
Abstract
In this research, boron nitride (BN) acting as both nucleating agent and thermally conductive filler was melt-mixed with poly(3-hydroxybutyrate) (PHB). It was assumed that the introduction of BN not only formed thermally conductive pathways to increase the thermal conductivity of PHB, but also reduced the interfacial thermal resistance due to the interaction between BN and PHB. The assumption was confirmed by density test, morphological observation, and rheological tests. Besides, the introduction of BN improved the thermal stability of PHB, as well. Poly(3-hydroxybutyrate-co-4-hydroxybutyrate) (P3,4HB) was introduced as a comparison with PHB to illustrate the effect of crystallinity on the thermal conductivity of PHB/BN composites. According to the DSC tests, BN was proved to be a good nucleating agent for both PHB and P3,4HB. As the wide-angle X-ray diffraction analysis results showed that the crystal structure of PHB/BN and P3,4HB/BN composites was the same, the reason that the thermal conductivity of PHB/BN composites was higher than that of P3,4HB/BN composites at all BN levels was mainly because of the difference of the degree of crystallinity.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Advance in electronic technologies results in the miniaturization of transistors [1], which allow more transistors to integrate into one single device. Besides, integration of transistors produced increasing heat in a smaller volume of space. It is well known that the working life of a device is closely related to the operating temperature [2]. Therefore, quick and effective dissipation of the heat generated in the high packing and power-density devices has become a critical issue to maintain the performance and stability of electronic devices and demand for the materials of the product to that possess good thermal conductivity [3, 4]. Meanwhile, the materials must possess low electrical conductivity [5]. In comparison with other materials, excellent electrical insulated properties and easily processing have made thermoset and thermoplastic polymers the basis of rather promising molding compounds for practical application. Unfortunately, because of the presence of defects such as polymer chain ends, entanglements, random orientation, and impurities, the thermal conductivity of typical polymers is usually between 0.1 and 0.4 W mK−1 [6], which is far below the requirement for effectively dissipating the heat in the devices.
To enhance the thermal transfer efficiency, a convenient method is to incorporate large amount of thermally conductive fillers such as AlN [7, 8], Al2O [9], SiC [10], and BN [11, 12] into the polymer matrix to form thermally conductive pathways. These thermally conductive fillers have attracted such attention primarily due to their fabulous high thermal conductivities and low electrical conductivities [13]. BN seems to be the most promising candidate among these thermally conductive fillers because of its low density (2.2 g cm−3), high mechanical strength (Young’s modulus 0.7–0.9 TPa), high thermal conductivity (320 W mK−1), and superior chemical stabilities [14]. A typical BN particle is inorganic with a hexagonal structure and with an atomically smooth surface, such that it is nearly free of dangling bonds and charge traps [15]. Usually, a BN crystal possesses high thermal conductivity in plane, but poor thermal transfer along the vertical direction [16]. Gu et al. prepared epoxy resin composites filled with BN and the thermal conductivity of up to 1.09 W mK−1 at 60 wt% BN was obtained [17]. Song et al. prepared PVA/BN films with a thermal conductivity as high as 30 W mK−1 using 50 vol% exfoliated BN [18]. Composites with extremely high thermal conductivity (32.5 W mK−1) filled with BN (the size of BN is about 225 μm) were achieved by Ishida at a filler loading of 78.5 vol% [19]. These results indicate that the incorporation of BN as thermally conductive filler is an efficient way to enhance the thermal conductivity of the composites.
At present, most of the polymers used for the thermally conductive composites are non-degradable, such as polyurethane [20], polyimide [21], polypropylene [22], or epoxy resin [23]. Problems like how to dispose them after the usage have aroused increasing attention. As the awareness of protecting the environment grows, eco-friendly and biodegradable plastics can be good substitutes and replace part of the conventional industrial polymers, used in many applications. PHB is one of the polyhydroxyalkanoate groups, produced in larger quantities by fermentation of agricultural waste like sugarcane, using micro-organisms as a source of energy [24]. In addition, PHB is a completely isotactic stereoregular polyester with a high tendency to crystallize [25]. Therefore, in this study, PHB is chosen as the matrix. Although there are many studies on thermally conductive polymer, most of the studies are based on amorphous polymers. The effect of crystallinity on the thermal conductivity of crystalline polymer composite is seldom investigated. In this research, BN is used as a good nucleating agent for PHB, meanwhile as one kind of desired thermally conductive filler. It is the aim to study the factors like crystallinity that influence the thermal conductivity of the composite. Furthermore, thermal behavior, crystalline structure, rheological properties, thermal stability, as well as the morphology of the composite are also investigated.
