
Abstract
The profound alterations in the structure, cellular composition, and function of synovial tissue in rheumatoid arthritis (RA) are the basis for the persistent inflammation and cumulative joint destruction that are hallmarks of this disease. In RA, the synovium develops characteristics of a tertiary lymphoid organ, with extensive infiltration of lymphocytes and myeloid cells. Concurrently, the fibroblast-like synoviocytes undergo massive hyperplasia and acquire a tissue-invasive phenotype. In this review, we summarize key components of these processes, focusing on recently-described roles of selected molecular markers of these cellular components of RA synovitis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Cellular composition of synovial tissue
Normal synovium is comprised primarily of two cell populations, termed type A and type B synoviocytes [1]. Type A synoviocytes are cells of the monocyte/macrophage lineage, while type B synoviocytes are of mesenchymal origin and are referred to as fibroblast-like synoviocytes (FLS). These two cell populations form the normal synovial lining, which is approximately two cell layers in thickness, lacks a true basement membrane, and is positioned above a poorly-demarcated connective tissue matrix that contains a modest vascular network, a few mast cells and few, if any, lymphocytes.
In RA, a massive increase in synovial cellularity occurs that represents a combination of cellular influx, hyperplasia of synovial cells (FLS), and activation/proliferation/differentiation of infiltrating immune system cells. Early disease in RA is associated with synovial vascular inflammation, followed by a robust infiltration of leukocytes into the synovium resulting in hyperplasia of the synovial lining, formation of lymph follicles, and development of the mature pannus. The three most abundant cell types in RA synovium are FLS, monocyte-lineage cells, and T lymphocytes (Table 1), but other cell populations, including B lymphocytes, plasma cells, dendritic cells, vascular endothelial cells, mast cells, and osteoclasts, are all very important in the pathogenesis of RA. Granulocytes (polymorphonuclear cells) are rare in RA synovium but are often the most abundant cellular component of RA synovial fluid. Growing appreciation of the molecular markers expressed in RA synovium not only provides insight into pathogenesis, but also offers targets for molecular imaging of RA synovitis [2].
FLS in RA
FLS are cells of mesenchymal origin that display many characteristics in common with fibroblasts, such as the expression of several types of collagens and the protein vimentin, a component of cytoskeletal filaments. FLS also secrete unique proteins that are normally absent in other fibroblast lineages, such as lubricin, a protein crucial for joint lubrication. Furthermore, these cells express a number of molecules important for the mediation of cell adhesion such as vascular cell adhesion molecule-1 (VCAM-1), various integrins and their receptors, and cadherin-11 [1]. Cadherins are particularly important for intercellular adhesion and coordinate morphogenesis during tissue development. They also support tissue integrity and architecture [3]. FLS express cadherin-11 in vivo, and this molecule plays a key role in homotypic adhesion of FLS and in synovial tissue organization (Table 2) [3]. Cadherin-11 can also generate activation signals in FLS, and its function may be regulated by controlled shedding of its N-terminal domain from the FLS surface [3, 4].
Rheumatoid Arthritis Fibroblast-like Synoviocytes (RA FLS) are important mediators of joint destruction, since they are able to invade adjacent collagenous structures, including articular cartilage. Using a severe combined immunodeficient (SCID) mouse chimera model in which human synovium is engrafted into mice, it was shown that inhibition of endogenous p53 leads to increased invasiveness and cellularity of FLS, highlighting the possibility that somatic mutations in the p53 tumor suppressor gene may contribute to synovial hyperplasia and cartilage damage in RA [5]. Using a similar SCID chimera model, it was shown that RA FLS display active recruitment to naïve cartilage via the neovasculature, independent of the site of application of the RA fibroblasts in vivo, leading to profound destruction of the target cartilage. These experimental findings support the idea that destructive arthritis can spread between joints, which can be partly explained by the transmigration of activated RA FLS [6].
