
Abstract
The study reports on the flammability, thermal stability, impact properties and thermal conductivity of shape-stabilized phase change materials based on a soft Fischer–Tropsch paraffin wax blended with polypropylene (PP). The blends were melt-mixed with expanded graphite (EG) up to 9 wt% to improve the thermal conductivity and flammability resistance of the material. The thermal stability and flammability results show an increase in thermal stability and flame resistance of PP in the presence of EG, with the flammability further increasing in the presence of wax, probably because of the smaller and better dispersed EG particles in the PP/wax/EG composite that gave rise to a more compact char layer. Although the thermal degradation mechanism did not change in the presence of EG, the EG particles retarded the evolution of the volatile degradation products. The storage modulus of the PP/wax/EG composite was lower than those of PP and PP/EG, and decreased with increasing in wax content because of the softening effect of the wax. The impact strength of the PP/wax/EG composites increased with increasing EG content in all the samples, but decreased with increasing wax content.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The cost of energy resources have become the main reason for the development of phase change materials [1–3]. By implementing proper energy storage methods, the discrepancy between the energy supply and demand can be overcome. Energy storage methods are generally classified as sensible storage or latent heat storage, or a combination of the two. Latent heat storage is based on the absorption or release of energy when a storage material undergoes a phase change. Sensible heat storage happens when energy is added to the material, thus increasing the temperature of the material without changing its phase [4, 5]. The advantages of latent heat storage in comparison to sensible heat storage are high heat storage densities, small system sizes, and a narrow temperature changes during charging and discharging processes.
Phase change materials are latent heat storage materials. Thermal energy transfer occurs when a material undergoes solid–liquid or liquid–solid phase changes [6]. In recent years, a new type of PCM, called a shaped-stabilized PCM, was developed. Shape-stabilized PCMs are composites consisting of paraffin wax encapsulated in a three-dimensional net structure formed by polymers, such as high-density polyethylene [7] or styrene–butadiene–styrene (SBS) copolymers [8]. The net structure of the shape-stabilized PCM composites prevents the leakage of liquid during the phase change of the paraffin. As long as the operating temperature is below the melting point of the supporting material, the shape-stabilized PCM can keep its shape even when the paraffin changes from solid to liquid.
Despite the many desirable properties of PCMs, their low thermal conductivity (generally below 0.3 W m−1 K−1) is a major drawback which leads to low heat storage/retrieval rates and which in turn limits its widespread utilization as an energy storage material. Many studies [9–13] have therefore been carried out with the aim of enhancing the thermal conductivity in PCM. Methods for the improvement of the thermal conductivity of PCMs involve the dispersion of metallic or nonmetallic materials with high thermal conductivity into the PCM. Carbon nanoparticles and nanofibers in particular attracted more interest because of their higher thermal conductivities. Another method used to improve the thermal conductivity is the impregnation of the PCM into a high thermal conductivity material with a porous structure, such as expanded graphite (EG) and carbon fibre brushes. For an example, Fukai and co-workers [14] inserted 2 vol% carbon fibre brushes into PCM, and reported a sixfold increase in thermal conductivity of the composites compared to that of the pure PCMs. Zhang and Fang [15] prepared EG/paraffin PCM composites with 15 wt% EG. When this system was used in latent heat thermal energy storage (LHTES) system, the durations for heat storage and retrieval were reduced by 27.4 and 56.4 %, respectively, compared with those of paraffin.
In the current study of a form-stable PCM, the polypropylene (PP) has excellent mechanical and physiochemical properties and is used in a wide variety of applications such as the automobile industry, electricity, engineering, housing materials, and transportation [16–18]. However, its inherent combustibility limits the range of applications. Intumescent flame retardants can be introduced into polyolefins because of the advantages of good safety and relatively high flame retarding efficiency [19]. According to literature, the following three components are incorporated into formulations to obtain intumescent flame retardancy: acid-source, char-forming agent and blowing agent [20].
Halogenated flame retardants are generally used in most engineering plastics because of their excellent retardant performance [21]. However, the severe environmental impact of the processing and combustion of various brominated flame retardants motivated the investigation into halogen-free flame retardants to replace brominated and chlorinated ones [22]. Graphite and metallic hydroxide flame retardants are the most interesting and promising halogen-free flame retardants [23]. Expanded graphite is widely used to increase char yield and promote the thermal stability of intumescent flame retardants (IFR) because of their low cost and simple preparation with most polymers [24]. Generally, the presence of graphite leads to the formation of a protective thermally stable surface layer, which limits the heat transfer from the flame to the substrate and mass transfer from the substrate to the flame. The overall rate of flame feeding by combustible products from polymer pyrolysis and thermo-oxidation is therefore decreased.
Chlorinated paraffins, ammonium polyphosphate (APP) and pentaerythritol were used as flame retardants for PP [16, 17, 25, 26]. Shao and co-workers [17] reported that the PP/ethylene diamine-modified APP systems showed more effective flame retardancy than the PP/unmodified APP systems. This was attributed to the formation of a stable char layer and the prevention of the flammable volatiles going into the flame zone, which leads to the formation of intumescent, compact and stable char layers, consequently leading to the better flame retardant performance of modified APP. Chen et al. [25] studied flame retardant polypropylene composites prepared by melt-mixing ammonium polyphosphate, pentaerythritol and hydroxyl silicone oil with the polymer. Cone calorimetry results showed that a synergistic effect occurred when hydroxyl silicone oil and an intumescent flame retardant (IFR) are both present in polypropylene composites. Hydroxyl silicone oil reacts with ammonium polyphosphate to form a silicon phosphate and ceramic-like structure, which increases the efficiency of the intumescent char shield. Apparently, solid acids formed through the reaction of ammonium polyphosphate and SiO2 on the surface of the burning composite, and their chemical catalytic action further reduced the heat release rate of the composite.