Materials and methods
PHB and P3,4HB (with 6.5 mol% 4-hydroxybutyrate (4HB) unit) were applied as the matrix and obtained from TianAn Biologic Materials Co. Ltd (Zhejiang, China). The density of PHB is 1.25 g cm−3. The BN powders (with diameter of 0.3–0.5 μm, purity of 99.5%, and density of 2.25 g cm−3) were purchased from Liaoning Pengda Technology Co., Ltd (Liaoning, China). They were used as received without any surface treatment.
PHB, P3,4HB, and BN powders were firstly dried in a vacuum at 80 °C for 12 h before melt mixing. Then, PHB, P3,4HB, and BN powders with different mass ratios were melt-mixed in a HAAK Rheomix 600 (Karlsruhe, Germany) at 180 °C for 5 min at a speed of 60 rpm. The PHB/BN composites were prepared with different BN loading ranging between 0 and 50% by weight. When the content of BN is over 50 wt%, the viscosity of the PHB/BN composites will increase significantly which is inconvenient for melt processing. Besides, higher content of BN will cost longer time to disperse them uniformly; the longer melting time will cause PHB to degrade. Consequently, 50 wt% BN is chosen as the maximum ratio in this experiment. PHB/BN composites and P3,4HB/BN composites were then compressed in a stainless steel mold at 180 °C with pressure of 10 MPa for 3 min. After that, both composites were quenched to room temperature to obtain samples.
Thermal diffusivity of the composites was determined at 25 °C by the laser flash method (ASTM E1461) using a Netzsch laser flash thermal diffusivity apparatus (LFA 427) on a cylindrical sample (disks of 12.7 mm diameter and 2 mm thickness, the entire surface was coated with a carbon layer by a carbon sprayer). At least three specimens were tested for each sample to get an average value. Thermal conductivity was then calculated from the multiplication of thermal diffusivity (mm2 s−1), density (g cm−3), and specific heat (Jg−1 k−1). The density was calculated by dividing mass by volume and the specific heat was calculated by DSC.
In SEM analysis, the specimens were first immersed in liquid nitrogen for about 5 min and then cryo-fractured. The fractured surfaces were coated with a thin layer of gold prior to the examination in a scanning electron microscope (model Japan JXA-840 ESEMFE).
Rheological behavior of PHB/BN composites was performed on a Physica MCR2000 rheometer (AR 2000ex USA) at 180 °C. Testing samples were about 1 mm in thickness. The gap distance between the parallel plates was set at 0.9 mm for all the tests. Frequency sweep for the all samples was carried out under nitrogen using 25 mm plate–plate geometry. A strain sweep test was initially carried out to determine the linear viscoelastic region of the materials at 180 °C, 1.25% strain was enough to make sure that the melt flow of the samples was at the linear viscoelastic zone. The angular frequency range used during the test was 0.1–100 rad s−1.
Thermogravimetric analysis (TGA) was performed with a Netzsch STA 409 PC simultaneous thermal analysis instrument. All samples with weight of 10 ± 0.2 mg were heated from room temperature to 500 °C at 10 °C min−1 under nitrogen.
WAXD experiments were performed on a D8 advance X-ray diffractometer (Bruker, Germany) in the range of 10°–40° at a scanning rate of 4° min−1. The Cu Kα radiation (λ = 0.15418 nm) source was operated at 40 kV and 200 mA.