In RA, FLS also secrete factors that promote inflammation, neovascularization, and cartilage degradation [7]. For example, RA FLS express high levels of cyclooxygenase-2 (COX-2) protein and synthesize prostaglandin E2 (PGE2) in vitro, and in rodent models of arthritis [8, 9]. Moreover, these same cells secrete pro-inflammatory and angiogenic chemokines and cytokines [10]. In response to cytokines produced by macrophages such as tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β), RA FLS secrete matrix-degrading enzymes, such as matrix metalloproteinases (MMPs), aggrecanases, and cathepsins [7]. MMPs released from RA FLS can modulate activity of cytokines and chemokines, release pro-apoptotic ligands from cell surfaces, and promote FLS invasion of the cartilage [8]. MMP-2, -3, and -1 aid in tissue invasion and the breakdown of proteoglycans and collagen types I and II [8]. MMPs also activate cathepsins that are potent collagen-degrading proteases produced in the synovium [11]. Moreover, MMPs from RA FLS invade more aggressively in a Matrigel matrix than FLS from osteoarthritis (OA) or avascular necrosis (AVN) synovium [12]. Despite these properties of secreted MMPs, recent work has identified a more crucial role for the membrane-anchored MMP-14 in invasion of types I and II collagen matrices and articular cartilage [13]. Moreover, MMP-14 is also required for the angiogenic response to FLS invasion through articular cartilage [13], and is highly expressed in RA synovium [14].
Chemokines, largely derived from activated fibroblasts, are responsible for recruitment of neutrophils (PMNs) as well as lymphocytes and monocytes. There is evidence for roles of IL-8/CXCL8 [15, 16], epithelial neutrophil-activating protein-78 (ENA-78/CXCL5) [17], monocyte chemoattractant protein-1 (MCP-1/CCL2) [18], macrophage inflammatory protein 1-alpha (MIP-1α/CCL3) [19], CX3CL1 (fractalkine) [20], MIP-3α [21], CXCL16 [22] and regulated on activation, and normal T cell expressed and secreted (RANTES/CCL5) [23, 24] in RA pathogenesis. Roles for some of these cytokines in RA initiation are possible, as MCP-1/CCL2 and MIP-1α/CCL3 are both increased in RA serum prior to disease onset [25]. Specific chemokines, including MCP-1, RANTES, growth-regulated protein alpha (Gro-α/CXCL1), and IL-8 are produced by RA FLS via distinct signaling pathways that could become targets for new therapeutic approaches [26].
Other chemoattractant proteins, distinct from conventional chemokines, also may have important roles in RA. For example, aminopeptidase N/CD13, an ecto-peptidase expressed by myeloid cells and FLS, has recently been examined in RA (Fig. 1). CD13 is released into synovial fibroblast culture supernatants by MMP-14 mediated shedding of CD13 from the FLS membrane [27, 28] and is found in high concentrations in RA synovial fluid. Using an as yet unidentified G-protein-coupled receptor and independent of its enzymatic activity, CD13 induces chemotaxis of cytokine-activated T cells, a T cell population similar to that found in RA synovium, at a concentration that is similar to the gradient between the CD13 concentration in RA serum versus RA synovial fluid, suggesting that CD13 could play an important role as a T cell chemoattractant in a positive feedback loop that contributes to RA synovitis [27].

Strong expression of CD13 in RA synovium. CD13 (green) in RA synovium, co-localized (yellow) with cadherin-11 (red), a marker of FLS. CD13 is also extensively expressed on monocyte-macrophage lineage cells in RA synovial tissue, which do not express cadherin-11
Angiogenesis is also considered to be an invasive process that requires proteolysis of the extracellular matrix, proliferation and migration of endothelial cells, as well as synthesis of new matrix components. Angiogenesis is a key event in the expansion of the synovial lining of joints. CXC chemokines as well as some CC chemokines, that are potent inducers of angiogenesis, are primarily produced in the RA joint by FLS [16, 20, 22, 29]. Typically, CXC chemokines are pro-angiogenic if they possess the ELR motif, but anti-angiogenic if they do not [29].
Citrullination of various proteins in RA, mediated by various isoforms of peptidyl arginine deiminase (PAD), is increasingly recognized as not only a mechanism for generation of RA-specific autoantigens, but also as a pathway for altering the function of various inflammatory mediators and receptors. PAD4 is expressed by FLS, supporting strong evidence for citrullination of FLS proteins being integral to inflammation and the pathogenesis of RA [30,31,32,33]. For example, citrullination of ENA-78 converts the primary target of this chemokine from the neutrophil to the monocyte and changes its pattern of chemokine receptor engagement [33]. A second example is the citrullination of calreticulin which enhances the ability of this molecule to function as a novel cell-activating receptor for the shared epitope MHC allele that is associated with susceptibility to and severity of RA [34].