A fair amount of studies investigated the flammability of shape-stabilized PCMs [19, 27, 28]. In these studies, the shape-stabilized PCMs were mostly based on paraffin, HDPE, expanded graphite and an intumescent flame retardant. Zhang et al. [19] showed an improvement in the flame retardancy of paraffin/HDPE/IFR (ammonium polyphosphate and pentaerythritol) with the addition of EG. Intumescent char layers were formed in the case of intumescent flame retardants (ammonium polyphosphate and pentaerythritol), but with the addition of EG into the IFR system the strength and stability of the intumescent char layer further increased. Better flammability properties were also found for a paraffin/HDPE/zinc borate + EG system compared to a paraffin/HDPE/APP/EG system [27]. No reports were found on the flammability of PP/wax/EG systems.
Based on the above literature studies, EG was added as flame retardant to the PP/paraffin wax blends to decrease the flammability of the form-stable PCM. Expanded graphite was also added to improve the thermal conductivity of the blends since both polypropylene (PP) and paraffin wax have low thermal conductivities. The prepared form-stable PCM blends, with and without flame retardants, were studied by means of cone calorimetry, impact testing, thermogravimetric analysis (TGA), and dynamic mechanical analysis (DMA).
Materials and methods
Materials
Soft Fischer–Tropsch paraffin wax (M3 wax) was supplied in powder form by Sasol Wax, South Africa. It is a paraffin wax consisting of approximately 99 % of straight-chain hydrocarbons and few branched chains, and it is primarily used in the candle industry. It has an average molar mass of 440 g mol−1 and a carbon distribution of C15–C78. Its density is 0.90 g cm−3 and it has a melting point of 57 °C. Isotactic polypropylene was supplied as pellets by Sasol Polymers, South Africa. It has an MFI of 10 g min−1, specific enthalpy of melting of 90 J g−1, melting temperature of 163–165 °C, and a density of 0.901 g cm−3. Expandable graphite ES 250 B5 was supplied by Qingdao Kropfmuehl Graphite (Hauzenberg, Germany).
Preparation of expanded graphite
The expandable graphite was first dried in an oven at 60 °C for 10 h. The expandable graphite was then heated in a furnace to 600 °C using a glass beaker and maintained at that temperature for 15 min to form expanded graphite.
Preparation of PP/wax blends and PP/wax/EG composites
All the samples (Table 1) were prepared by a melt-mixing process using a Brabender Plastograph 50 mL internal mixer at 180 °C and 60 rpm for 20 min. For the blends, the components were physically premixed and then fed into the heated mixer, whereas for the composites, the EG was added into the Brabender mixing chamber within 5 min after adding the PP or premixed PP/wax blends. The samples were then melt-pressed at 180 °C for 5 min under 50 kPa pressure using a hydraulic melt press to form 15 × 15 × 2 cm3 sheets.
Sample analysis
The optical microscopy images were captured using a CETI (Belgium) optical microscope and the samples were cut using a microtome knife (Microtome American optical model 820).
The DSC analyses were done in a Perkin Elmer Pyris-1 differential scanning calorimeter (USA) under flowing nitrogen (flow rate 20 mL min−1). Samples of mass 5–10 mg were sealed in aluminium pans and heated from −30 to 200 °C at a heating rate of 10 °C min−1, and cooled and re-heated under the same conditions. The peak temperatures of melting and melting enthalpies were determined from the second heating scans. The DSC measurements were repeated on three different samples of the same composition. The temperatures and enthalpies are reported as average values with standard deviations.
A Ceast Impactor ΙΙ (Italy) was used to investigate the impact properties of the blends and composites to establish whether EG gives rise to improved impact properties. The samples were rectangular with a width of 10 mm, a thickness of 3 mm and length of 83 mm, and were V-notched (2 mm deep) edgewise. The pendulum hammer was situated at an angle of 50° from the release spot and the samples were tested at an ambient temperature of 24 °C. Five samples of each composition were tested and the average and standard deviation values are presented.
Dynamic mechanical analysis was performed in a Perkin Elmer Diamond DMA (USA) from −90 to 90 °C in bending (dual cantilever) mode at a heating rate of 3 °C min−1, a frequency of 1 Hz, and an amplitude of 10 μm.
Cone calorimetry measurements were performed on a Fire Testing Technology dual analysis cone calorimeter using a cone shaped heater at an incident heat flux of 35 kW m−2. The specimens, with dimensions of 6 × 100 mm × 100 mm3, were prepared by compression moulding. The following quantities were measured using the cone calorimeter: Peak heat release rate, time to ignition, mass loss rate, and carbon monoxide and carbon dioxide yields.
The thermogravimetric (TGA) analyses were carried out in a Perkin Elmer Pyris-1 thermogravimetric analyzer. Samples ranging between 5 and 10 mg were heated from 30 to 650 °C at a heating rate of 10 °C min−1 under nitrogen flow (20 mL min−1). The TGA Fourier-transform infrared (TGA-FTIR) analyses were performed in a Perkin Elmer STA6000 simultaneous thermal analyser from Waltham, Massachusetts, USA. The analyses were done under flowing nitrogen at a constant flow rate of 20 mL min−1. Samples (20–25 mg) were heated from 30 to 650 °C at 10 °C min−1 and held for 4 min at 650 °C. The furnace was linked to the FTIR (Perkin Elmer Spectrum 100, Massachusetts, USA) with a gas transfer line. The volatiles were scanned over a 4000–400 cm−1 wavenumber range at a resolution of 4 cm−1. The FTIR spectra were recorded in the transmittance mode at different temperatures during the thermal degradation process.
Thermal conductivity measurements were performed on circular discs 5 mm thick and 12 mm in diameter using a ThermTest Inc. Hot Disk TPS 500 thermal constants analyzer. The instrument uses the transient plane source method. A 3.2 mm Kapton disk type sensor was selected for the analysis. The sensor was sandwiched between two sample discs. Three measurements were performed for each composition.
Results and discussion
Morphology
Figure 1 shows the optical microscopy images of some of the investigated samples. Comparison of the two images in this figure shows that in the absence of wax the EG particles are more agglomerated. The low molecular weight wax obviously contributes to a better dispersion of EG in the PP matrix, probably because the wax penetrates in between the EG layers and separates the layers, giving rise to smaller and better dispersed EG particles, which improves the thermal conductivity and flame resistance properties.