Thermal behavior of PHB/BN composites was performed in a differential scanning calorimeter DSC (TA instrument, Q20, USA). Indium was used for temperature and enthalpy calibration. The specimens varied between 5 and 8 mg were cut from the compressed molded samples, and then placed in a sealed aluminum pan. All the measurements were performed in nitrogen atmosphere. The specimens were heated from room temperature to 190 °C at a heating rate of 10 °C min−1, held for 2 min to eliminate the thermal history, then cooled to 0 °C at a cooling rate of 10 °C min−1. The degree of crystallinity, χ c, was calculated from the areas of the corresponding DSC melting peaks according to:
where χ PHB is the weight fraction of PHB in the composite, ΔH 0 is the heat of fusion of 100% crystalline PHB (146 J g−1), and ΔH m is the peak area of the melting range of consideration.
Results and discussion
Thermal conductivity of PHB/BN composites
Thermal conductivity of PHB/BN composites is presented in Fig. 1. It can be seen that the thermal conductivity of PHB/BN composites increases gradually with increasing filler loading. The thermal conductivity of neat PHB is about 0.26 W mK−1 at room temperature. The highest thermal conductivity of 1.37 W mK−1 was obtained when the content of BN was 50 wt%, about five times higher than neat PHB. The heat transport between the BN particles takes place via lattice vibration waves (phonons) [26]. It has been known that the polymer composites having high thermal conductivity can be obtained by maximizing the formation of conductive networks [27]. At low filler fraction, the thermally conductive paths may not steadily form completely thermally conductive pathways. This is because the polymer between the BN gaps will cause phonon scattering at the interface known as Kapitza resistance, which is one of the main reasons of reducing heat transfer [28]. While it can be assumed that at higher filler fraction, continuous thermally conductive pathways are formed, so that a higher thermal conductivity is obtained.

Thermal conductivity of PHB/BN composites
Density of PHB/BN composites
Figure 2 shows the density curves as a function of weight fraction of BN. The theoretical density is calculated from the densities of PHB and BN. The density of the PHB/BN composites increases with the increasing BN content. In addition, the real density of PHB/BN composites matches well with the theoretical value. It can be concluded from the density curves that the interaction between BN and PHB is strong that very few voids exist in the PHB/BN composites.

Density of PHB/BN composites fabricated by different methods
Morphological observation of PHB/BN composites
The cryo-fractured surface morphologies of neat PHB and PHB/BN composites with 50 wt% BN are presented in Fig. 3. A typical brittle fracture can be observed in the surface of PHB (Fig. 3a) which is smooth. The fractured surface of the neat PHB revealed its nature of weak resistance to crack initiation and propagation [29], while for composites with 50 wt% BN, the fracture surface exhibits quite rough morphology, which is originated from the pulling-out of PHB, implying the inhibition ability of the BN fillers on the propagation and advancing of cracks in the interface [30]. The increasing high viscosity usually will result in the formation of voids [31], which are detrimental to the thermal conductivity of the composites. However, no obvious voids are observed for PHB/BN composites in Fig. 3b. In addition, when 50 wt% BN was added into the matrix, complete thermal conductive pathways should be observed in the figure. However, the interfaces between BN particles and PHB are quite indistinct and BN particles can hardly be distinguished from the matrix in Fig. 3b even at 50 wt% content. This result is in accordance with the result obtained from Fig. 2, which also means that the interaction between BN particles and PHB matrix is quite strong. As BN is a good nucleating agent for PHB, the interfacial thermal resistivity between BN particles and PHB can be minimized and improve the thermal conductivity [32].