RA FLS also display an altered phenotype compared to cells present in normal synovium. This can be easily observed in culture in which RA FLS lose contact inhibition and dependency, thus contributing to the increase in the number of fibroblasts in the inflammatory tissue [10]. This is remarkably similar to the growth of progenitor like cells. Hyperproliferating synoviocytes in the RA joint have many features of progenitor cells and display proteins that are uniquely attributable to both cell phenotypes. For example, Inhibitor of DNA binding 1 (Id1) is a nuclear protein containing a basic helix-loop-helix (bHLH) domain that regulates cell growth and is expressed in endothelial progenitor cells [35]. Histologic analysis of synovial tissue reveals that Id1 is also highly expressed in the vasculature of RA synovial tissue [35], and by FLS [36], suggesting that Id1 displays pleiotropic properties in cells that exhibit hyperproliferative responses. RA synovial fluid also contains abundant amounts of Id1 with the primary source being from activated FLS [36]. Once released, Id1 acts as a potent inducer of angiogenesis [35]. Id1 is also packaged into FLS exosomes and released, setting the stage for the intriguing possibility that Id1 (and possibly other nuclear proteins) may be delivered to other cells within the RA synovium that do not themselves produce Id1 [36]. Similarly, alveolar macrophages secrete the STAT-induced STAT signaling inhibitors SOCS1 and SOCS3 in exosomes and microparticles for uptake by alveolar epithelial cells and subsequent inhibition of STAT activation in vitro and in vivo [37].
FLS may also amplify joint inflammation by cognate cell–cell interactions within the rheumatoid synovium. Such interactions may alter the phenotype of FLS, and also provide additional pathways for FLS to exert control over the behavior of the other cell types in RA synovium. Interactions between FLS and T lymphocytes are considered in more detail below. The multiple interactions between FLS and other key cell populations in RA, through secreted mediators, microparticles, and direct cell–cell contact, create numerous positive feedback loops that enhance the inflammatory environment within the RA synovium, attracting and activating more immune cells and ultimately contributing to joint destruction [38,39,40].
Monocytic cells in RA
Monocyte (MN)/macrophage recruitment into and activation within the synovial membrane are critical steps in the pathogenesis of RA. An early hallmark of active RA is an increased number of synovial MNs/macrophages which correlates with the degree of joint erosion while their depletion from inflamed synovium results in therapeutic benefit [41, 42]. Upon recruitment and activation in the synovial tissue (ST), these MNs/macrophages secrete pro-inflammatory cytokines such as TNF-α, IL-1β, and IL-6 which contribute to RA pathogenesis [43]. Biologic therapies targeting these mediators highlight their importance in RA. In this section, we will discuss the heterogeneity of human and murine MNs/macrophages and their characteristics and functions in RA.
Macrophages are co-localized with FLS in the normal ST membrane where the predominant cells are FLS. In RA, a greater fraction of the cells in ST are MNs/macrophages [44]. Resident tissue macrophages are typically derived from circulating MNs, which originate from CD34+ bone marrow progenitors. Recent lineage-tracing studies indicate that some tissue-resident macrophages originate from embryonic precursors instead of CD34+ bone marrow progenitors [45] and maintain themselves by self-renewal [46]. For example, intestinal lamina propria and dermal macrophages originate from CD34+ bone marrow progenitors and are continually replaced by MNs from the circulation [47, 48]. The sources of ST macrophages and their self-renewal capacity have not been characterized in humans.
Murine MN subset classification and functions have been well characterized and accepted [49]. However, classification of human MN subsets linked to their functional phenotypes remains only partially defined. Two major MN subsets have been identified in mice: “classical” (Ly6Chi CX3CR1int CCR2+); and “non-classical” (Ly6Clow CX3CR1hi CCR2−) [50]. Ly6C+ is further divided into Ly6Chi and Ly6Cint. The surface markers for Ly6C+ subsets are CD11b+CD115+ and chemokine receptors are CCR2hiCX3CR1low. The surface markers and chemokine receptors for Ly6C− (Ly6Clow) MNs are CD11b+CD115+ and CCR2lowCX3CR1hi [51].