Optical microscopy images of a 94/6 w/w PP/EG, and b 65.8/28.2/6 w/w PP/wax/EG
Differential scanning calorimetry (DSC)
The DSC results in Fig. 2 show an endothermic peak shoulder and peak for the wax between 20 and 60 °C. The peak shoulder corresponds to a solid–solid transition, and the peak is due to wax melting. The peak around 160 °C is due to PP melting. The presence of two peaks in the blends and composites indicates that PP and wax are immiscible in the crystalline phase. The melting temperature of PP decreased when wax is present in both the blends and composites. This reduction could be the result of a decrease in the lamellar thickness of PP in the presence of wax [29], or because of some co-crystallization of the wax and PP.

DSC heating curves of PP and wax, as well as their 60/40 w/w blend and the related composites
Fire-retardant properties
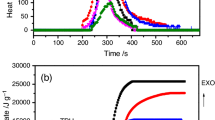
The heat release rate (HRR) is a very important parameter and can be used to express the intensity of fire [30]. A highly flame retardant system normally shows a low HRR peak. The HRR plots for the PP, PP/wax blend and their EG composites are shown in Fig. 3. When EG is introduced into the neat PP and PP/wax blend systems, the peak HRRs are dramatically reduced. HRR is a function of heat generation rate and heat transfer, and the heat generation rate is related to oxygen transfer during combustion, so the decrease in heat release rate is mostly the result of a decrease in oxygen transfer [31]. The addition of EG into PP and PP/wax blends therefore enhances the barrier properties of the char layer so that the heat transfer rate is reduced. It is interesting that the HRR of the PP/wax/EG composite is lower than that of PP/EG.