SEM micrographs of PHB/BN composites. a Neat PHB; b 50 wt% BN
Rheological properties of PHB/BN composites
Rheological analysis is regarded as an effective way to evaluate the melting parameters of polymers. The presence of the BN particles has a major influence on the linear viscoelasticity of PHB matrix. Figure 4 exhibits the storage modulus (G′), loss modulus (G″), and complex viscosity (η*) versus frequency for the PHB/BN composites at 180 °C. In Fig. 4c, neat PHB showed typical Newtonian fluid behavior when the frequency was around 1 rad s−1. The Newtonian behavior even existed when 10 wt% BN was added. With more BN loading fractions in PHB, the Newtonian behavior gradually disappeared and the pseudo-plastic shear thinning behavior became obvious. The storage modulus (G′) and the loss modulus (G″) versus frequency are shown in Fig. 4a, b. G′ and G″ increased with increasing the BN content, which is due to the reinforcement of BN. In Fig. 4a, it can be seen that the neat PHB displays a typical terminal behavior at low frequencies. However, this behavior gradually disappears with the increasing BN. When the BN content reaches up to 20 wt%, the PHB/BN composites showed solid-like response in the low frequency, indicating an elastic deformation-dominated flow happened. At this loading, the interactions between BN particles are strong enough, leading to the formation of percolated BN network [33]. In this situation, a higher shear force and a longer relaxation time are needed for the composites to flow.

Rheological properties of PHB/BN composites. a Storage modulus (G′); b loss modulus (G″); c complex viscosity (η*)
Thermal stability
Thermal stability is a key factor for polymer materials both in processing and applications. Figure 5 shows the thermal stability of PHB/BN composites and the corresponding thermal stability parameters are presented in Table 1. It can be seen that PHB decomposes in a single step. PHB composites exhibit the similar degradation characters in the whole temperature range and start at higher temperature, which means that all the PHB composites exhibit the same degradation mechanism. The 5% weight loss temperature (T −5%) and the temperature corresponding to the maximum rate of weight loss (T max) were used to illustrate the thermal degradation process of the blends. T −5% and T max of PHB are 272.4 and 289.1 °C. However, it was obviously to notice that the PHB decomposed at a higher temperature after the introduction of BN. The T −5% and T max of PHB composites increased by about 11 and 30 °C, respectively, indicating the improvement of the thermal stability of PHB. At the tested temperature, BN could not be decomposed. The weight loss of the PHB/BN composites was totally due to the thermal degradation of the PHB. The thermal stability improvement of the PHB/BN composites could be explained by the barrier effect of fillers [34], which improved the resistance to thermal degradation and restricted the diffusion of the decomposition products from the bulk polymer into the gas phase. Consequently, the thermal decomposition temperature of the PHB/BN composites shifted to a higher level and the thermal stability of the composites was improved.

a TGA curves and b DTG curves of PHB/BN composites
Thermal behavior of PHB/BN and P3,4HB/BN composites
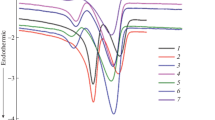
To discuss the effect of crystallinity on the thermal conductivity of PHB/BN composites, thermal behavior should be under investigation at first. P3,4HB [35] (one kind of PHB-based copolymers) was chosen as a comparison in this experiment. Figure 6 shows the DSC thermograms of PHB/BN and P3,4HB/BN composites. Figure 6a, c gives the DSC heating thermograms of PHB/BN and P3,4HB/BN composites of the first heating run from 0 to 190 °C at a heating rate of 10 °C min−1. The crystallinity of PHB/BN and P3,4HB/BN composites calculated from the melting peaks according to Eq. (1) obtained from DSC tests is summarized in Table 2. In Fig. 6a, the melting temperature (T m) of PHB increases with the addition of BN, suggesting that the nucleating agent BN enhances the formation of more perfect crystals. As can be seen from Table 2, with the addition of 20 wt% BN, the crystallinity of PHB/BN composites reaches the highest. All these results indicated that the addition of BN increased not only the crystallization but also the crystal perfection. However, when more BN was added into PHB, it could be clearly seen from both Fig. 6a and Table 2 that the ΔH m and crystallinity of PHB/BN composites slightly decreased. The T m of the composites with 50 wt% BN is about 3.2 °C lower than that of 10 wt% BN. This decrease can be associated with the deterioration of the crystal perfection in the presence of fillers. When more BN was added into the PHB, there were more nucleating sites. Consequently, the thickness of the crystals became thinner compared with the composites containing lower BN, leading to the decrease of T m. DSC thermograms of P3,4HB/BN composites are shown in Fig. 6c. Two clearly separated melting peaks are detected for neat P3,4HB and P3,4HB/BN composites. The lower one corresponds to the heterogeneous crystallization, while the higher one is due to the melting of reorganized crystals during the subsequent heating. Crystallinity of P3,4HB/BN composites presented in Table 2 is calculated from the integration of both melting peaks. Neat P3,4HB exhibits the lowest melting peaks at about 134 and 148 °C, indicating imperfect crystals formed in the matrix. With the introduction of BN, both melting peaks shifted to higher temperatures and the crystallinity followed the same trend as PHB/BN composites.