Ly6Chi MNs, referred as inflammatory MNs, require CCR2 expression for egress from the bone marrow [52, 53]. Granulocyte-macrophage colony-stimulating factor (GM-CSF), a pro-inflammatory cytokine involved in myeloid cell differentiation, plays an important role in many autoimmune diseases, including RA, by regulating the inflammatory signature and progeny of Ly6Chi MNs [54]. Ly6Chi MNs contribute to the initiation of tissue inflammation [55], secrete TNF-α, nitric oxide, and IL-1β upon bacterial infection [56] and type 1 interferon (IFN) in response to viruses [57]. The interaction between CCR2-CCL2 in Ly6Chi MNs alters the conformation of very late antigen-4 (VLA-4), which results in increased MN transmigration into inflamed tissue [58]. Ly6Chi MNs also differentiate into M1 macrophages, which secrete pro-inflammatory cytokines such as TNF-α and IL-6 and contribute to tissue degradation and T cell activation.
CD14 is highly expressed on the surface of human MNs and macrophages and is a pattern recognition receptor. Three subsets of MNs have been defined in humans: classical (CD14++CD16−), intermediate (CD14++CD16+), and non-classical (CD14+CD16++) [59]. The classical and non-classical human MNs are homologous to their classical and non-classical counterparts in mice. Functionally, classical (CD14++CD16−) and intermediate (CD14++CD16+) MNs possess more phagocytic and pro-inflammatory properties than non-classical MNs [60].
Intermediate (CD14++CD16+) MNs express CCR2 and selectively CCR5, which bind, respectively, MIP-1α, which is chemotactic for macrophages, and CCL5/RANTES. Intermediate MNs produce significantly more TNF-α and IL-1β compared to other populations in response to lipopolysaccharide (LPS); however, production of IL-6, IL-8, and IL-10 are not different in intermediate and classical monocytes when compared to non-classical monocytes [60, 61]. Human CD14++CD16+ intermediate monocytes display pro-inflammatory functions and are involved in antigen presentation and T cell activation, as indicated by gene signature studies [61]. These MNs produce TNF-α and IL-1β in high concentration in response to LPS but have low peroxidase activity [60]. During inflammation, classical and intermediate monocytes adhere to and invade tissue by interaction of CCR2/CCL2 (MCP-1/CCL2) and/or CCR5/CCL5 in a very late antigen 1 (VLA1)/VCAM1 dependent manner. The frequency of intermediate MNs is increased in peripheral blood of patients with chronic RA compared to sex- and age-matched healthy donors, while the frequency of non-classical CD14+CD16++ MNs does not differ between patients and controls [62, 63]. However, another study found that absolute cell counts of both intermediate and non-classical MN populations are increased in RA patient blood [64].
Human CD14+CD16++ non-classical MNs patrol the vessel wall and invade tissues when triggered by interaction of CX3CR1 with CCL3 in a lymphocyte functional antigen-1 (LFA-1)/intercellular adhesion molecule-1 (ICAM-1)-dependent manner. This subset releases IL-1β and TNF-α in response to ligation of toll-like receptor 7 (TLR7) or TLR8, suggesting a pathological role in autoimmune disease [60]. Classical MNs represent ~90% of blood MNs while the number of CD16 expressing cells is very small in healthy individuals but the number of CD16 expressing cells increases in infection and inflammatory conditions [59]. Classical (CD14++CD16−) and intermediate CD14++CD16+ MNs resemble mouse Ly6C+ inflammatory MNs, while non-classical (CD14+CD16++) MNs act like Ly6C− anti-inflammatory MNs and patrol vascular endothelium [50]. However, some studies suggest a pro-inflammatory role of CD14+CD16++ cells because of the production of inflammatory cytokines.