Heat release rate curves for PP, the PP/wax blend, and the PP/EG and PP/wax/EG composites
It seems as if the low molecular weight wax contributed to a better dispersion of EG in the PP matrix. This is probably because the wax penetrates in between the EG layers and separates the layers, giving rise to smaller and better dispersed EG particles. This enhanced the stability of the char layer formed, so that it was more difficult for the heat and oxygen to reach the PP matrix. The presence of wax accelerated the char layer formation and improved the compactness of the char layer. It has been reported that the rate of char layer formation and the compactness of a char layer have a strong influence on the reduction of the heat release rate and improvement in the flame retardancy [16], and that low peak heat release rate (PHRR) values correlate with well-dispersed systems [32, 33]. It is further clear from Fig. 3 that the heat release rate peak of the PP/wax blend is less intense than that of the neat PP, which means that the presence of wax in some way reduces the flammability of PP. However, the time to ignition (TTI) values in Table 2 show that ignition starts earlier for the PP/wax blend and the PP/wax/EG composite than for the neat PP and the PP/EG composites. This is probably because of the lower pyrolysis temperature of the wax compared to PP and PP/EG. Because of its lower peak heat release rate, the PP/wax/EG composite also shows a better fire performance index value. Fire performance index is considered to be the best individual indicator of overall fire hazard, and therefore smaller fire performance index values are an indication of a smaller fire hazard. The two composites further show better fire growth index values (an index for estimating fire growth, thus a lower fire growth index value may indicate a better flame retardancy) than PP and the PP/wax blend.
Figure 3 also shows that the peak heat release rate values decreases in the order PP > PP/wax > PP/EG > PP/wax/EG. The reason for this is that heat and flammable volatiles penetrated the PP and PP/wax blend in the absence of a flame retardant material (in this case EG), and therefore no char residues were formed. The PP/EG and PP/wax/EG composites show lower peak heat release rate values because of the formation of a char layer. The char layer prevented heat and oxygen from penetrating the PP and wax, and the flammable volatiles from entering the flame zone, leading to an improved flame resistance. The PP/EG composite shows a higher peak heat release rate value than the PP/wax/EG composite. This is attributed to the more compact and dense char layer formed in the presence of wax in the case of the PP/wax/EG composites (see discussion further on). Because of this, there was a much slower diffusion of decomposition products out of the system, and a slower penetration of heat and oxygen into the system.
Li et al. [34] reported that an intumescent flame retardant consisting of a char-foaming agent (CFA), ammonium polyphosphate and lanthanum oxide is very effective in improving the flame retardance of PP. They observed that the neat PP burned very fast after ignition, showing one sharp heat release rate peak with a maximum at 1025 kW m−2, which indicates a higher flammability than what we observed. They further observed that the addition of lanthanum oxide and intumescent flame retardants into PP improved the flame resistance of PP by 76 %, which is better than the about 50 % improvement we observed when mixing PP with wax and EG. Xu et al. [16] observed improvements smaller than or similar to ours with the addition of 25 % ammonium polyphosphate, poly[N 4-bis(ethylenediamine)-phenyl phosphonic-N 2, and N 6-bis(ethylenediamine)-1,3,5-triazine-N-phenyl phosphate] (PTPA). However, when combining ammonium polyphosphate and PTPA, a much more significant improvement in flame retardancy of 85 % was obtained.
The physical structure of the char layer plays a significant role in the performance of an intumescent flame retardant material. The charred residues were photographed at the end of the combustion process (Fig. 4). The PP and PP/wax blend did not show any char residue, while intumescent char layers were formed in the case of the PP/EG and PP/wax/EG composites. Heat and flammable volatiles must have penetrated the charred PP/EG and PP/wax/EG composites less than the PP and the PP/wax blend. This is because of the continuous, compact and thick carbonaceous layer in the presence of EG. This prevented the decomposition products from escaping out of the system, and the penetration of heat and oxygen into the system. A char layer will become fragmented when a lot of gaseous decomposition products escape, which will make it easier for heat and oxygen to enter the system. Careful inspection of these photos shows a much more compact char layer in the case of PP/wax/EG, which explains the improved flame retardancy of this system.

Photos of the a PP/EG and b PP/wax/EG char residues obtained at the end of the combustion process
One important reaction to fire parameter is the formation of carbon monoxide and carbon dioxide. CO and CO2 are present in large amounts in all fires. It is seen in Fig. 5 that both CO and CO2 production of PP increase and reach a maximum around 300 s, after which it decreases until it reaches zero above 400 s. The CO and CO2 production for the composites increased and reached a much lower maximum around 200 s, but this value was maintained over a much longer period of time. This is probably because the insulating char layer trapped these gases, and they only gradually escaped through micro-cracks over a longer period of time. The PP/wax blend and PP/EG composite also reached almost the same value for CO production, although the PP/wax blends released the gases over a much shorter period of time. The reason for this is the presence of EG agglomerates which resulted in the formation of an ineffective char barrier which allowed the slower release of CO over a longer period of time. The presence of wax in the PP/wax/EG composite resulted in a better dispersed system than PP/EG, and a more effective char was formed which allowed the release of smaller amounts of CO and CO2 over a longer period of time.

a Carbon monoxide and b carbon dioxide production plots of PP, PP/wax blend, as well as PP/EG and PP/wax/EG composites
Thermogravimetric analysis (TGA)
The TGA curves in Fig. 6 show that EG is thermally stable up to temperatures higher than the decomposition temperatures of wax and PP. The wax decomposes at temperatures lower than that of PP. The degradation of PP generally produces major products such as pentane (24.3 %), 2-methyl-1-pentene (15.4 %), and 2,4-dimethyl-1-heptene (18.9 %) with the minor product being propane [35]. All these products are simultaneously released as is evident from the single TGA mass loss step in Fig. 6.

TGA curves of PP, wax and EG
The TGA curves of neat PP, the PP/wax blend, as well as the PP/EG and PP/wax/EG composites, are shown in Figs. 7 and 8. The mass loss temperatures of PP increased significantly with the addition EG, irrespective of the filler content. This can be associated with the two-dimensional planar structure of EG in the PP matrix, which served as a barrier preventing further degradation of the underlying PP matrix. It probably also trapped the volatile decomposition products, which could only escape at much higher temperatures. Therefore, the higher mass loss temperatures do not necessarily indicate an increase in the thermal stability of the polymer, but are probably the result of retarded evolution of the degradation products, although there could have been a delay in the actual polymer degradation, which is something we could not monitor with the experimental techniques available to us. Contrary to our own observations, Kim et al. [36] observed slightly lower mass loss temperatures for exfoliated graphite nanoplatelets/LLDPE nanocomposites compared to those of the neat LLDPE matrix. They attributed this to the physisorbed water evaporating at 450 °C.