First heating run of DSC thermograms of PHB/BN and P3,4HB/BN composites
In Fig. 6b and d, both PHB/BN and P3,4HB/BN composites were cooled to −25 °C from the melting temperature at a cooling rate of 10 °C min−1. Usually, the addition of nucleating agent may effectively accelerate the crystallization of polymers. Besides, a high melt crystallization temperature and a narrower crystallization temperature range are considered to be the indicators of fast crystallization. From Fig. 6b, the crystallization from the melt of nucleated PHB occurred at higher temperatures with respect to neat PHB (about 45 °C higher) and this phenomenon could be observed for all PHB/BN composites. In addition, the crystallization peak became sharper and narrower after BN was introduced implying that the crystallization became faster and the diversity of crystalline morphologies became narrower [36]. Similar results can also be found for P3,4HB/BN composites in Fig. 6d. As expected, the particle surfaces of BN acted as crystalline substrates onto which the polymer chains were more easily adsorbed and folded. Heterogeneous nucleation enhanced the nucleation rate in a higher temperature region, thus resulting in a shifting of the crystallization temperature (T c) value. Therefore, BN is a good nucleating agent for both PHB/BN and P3,4HB/BN composites. The crystallization parameters such as the melting enthalpy and the crystallinity are summarized in Table 2. From Table 2, it can be clearly seen that a small amount of BN incorporated into the matrix can increase the crystallinity of the composites. However, the crystallinity of the composites tends to decrease with the increasing BN content. This is because the increasing BN particles in the PHB and P3,4HB hinder the movement of polymer molecular chains, consequently leading the crystallinity of the composites to decrease.
Wide-angle X-ray diffraction
It should be noticed that DSC shows no direct information on the structure formation of the crystallized PHB/BN and P3,4HB/BN composites. For that purpose, WAXD analysis, which provides direct information on the crystalline phases, is applied to the samples under investigation. Figure 7 shows the WAXD patterns for PHB/BN composites and P3,4HB/BN composites. In Fig. 7a, all the PHB/BN composites showed the same characteristic diffraction peaks. The peaks observed at 13.4°, 16.8°, and 22.5° are characteristics of the (020), (110), and (101) crystalline planes of PHB unit cell [37]. The diffraction peak at 26.1° is due to the presence of BN crystals and as it can be seen that the magnitude of the peak increased with the increase of BN concentration in the PHB/BN composites. Apparently, the diffraction patterns of the PHB/BN blends were similar to one another, implied that the structure of PHB crystals was not modified by the introduction of BN particles. In Fig. 7b, one can see that the characteristic diffraction peaks of P3,4HB/BN composites remain unchanged when compared with those of PHB/BN. This phenomenon indicated that P3,4HB/BN composites and PHB/BN composites share the same crystal structure.