In RA MNs/macrophages produce pro-inflammatory cytokines, including TNF-α, IL-6 and IL-1, and MMPs, leading to endothelial cell activation and cartilage damage (49.50). These cells also secrete various chemokines which help to recruit more leukocytes into the inflamed joint. CD14+CD68+ monocytes have an activated phenotype with increased expression of human leukocyte antigen (HLA-DR; involved in antigen presentation to CD4+ T cells), costimulatory molecules (CD80, CD86, and CD40), adhesion molecules such as ICAM-1/CD54, and some chemokine receptors [65,66,67,68,69]. Depletion of activated MNs by repeated leukapheresis in patients with active RA leads to improvements in disease activity that persist for 10–12 weeks [70, 71].
CD68 and CD163 are two commonly used markers for identification of synovial macrophages in RA synovium. CD68 is a scavenger receptor that binds to oxidized low-density lipoprotein and is involved in cell–cell interactions [72]. The number of synovial CD68+ macrophages correlates with disease activity, indicating the importance of these cells in RA pathogenesis [41, 73, 74]. CD68 is present on both cell surface and lysosomal membranes [75]. CD68 is a sensitive biomarker to predict the possible efficacy of new anti-rheumatic treatments, as the changes in the number of synovial sublining CD68+ macrophages correlate with clinical improvement [76]. The synovial sublining CD68+ macrophage count is correlated significantly with radiologic outcome and radiologic progression in RA patients [41]. CD163 is a type I transmembrane protein that belongs to the group B scavenger receptor cysteine-rich superfamily [77]. CD163 may be a better macrophage marker as compared with CD68 in RA synovium, because it discriminates between synovial macrophages and synovial intimal fibroblasts, some of which also stain positive for CD68 in diseased tissue [78]. Additionally, soluble CD163 in sera is a promising diagnostic marker for untreated new-onset systemic juvenile idiopathic arthritis and macrophage activation syndrome [79].
Polarized macrophages are referred to as M1 or M2 cells. Classically activated M1 macrophages are induced by IFN-γ, LPS, GM-CSF, and TNF-α, whereas alternatively activated M2 macrophages are induced by IL-4, IL-13, monocyte colony stimulating factor (M-CSF), immune complexes, IL-10, and glucocorticoids [80]. Transcription factors involved in M1 macrophage polarization include NF-κB, STAT1, and interferon regulatory factor 5 (IRF5), while IRF4, STAT6, c-Myc, PPARγ, and Kruppel-like factor 4 are involved in M2 polarization [81]. M1 macrophages express a high level of TNF-α, IL-1, IL-6, IL-23, IL-12, type I IFN, reactive nitrogen intermediate (RNI), reactive oxygen intermediate (ROI), and CXCL9, 10, and 11, while M2 macrophages express a high level of IL-4, IL-10, CD163, CD206, and CCL16, 17, 18, 22, and 24 [80, 82]. M1 polarized macrophages are generally involved in resistance to intracellular pathogens and to tumors in the context of Th1-driven responses, whereas M2 polarized macrophages are effectors of resistance to parasites, have immunoregulatory properties, promote tumor growth and invasiveness and orchestrate tissue repair and remodeling (including fibrosis) [83, 84]. The M1/M2 model is a useful scheme, but it may not fully reflect the flexibility and the diversity of the monocyte/macrophage activation process in various organs involved in immune-mediated diseases, in which many intermediate and diverse phenotypes have been demonstrated [85, 86]. The polarization state of synovial macrophages in RA is not yet well understood.
Expression of GM-CSF and M-CSF is increased in synovial fluid from RA patients. TNF-α and IL-1 induce production of GM-CSF and M-CSF by synovial fibroblasts and chondrocytes, suggesting that GM-CSF and M-CSF play a pathogenic role in arthritis. Both GM-CSF-deficient and Csf1op/op mice are resistant to collagen-induced arthritis [87]. Consistently, blockade of GM-CSF or M-CSF inhibits the development of arthritis [87, 88]. Phase 1 or phase 2 clinical trials and preclinical studies for inflammatory arthritis have employed specific antibodies against GM-CSF, M-CSF or their receptors [54, 89].