TGA curves of neat PP and the PP/EG composites

TGA curves of the PP/wax blend and the PP/wax/EG composites
All the wax-containing samples degraded in two steps (Fig. 8). The first step between 250 and 300 °C corresponds to the degradation of the wax, while the second step between 250 and 450 °C corresponds to the degradation of the PP. The 70/30 w/w PP/wax and 50/50 w/w PP/wax blends and their EG containing composites (not shown) showed similar behaviour. The thermal stability of the blend apparently improved in the presence of and with increasing graphite content, because the TGA curves in Fig. 8 show an increase in mass loss temperatures of the wax and PP in the composites. This could have been the result of the interaction between the PP and wax in the PP/wax blend and the graphite particles, which reduced the free radical chain mobility and thus slowed down the degradation process. However, as discussed earlier in this paper, the volatile degradation products were more probably trapped in the EG network (because of a relatively strong interaction between these products and EG) and only started evaporating at higher temperatures. Mhike et al. [37] considered that the interaction between the wax or LDPE chains and the graphite particles reduced the free radical chain mobility and thus slowed down the degradation process. Which of these two effects, or a combination of them, caused the observed increase in the TGA mass loss temperatures, is a topic that should be researched in more depth.
For all the composites there is a good correlation between the % residue at 550 °C and the amount of graphite originally mixed into the composite, which confirms that the graphite was generally well dispersed into the PP/wax blend.
Fourier-transform infrared (FTIR) analysis of volatiles from TGA analysis
TGA-FTIR analyses were done to establish the nature of the degradation products of PP and wax in the different samples. The degradation of PP normally occurs via a random chain scission mechanism; it does not involve crosslinking or chain branching. Because of the presence of tertiary carbons in PP, the bonds are ruptured through a β-scission mechanism followed by a radical transfer process which leads to the formation of the degradation products [38]. All these processes occur simultaneously as evident by a single TGA mass loss step between 250 and 450 °C (Fig. 6), which is in line with previously reported data [35]. The paraffin wax releases volatiles at temperatures around 290 °C (Fig. 6).
It can be seen that the main volatiles released are CO2, which is more visible in the PP/EG composite, and aliphatic or unsaturated alkanes (C–H bending vibration, in-plane rocking of CH2—Figs. 9, 10, 11, 12 and Table 3). It has been reported that during intercalation of natural graphite by strong acids some of the carbon double bonds are oxidized, which leads to the formation of oxygen-containing functional groups like carboxyls (C=O), which will form CO2 during decomposition [39]. It was also reported in the literature [19] that the CO2 evolved during PCM degradation increased when EG was introduced. There was no shift in the positions of the absorption peaks of all the samples, which indicates that the presence of wax, EG and/or wax + EG did not change the decomposition mechanism of the blends and composites.

FTIR curves of a wax and b PP at different temperatures during the thermal degradation in a TGA at a heating rate of 10 °C min−1

FTIR curves of a 70/30 w/w PP/wax and b 65.8/28.2/6 w/w PP/wax/EG at different temperatures during the thermal degradation in a TGA at a heating rate of 10 °C min−1

FTIR curves of a 70/30 w/w PP/wax and b 65.8/28.2/6 w/w PP/wax/EG during the thermal degradation in a TGA at a heating rate of 10 °C min−1

FTIR curves of 94/6 w/w PP/EG at different temperatures during the thermal degradation in a TGA at a heating rate of 10 °C min−1
Dynamic mechanical analysis (DMA)
The storage and loss modulus of the investigated samples are shown in Figs. 13 and 14. It was not possible to analyse pure wax and the PP/wax blends, because they were so brittle that they cracked or broke during the process of mounting in the DMA. The storage modulus of neat PP and the PP/EG composite is higher than those of the PP/wax/EG composites, especially above 60 °C which is after the melting of the wax in the composites. This is because of the softening effect of the wax on the composite, especially when the wax is in the molten state. A soft or flexible material typically creates more free volume, allowing the polymer chains a higher degree of mobility through Brownian movement. This lowers the temperature where segmental mobility can occur and makes the material more elastic.