WAXD patterns of a PHB/BN and b P3,4HB/BN composites
Effect of crystallinity on the thermal conductivity
The comparison of the thermal conductivity of PHB/BN and P3,4HB/BN composites is shown in Fig. 8. The thermal conductivity of neat P3,4HB is found to be about 0.23 W mK−1. Thermal conductivity of 0.92 W mK−1 could be acquired when the BN content was 50 wt%, about four times higher than neat P3,4HB. However, it is worth noted that the thermal conductivity of the P3,4HB/BN composites is lower than the PHB/BN composites at all BN levels. The difference of thermal conductivity between the PHB/BN composites and the P3,4HB/BN composites is small at a relatively low content of BN. However, when the BN fraction loading is higher than 20 wt%, the gaps between the PHB/BN composites and the P3,4HB/BN composites increasingly enlarge. As the crystal structures of P3,4HB/BN composites are almost the same as the crystal structures of PHB/BN according to the XRD results. The main reason that causes the difference between the two composites consequently can be attributed to the difference of crystallinity. One can see a significantly different crystallinity between PHB/BN and P3,4HB/BN composites as summarized in Table 2. This difference can be due to the existence of 4HB units in PHB. The 4HB units in PHB break the regularity of PHB crystals, acting as defects which restrict the crystallization of PHB. As the better heat transport can be obtained in the crystalline phase, phonon scattering in the matrix will become severe because of the increasing amorphous phase. Consequently, the thermal conductivities of the P3,4HB/BN composites are lower than those of PHB/BN composites.

Thermal conductivity of PHB/BN and P3,4HB/BN composites
Conclusions
In summary, a facile melt mixing process is developed to fabricate thermally conductive PHB/BN composites with various filler fractions. It was found that the thermal conductivity of PHB/BN composites was obviously enhanced with the incorporation of BN particles. The enhancement was attributed to the formation of thermally conductive pathways, which facilitate the heat to conduct more easily. SEM images provided that no visible voids are found in the composites. BN particles could not be clearly identified until using the chloroform to etch the PHB at the surface, which indicated that the interaction between PHB and BN particles was quite strong. Rheological properties indicated that the viscosity of PHB/BN composites increased with the introduction of BN. In the meantime, the thermal stability of PHB was also improved. Besides, thermally conductive pathways were formed according to the solid-like response in the low frequency. In this research, crystallinity that influenced the thermal conductivity of PHB/BN composites was considered. The comparison between P3,4HB/BN composites and PHB/BN composites showed the relation between crystallinity and thermal conductivity. BN was proved to act as strong nucleating agent in both PHB and P3,4HB based on the DSC results. Even though the thermal conductivities of both PHB/BN and P3,4HB/BN composites increased with BN content, the thermal conductivity of PHB/BN composites was higher than that of P3,4HB/BN composites. As there was no difference between the crystal structures of both composites from the WAXD reflections, crystallinity was the main reason that caused the difference of the thermal conductivity. This is because heat is preferred to transfer in the bulk with higher crystallinity.
References
Luyt AS, Molefi JA, Krump H (2006) Thermal, mechanical and electrical properties of copper powder filled low-density band linear low-density polyethylene composites. Polym Degrad Stab 91:1629–1636. doi:10.1016/j.polymdegradstab.2005.09.014
Sim LC, Ramanan SR, Ismail H, Seetharamu KN, Goh TJ (2005) Thermal characterization of Al2O3 and ZnO reinforced silicone rubber as thermal pads for heat dissipation purposes. Thermochim Acta 430:155–165. doi:10.1016/j.tca.2004.12.024
