org Electron 47:194–199
Crossley DL, Urbano L, Neumann R, Brourke S, Jones J, Dailey LA, Green M, Humphries MJ, King SM, Turner ML (2017) Post-polymerization C–H borylation of donor–acceptor materials gives highly efficient solid state near-infrared emitters for near-IR-OLEDs and effective biological imaging. ACS Appl Mater Interfaces 9:28243–28249
Altınok E, Smith ZC, Thomas SW (2015) Two-dimensional, acene-containing conjugated polymers that show ratiometric fluorescent response to singlet oxygen. Macromolecules 48:6825–6831
Nakamura M, Yamabuki K, Oishi T, Onimura K (2013) Synthesis and fluorescent properties of conjugated copolymers containing maleimide and fluorene units at the main chain. J Polym Sci Part A Polym Chem 51:4945–4956
Yang J, Jiang C, Zhang Y, Yang R, Yang W, Hou Q, Cao Y (2004) High-efficiency saturated red emitting polymers derived from fluorene and naphthoselenadiazole. Macromolecules 37:1211–1218
Wang F, Bazan GC (2006) Aggregation-mediated optical properties of pH-responsive anionic conjugated polyelectrolytes. J Am Chem Soc 128:15786–15792
Yang R, Tian R, Yan J, Zhang Y, Yang J, Hou Q, Yang W, Zhang C, Cao Y (2005) Deep-red electroluminescent polymers: synthesis and characterization of new low-band-gap conjugated copolymers for light-emitting diodes and photovoltaic devices. Macromolecules 38:244–253
Guo Z-S, Pei J, Zhou Z-L, Zhao L, Gibson G, Lam S, Brug J (2009) Amine groups-functionalized alcohol-soluble polyfluorene derivatives: synthesis, photophysical properties, and self-assembly behaviors. Polymer 50:4794–4800
Brookins RN, Schanze KS, Reynolds JR (2007) Base-free Suzuki polymerization for the synthesis of polyfluorenes functionalized with carboxylic acids. Macromolecules 40:3524–3526
Hong JW, Hemme WL, Keller GE, Rinke MT, Bazan GC (2006) Conjugated-polymer/DNA interpolyelectrolyte complexes for accurate DNA concentration determination. Adv Mater 18:878–882
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ando, D., Ijichi, J., Uno, T. et al. Preparation of donor–acceptor polyfluorenes with pendant carboxyl or amine functionalities and their photoluminescence properties. Polym. Bull. 76, 6137–6151 (2019). https://doi.org/10.1007/s00289-019-02701-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00289-019-02701-6