Storage modulus curves of PP, as well as the PP/EG and PP/wax/EG composites

Loss modulus curves of PP, as well as the PP/EG and PP/wax/EG composites
Two transitions are observed in the loss modulus curves in Fig. 14. The relaxation between −20 and 30 °C, which is the β-relaxation, is attributed to the glass transition of the amorphous phase of PP. The relaxation between 40 and 60 °C is because of the melting of wax, which is the reason why this transition is not observed for PP and PP/EG. The glass transition of PP in PP/EG is at the same temperature than that of PP, which confirms that there is a fairly weak interaction between the PP chains and the EG particles. The temperature of this transition decreased by about 15 °C in the PP/wax/EG composites. As mentioned above, the wax has a strong softening effect on PP, and this is also the reason for the observed decrease in the glass transition temperature of the PP. The PP/wax blend samples are very brittle, despite the softening effect of the wax, while the PP/wax/EG composites are not so brittle. This is because of a large amount of highly crystalline and brittle wax in the PP/wax samples. In the case of the PP/wax/EG composites, the porous EG structure absorbs the wax resulting in fewer wax crystals in the PP matrix. The graphite therefore reinforced the samples and countered the brittle effect of the wax.
Impact properties
The impact properties of a polymer may be modified by adding some fillers. Polymeric impact modifiers may be incorporated to act as barriers or crack blunting regions to the advancing crack. The impact strengths of neat PP, PP/wax blends and the PP/wax/EG composites are shown in Figs. 15 and 16. The addition of wax decreased the impact strength of both PP and the PP/EG composites. The low molar mass wax crystals acted as internal flaws with high stress concentrations, and therefore as defects points for the initiation and propagation of stress cracking. It is known that the polymer matrix adds toughness to a composite, and therefore an increase in wax content will result in a decrease in the polymer content, resulting in a smaller amount of matrix to withstand the applied load. The impact strength of the PP/wax/EG composites increased in the presence of and with increasing expanded graphite content in all the investigated samples. One could expect a reduction in toughness with the addition of stiff graphite platelets due to the limited plastic flow of the PP matrix, because the more ductile matrix was replaced by the more rigid dispersed particles. Different factors, like filler particle size and interaction between the polymer and the other components, may have contributed to this improvement in the impact strength of the composites. In this case, the wax on the EG surface improved the interaction between the EG and the PP, giving rise to a good dispersion and effective stress transfer. The particle dispersion is important in controlling the mechanical properties of composites; large particles act as crack nucleation sites [40] and if the agglomerated particles are far away from each other, it is easier for crazes to develop into cracks and for cracks to propagate. The smaller the size of the filler particles and the better they are dispersed, the easier it is for a craze to terminate at a neighbouring particle without developing into a crack.

Impact strengths of PP and PP/wax blends

Impact strengths of PP/wax blends containing different contents of expanded graphite
Thermal conductivity
Phase change materials are used for the storage and release of thermal energy. One of the performance indicators is the rate of energy storage and release, which strongly depends on thermal conductivity of the PCMs. Since PP and wax both have very low thermal conductivities (Fig. 17), it is important to investigate how the thermal conductivity of the PCM system can be improved through the addition of conductive fillers. In our investigation, the thermal conductivity of both PP and PP/wax blends increased with increasing graphite content (Fig. 17). This can be attributed to high thermal conductivity of the EG [13]. However, it was observed that the thermal conductivities of the PP/wax/EG composites were higher than those of the PP/EG composites. It has been reported in the literature that the heat is conducted predominantly by phonons in the nanographite materials [41]. To improve the overall thermal transport in filled polymers, the acoustic impedance mismatch (a function of the acoustic speed and density of the medium) at the interface between the filler and the polymer has to be reduced. Because of the smaller and better dispersed EG particles in the PP/wax/EG composites, and their strong affinity for wax which sits at the interface between PP and the EG particles, the interfacial heat transfer between the graphite platelets and the PP matrix was obviously improved. For the PP/EG composites, the weak interaction between EG and PP increases the acoustic impedance which results in a large thermal contact resistance at the filler-–PP interface and a reduction in the thermal conductivity of the overall system.