Xu Y, Luo X, Chung DDL (2002) Lithium doped polyethylene–glycol-based thermal interface pastes for high thermal contact conductance. J Electron Packag 124:188–191. doi:10.1115/1.1477191
Wolff EG, Schneider DA (1998) Prediction of thermal contact resistance between polished surfaces. Int J Heat Transf 41:3469–3482. doi:10.1016/S0017-9310(98)00067-2
Yu J, Mo H, Jiang P (2015) Polymer/boron nitride nanosheet composites with high thermal conductivity and sufficient dielectric strength. Polym Adv Technol 26:514–520. doi:10.1002/pat.3481
Wang Z, Fu Y, Meng W, Zhi C (2014) Solvent-free fabrication of thermally conductive insulating epoxy composites with boron nitride nanoplatelets as fillers. Nanoscale Res Lett 9:643–649. doi:10.1186/1556-276x-9-643
Shi Z, Radwan M, Kirihara S, Miyamoto Y, Jin Z (2009) Enhanced thermal conductivity of polymer composites filled with three-dimensional brushlike AlN nanowhiskers. Appl Phys Lett 95:22410422. doi:10.1063/1.3271028
Huang X, Iizuka T, Jiang P, Ohki Y, Tanaka T (2012) Role of interface on the thermal conductivity of highly filled dielectric epoxy/AlN composites. J Phys Chem C 116:13629–13639. doi:10.1021/jp3026545
Esfe H, Karimipour A, Yan W, Dahari M (2015) Experimental study on thermal conductivity of ethylene glycol based nanofluids containing Al2O3 nanoparticles. Int J Heat Transf 88:728–734. doi:10.1016/j.ijheatmasstransfer.2015.05.010
Zhou T, Wang X, Mingyan GU, Liu X (2008) Study of the thermal conduction mechanism of nano-SiC/DGEBA/EMI-2,4 composites. Polymer 49:4666–4672. doi:10.1016/j.polymer.2008.08.023
Ahn K, Kim K, Kim M, Kim J (2015) Fabrication of silicon carbonitride-covered boron nitride/Nylon 6,6 composite for enhanced thermal conductivity by melt process. Ceram Int 41:2187–2195. doi:10.1016/j.ceramint.2014.10.018
He Y, Wang Q, Liu W, Liu Y (2014) Functionalization of boron nitride nanoparticles and their utilization epoxy composites with enhanced thermal conductivity. Phys Status Solid A 211:677–684. doi:10.1002/pssa.201330305
Yao Y, Zeng X, Guo K, Sun R, Xu J (2015) The effect of interfacial state on the thermal conductivity of functionalized Al2O3 filled glass fibers reinforced polymer composites. Compos A 69:49–55. doi:10.1016/j.compositesa.2014.10.027
Lee D, Song SH, Hwang J, Jeon S (2013) Enhanced mechanical properties of epoxy nanocomposites by mixing noncovalently functionalized boron nitride nanoflakes. Small 9:2602–2610. doi:10.1002/smll.201203214
Liem H, Choy HS (2013) Superior thermal conductivity of polymer nanocomposites by using graphene and boron nitride as fillers. Solid State Commun 163:41–45. doi:10.1016/j.ssc.2013.03.024
Zhi C, Bando Y, Terao T, Tang C, Kuwahara H, Golberg D (2009) Towards thermoconductive, electrically insulating polymeric composites with boron nitride nanotubes as fillers. Adv Funct Mater 19:1857–1862. doi:10.1002/adfm.200801435
Gu J, Zhang Q, Dang J, Xie C (2012) Thermal conductivity epoxy resin composites filled with boron nitride. Polym Adv Technol 23:1025–1028. doi:10.1002/pat.2063
Song W, Wang P, Cao L, Anderson A, Meziani M, Farr AJ, Sun Y (2012) Polymer/boron nitride nanocomposite materials for superior thermal transport performance. Angew Chem Int Ed 51:6498–6501. doi:10.1002/anie.201201689
Ishida H, Rimdusit S (1998) Very high thermal conductivity obtained by boron nitride-filled polybenzoxazine. Thermochim Acta 320:177–186. doi:10.1016/s0040-6031(98)00463-8
Kim K, Kim M, Kim J (2014) Enhancement of the thermal and mechanical properties of a surface-modified boron nitride–polyurethane composite. Polym Adv Technol 25:791–798. doi:10.1002/pat.3291
Tsai M, Tseng H, Chiang J, Li J (2014) Flexible polyimide films hybrid with functionalized boron nitride and graphene oxide simultaneously to improve thermal conduction and dimensional stability. ACS Appl Mater Interfaces 6:8639–8645. doi:10.1021/am501323m