Thermal conductivities of PP and the PP/wax blends with different amounts of expanded graphite
Conclusions
The effect of expanded graphite on the flammability, thermal stability, thermomechanical behaviour and thermal conductivity of PP and PP/wax composites was investigated. Improvements in thermal stability, thermal conductivity and flame resistance were observed for the PP/EG, and these properties further improved for the PP/wax/EG composites. The improvement of the flame resistance in the presence of wax is especially interesting, and was attributed to the formation of a more compact char layer that more effectively prevented the penetration of heat and oxygen and the release of flammable volatiles. The thermomechanical results confirmed the softening effect of the wax in the PP/wax/EG composites. It is interesting that the PP/wax blends could not be analysed because of their brittleness, but when the same blends contained EG, their brittleness significantly decreased because of the wax penetrating into the EG layered structure. The thermal conductivities of the PP/wax/EG composites were higher than those of the PP/EG composites, which was attributed to the smaller and better dispersed EG particles in the PP/wax/EG composites. TGA-FTIR analyses confirmed that the presence of wax, EG and/or wax + EG did not change the thermal decomposition mechanism of the blends and composites.
References
Song G, Ma S, Tang G, Yin Z, Wang X (2010) Preparation and characterization of flame retardant form-stable phase change materials composed by EPDM, paraffin and nano magnesium hydroxide. Energy 35:2179–2183. doi:10.1016/j.energy.2010.02.002
Zalba B, Marin JM, Cabeza LF, Mehling H (2003) Review on thermal energy storage with phase change materials, heat transfer analysis and applications. Appl Therm Eng 23:251–283. doi:10.1016/S1359-4311(02)00192-8
Kenisarin M, Mahkamov K (2007) Solar energy storage using phase change materials. Renew Sustain Energy Rev 11:1913–1965. doi:10.1016/j.rser.2006.05.005
Farid MM, Khudhair AM, Razack SAK, Al-Hallaj S (2004) A review on phase change energy storage: materials and applications. Energy Convers Manag 45:1597–1615. doi:10.1016/j.enconman.2003.09.015
Sharma A, Tyagi VV, Chen CR, Buddhi D (2009) Review on thermal energy storage with phase change materials and applications. Renew Sustain Energy Rev 13:318–345. doi:10.1016/j.rser.2007.10.005
Luyt AS, Krupa I (2009) Phase change materials formed by UV curable epoxy matrix and Fischer-Tropsch paraffin wax. Energy Convers Manag 50:57–61. doi:10.1016/j.enconman.2008.08.026
Mngomezulu ME, Luyt AS, Krupa I (2011) Structure and properties of phase change materials based on high density polyethylene, hard Fischer-Tropsch paraffin wax, and wood flour. Polym Compos 32:1155–1163. doi:10.1002/pc.21134
Xiao M, Feng B, Gong K (2002) Preparation and performance of shape stabilized phase change thermal storage materials with high thermal conductivity. Energy Convers Manag 43:103–1078. doi:10.1016/S0196-8904(01)00010-3
Shaikh S, Lafdi K, Hallinan K (2008) Carbon nano-additives to enhance latent energy storage of phase change materials. J Appl Phys 103:094302. doi:10.1063/1.2903538
Elgafy A, Lafdi K (2005) Effect of carbon nanofiber additives on thermal behavior of phase change materials. Carbon 43:3067–3074. doi:10.1016/j.carbon.2005.06.042
Bonnet P, Siruede D, Garrier B, Chauvet O (2007) Thermal properties and percolation in carbon nanotubes-polymer composites. Appl Phys Lett 91:201910. doi:10.1063/1.2813625
Wang JF, Xie HQ, Zhong X (2008) Thermal properties of heat storage composites containing multiwalled carbon nanotubes. J Appl Phys 104:113537. doi:10.1063/1.3041495
Sari A, Karaipekli A (2007) Thermal conductivity and latent heat thermal energy storage characteristics of paraffin/expanded graphite composite as phase change material. Appl Therm Eng 27:1271–1277. doi:10.1016/j.applthermaleng.2006.11.004
Fukai J, Morozumi Y, Miyatake O (2003) Improvement of thermal characteristics of latent heat thermal energy storage units using carbon-fiber brushes: experiment and modelling. Int J Heat Mass Transf 46:4513–4525. doi:10.1016/S0017-9310(03)00290-4
Zhang ZG, Fang XM (2006) Study on paraffin/expanded graphite composite phase change thermal energy storage material. Energy Convers Manag 47:303–310. doi:10.1016/j.enconman.2005.03.004
Xu ZZ, Huang JQ, Chen MJ, Tan Y, Wang YZ (2013) Flame retardant mechanism of an efficient flame-retardant polymeric synergist with ammonium polyphosphate for polypropylene. Polym Degrad Stab 98:2011–2020. doi:10.106/j.polymdegradstab.2013.07.010
Shao ZB, Deng C, Tan Y, Chen MJ, Chen L, Wang YZ (2014) Flame retardation of polypropylene via a novel intumescent flame retardant: ethylenediamine-modified ammonium polyphosphate. Polym Degrad Stab 106:88–96. doi:10.1016/j.polymdegrastab.2013.10.005
Lei ZQ, Cao YM, Xie F, Ren H (2012) Study on surface modification and flame retardants properties of ammonium polyphosphate for polypropylene. J Appl Polym Sci 124:781–788. doi:10.1002/app.35064
Zhang P, Hu Y, Song L, Ni J, Xing W, Wang J (2010) Effect of expanded graphite on properties of high-density polyethylene/paraffin composite with intumescent flame retardant as a shape-stabilized phase change material. Sol Energy Mater Sol Cells 94:360–365. doi:10.1016/j.solmat.2009.10.014
Lecouvet B, Sclavons M, Bailly C, Bourbigot S (2013) A comprehensive study of the synergistic flame retardant mechanisms of halloysite in intumescent polypropylene. Polym Degrad Stab 98:2268–2281. doi:10.1016/j.polymdegrastab.2013.08.024
Wu X, Wang L, Wu C, Yu J, Xie L, Wang G, Jiang P (2012) Influence of char residues on flammability of EVA/EG, EVA/NG and EVA/GO composites. Polym Degrad Stab 97:54–63. doi:10.1016/j.polymdegrastab.2011.10.011
Ramani A, Dahoe AE (2014) On the performance and mechanism of brominated and halogen free flame retardants in formulations of glass fibre reinforced poly(butylene terephthalate). Polym Degrad Stab 104:71–86. doi:10.1016/j.polymdegradstab.2014.03.021
Li Z, Qu B (2003) Flammability characterization and synergistic effects of expandable graphite with magnesium hydroxide in halogen-free-retardant EVA blends. Polym Degrad Stab 81:401–408. doi:10.1016/S014-3910(03)00123-X
Bai G, Guo C, Li L (2014) Synergistic effect of intumescent flame retardant and expandable graphite on mechanical and flame-retardant properties of wood flour-polypropylene composites. Constr Build Mater 50:148–153. doi:10.1016/j.conbuildmat.2013.09.028
Chen X, Jiao C (2009) Synergistic effects of hydroxyl silicone oil on intumescent flame retardant polypropylene system. Fire Saf J 44:1010–1014. doi:10.1016/j.firesaf.2009.06.008
Xia Y, Jing F, Mao Z, Guan Y, Zheng A (2014) Effects of ammonium polyphosphate to pentaerythritol ratio on composition and properties of carbonaceous foam deriving from intumescent flame-retardant polypropylene. Polym Degrad Stab 107:64–73. doi:10.1016/j.polymdegradstab.2014.04.016
Cai Y, Song L, He Q, Yang D, Hu Y (2008) Preparation, thermal and flammability properties of a novel form-stable phase change materials based on high density polyethylene/poly(ethylene-co-vinylacetate)/organophilic montmorillonite nanocomposites/paraffin compounds. Energy Convers Manag 49:2055–2062. doi:10.1016/j.enconman.2008.02.013
Cai Y, Wei Q, Huang F, Gao W (2008) Preparation and properties studies of halogen-free flame retardant form-stable phase change materials based on paraffin/high density polyethylene composites. Appl Energy 85:7654–7775. doi:10.1016/j.apenergy.2007.10.017
AlMaadeed MA, Labidi S, Krupa I, Karkri M (2015) Effect of expanded graphite on the phase change materials of high density polyethylene/wax blends. Thermochim Acta 600:35–44. doi:10.1016/j.tca.2014.11.023
Ye L, Qu B (2008) Flammability characteristics and flame retardant mechanism of phosphate-intercalated hydrotalcite in halogen-free flame retardant EVA blends. Polym Degrad Stab 93:918–924. doi:10.1016/j.polymdegradstab.2008.02.002
Zhang P, Song L, Lu H, Wang J, Hu Y (2010) The influence of expanded graphite on thermal properties for paraffin/high density polyethylene/chlorinated paraffin/antimony trioxide as a flame retardant phase change material. Energy Convers Manag 51:2733–2737. doi:10.1016/j.enconman.2010.06.003
Fawn UM, Yao Q, Nakajima H, Manias E, Wilkie CA (2005) Expandable graphite/polyamide-6 nanocomposites. Polym Degrad Stab 89:70–84. doi:10.1016/j.polymdegradstab.2005.01.004
Wang X, Rathore R, Songtipya P, Gasco MDMJ, Manias E, Wilkie CA (2011) EVA-layered double hydroxide (nano) composites: mechanism of fire retardancy. Polym Degrad Stab 96:301–313. doi:10.1016/j.polymdegradstab.2011.10.011
Li Y, Li B, Dai J, Jia H, Gao S (2008) Synergistic effects of lanthanum oxide on a novel intumescent flame retardant polypropylene system. Polym Degrad Stab 93:9–16. doi:10.1016/j.polymdegradstab.2007.11.002
Peterson JD, Vyazovkin S, Wight CA (2001) Kinetics of the thermal and thermo-oxidative degradation of polystyrene, polyethylene and polypropylene. Macromol Chem Phys 202:775–784. doi:10.1002/1521-3935(20010301)202:6<775::AID-MACP775>3.0.CO;2-G
Kim S, Do I, Drzal LT (2010) Thermal stability and dynamic mechanical behavior of exfoliated graphite nanoplatelets-LLDPE nanocomposites. Polym Compos 31:755–761. doi:10.1002/pc.20781
Mhike W, Focke WW, Mofokeng JP, Luyt AS (2012) Thermally conductive phase change materials for energy storage based on low-density polyethylene, soft Fischer-Tropsch wax and graphite. Thermochim Acta 527:75–82. doi:10.1016/j.tca.2011.10.008
Palacios J, Perera R, Rosales C, Albano C, Pastor JM (2012) Thermal degradation kinetics of PP/OMMT nanocomposites with mPE and EVA. Polym Degrad Stab 97:729–737. doi:10.1016/j.polymdegradstab.2012.02.009
Gogoi P, Boruah M, Bora C, Dolui SK (2014) Jatropha curcas oil alkyd/epoxy resin/expanded graphite (EG) reinforced bio-composite: evaluation of the thermal, mechanical and flame retardancy properties. Prog Org Coat 77:87–93. doi:10.1016/j.porgcoat.2013.08.006
Chen B, Evans JRG (2009) Impact strength of polymer-clay nanocomposites. R Soc Chem 5:3572–3584. doi:10.1039/b902073j
Shi JN, Ger MD, Liu YM, Fan YC, Wen NT, Lin CK, Pu NW (2013) Improving the thermal conductivity and shape-stabilization of phase change materials using nanographite additives. Carbon 51:365–372. doi:10.1016/j.carbon.2012.08.068
Acknowledgments
The National Research Foundation of South Africa is acknowledged for financial support of the project.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Mochane, M.J., Luyt, A.S. The effect of expanded graphite on the flammability and thermal conductivity properties of phase change material based on PP/wax blends. Polym. Bull. 72, 2263–2283 (2015). https://doi.org/10.1007/s00289-015-1401-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00289-015-1401-9