Takahashi S, Imai Y, Kan A, Hotta Y, Ogawa H (2014) Dielectric and thermal properties of isotactic polypropylene/hexagonal boron nitride composites for high-frequency applications. J Alloy Comp 615:141–145. doi:10.1016/j.jallcom.2014.06.138
Kim K, Kim M, Kim J (2014) Thermal and mechanical properties of epoxy composites with a binary particle filler system consisting of aggregated and whisker type boron nitride particles. Compos Sci Technol 103:72–77. doi:10.1016/j.compscitech.2014.08.012
Cai Z, Hou C, Yang G (2012) Characteristics and bending performance of electroactive polymer blend made with cellulose and poly(3-hydroxybutyrate). Carbohydr Polym 87:650–657. doi:10.1016/j.carbpol.2011.08.038
Naffakh M, Marco C, Ellis G (2014) Inorganic WS2 nanotubes that improve the crystallization behavior of poly(3-hydroxybutyrate). Cryst Eng Comm 16:1126–1135. doi:10.1039/C3CE41987H
Wurm A, Lellinger D, Minakov AA, Skipa T, Pötschke P, Nicula R, Alig I (2014) Crystallization of poly(-caprolactone)/MWCNT composites: a combined SAXS/WAXS, electrical and thermal conductivity study. Polymer 5:2220–2232. doi:10.1016/j.polymer.2014.02.069
Park HJ, Kim TA, Kim R, Kim J, Park M (2013) A new method to estimate thermal conductivity of polymer composite using characteristics of fillers. J Appl Polym Sci 129:965–972. doi:10.1002/app.38653
Kim K, Kim M, Hwang Y, Kim J (2014) Chemically modified boron nitride-epoxy terminated dimethylsiloxane composite for improving the thermal conductivity. Ceram Int 140:2047–2056. doi:10.1016/j.ceramint.2013.07.117
Tang L, Zhang H, Sprenger S, Ye L, Zhang Z (2012) Fracture mechanisms of epoxy-based ternary composites filled with rigid-soft particles. Compos Sci Technol 72:558–565. doi:10.1016/j.compscitech.2011.12.015
Huang L, Zhu P, Li G, Wong C (2015) Spherical and flake-like BN filled epoxy composites: morphological effect on the thermal conductivity, thermo-mechanical and dielectric properties. J Mater Sci Mater Electron 26:3564–3572. doi:10.1007/s10854-015-2870-1
Donnay M, Tzavalas S, Logakis E (2015) Boron nitride filled epoxy with improved thermal conductivity and dielectric breakdown strength. Compos Sci Technol 110:152–158. doi:10.1016/j.compscitech.2015.02.006
Haggenmueller R, Guthy C, Lukes JR, Fischer JE, Winey KI (2007) Single wall carbon nanotube/polyethylene nanocomposites: thermal and electrical conductivity. Macromolecules 40:2417–2421. doi:10.1021/ma0615046
Li Y, Han C, Bian J, Han L, Dong L, Gao G (2012) Rheology and biodegradation of polylactide/silica nanocomposites. Polym Compos 33:1719–1727. doi:10.1002/pc.22306
Gao Z, Zhao L (2015) Effect of nano-fillers on the thermal conductivity of epoxy composites with micro-Al2O3 particles. Mater Des 66:176–182. doi:10.1016/j.matdes.2014.10.052
Pan P, Liang Z, Nakamura N, Miyagawa T, Inoue Y (2009) Uracil as nucleating agent for bacterial poly[(3-hydroxybutyrate)-co-(3-hydroxyhexanoate)] copolymers. Macromol Biosci 9:585–595. doi:10.1002/mabi.200800294
Kai W, He Y, Inoue Y (2005) Fast crystallization of poly(3-hydroxybutyrate) and poly(3-hydroxybutyrate-co-3-hydroxyvalerate) with talc and boron nitride as nucleating agents. Polym Int 54:780–789. doi:10.1002/pi.1758
Jing X, Qiu Z (2012) Effect of low thermally reduced graphene loadings on the crystallization kinetics and morphology of biodegradable poly(3-hydroxybutyrate). Ind Eng Chem Res 51:13686–13691. doi:10.1021/ie3018466
Acknowledgements
This work was supported by the National Natural Science Foundation of China (51503204) and the Fund of Chinese Academy of Sciences (Changchun Branch) (No. 2015SYHZ0014).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Li, Z., Kong, J., Han, L. et al. Effect of crystallinity on the thermal conductivity of poly(3-hydroxybutyrate)/BN composites. Polym. Bull. 75, 1651–1666 (2018). https://doi.org/10.1007/s00289-017-2114-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00289-017-2114-z