T lymphocytes in RA
The T cell population in RA is polyclonal [90] and includes increased numbers of activated and memory cells compared to peripheral blood T cells (Fig. 2). Activated T cells in RA synovium express a combination of surface markers that characterize both early (CD69) and late (class II MHC) stages of T cell activation [91]. To what extent RA synovial T cells are activated elsewhere in the body, with subsequent migration to synovial tissue, versus stimulation by antigen and second signals from antigen-presenting cells locally, is not known, but both scenarios are likely. T cells can also be activated by cocktails of pro-inflammatory cytokines (including cytokines present in RA synovium), and such T cells display an array of surface structures similar to T cells isolated directly from the RA joint [91]. Moreover, these cytokine-activated T cells (Tck) are especially potent in inducing macrophages to secrete TNF [92], and interact closely with FLS in vitro, which results in activation of both cell types [93, 94]. Surface structures involved in these interactions include membrane-anchored TNF on the Tck [93] and B7-H3 on the FLS [94]. FLS lack robust expression of CD80 (B7.1) and CD86 (B7.2), along with most other members of the B7 family, but express B7-H3 constitutively, as do fibroblasts from other tissues [94]. The identity of the receptor(s) for B7-H3 on the T cell surface remains controversial. While many “professional” antigen-presenting cell populations are present in RA synovium, including dendritic cells, macrophages, and B lymphocytes, RA FLS also can present superantigen to naïve T cells [95], and arthritogenic, HLA-DR4-restricted peptide autoantigens to previously activated T cells [96]. Interactions between CD4 T cells and monocytes in RA synovium also have multiple important consequences, which can include induction of differentiation of pathogenic polarized T cell and macrophage subpopulations, differentiation of osteoclasts in synovial tissue, secretion of pro-inflammatory mediators, and induction of or protection from apoptosis [97].

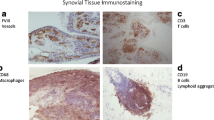
A dense infiltrate of activated T cells in RA synovium. The CD30 molecule (upper right panel) is expressed on activated T and B lymphocytes, and the CD3 complex (lower left panel) is present on all T cells. CD30, which is found on a very small proportion of circulating lymphocytes, is expressed by >90% of the synovial T cells. The lower right panel, showing merged red and green fluorescence, which generates a yellow color, indicates that the great majority of the CD30+ cells in the RA synovium are T cells. The blue color is DAPI, which identifies nuclei
RA synovium is a site of autoantibody production and some RA synovia contain structures that resemble germinal centers. A recent report describes a novel CD4+ T cell subset that is distinct from T follicular helper cells and that appears to be the principal T cell subset that drives B cell to plasma cell differentiation in RA synovium [98]. These cells express PD-1 and lack CXCR5, but express other chemokine receptors that direct migration to the joint. Expression of surface SLAMF5 and secretion of IL-21 are important in the interaction of these cells with B lymphocytes [98]. Another group recently demonstrated that a subset of CD4+ IL-21-producing T cells was expanded in RA synovial fluid, and that some of these cells also produced TNF-α and RANK-ligand [99]. Either IL-21 or co-culture with these T cells augmented FLS production of MMPs and IL-6 [99]. Interleukin 21 also promotes FLS migration and tissue invasion [100], suggesting that this novel T cell subset and/or its key mediators may become an important therapeutic target in RA.
Regulatory T cells (Tregs) are essential for prevention of autoimmunity, and Treg defects have been sought in many autoimmune diseases, including RA. CD4+ Tregs, which express CD25 and the transcription factor FoxP3, are present in RA, both systemically and in the joint. While not found to be numerically deficient in most studies, they appear to have impaired function, a defect that can be reversed in some patients following successful treatment of RA [101].
Heterogeneity of RA synovium and the potential for optimization of targeted therapies
The extent of lymphoid tissue organization is known to be heterogeneous among RA patients [102]. Recent work has extended our understanding of RA synovial tissue heterogeneity by global analysis of synovial tissue gene expression [103]. This analysis led to molecular definition of four RA synovial subsets, termed lymphoid, myeloid, fibroid, and low inflammatory. These subsets were also distinct when analyzed by immunohistochemistry and by flow cytometry. T cells were present in all subsets, but B cells were found only in the lymphoid and myeloid subsets and were abundant only in the lymphoid subset. Based on the patterns of gene expression in the lymphoid and myeloid subsets, two circulating biomarkers were identified: soluble ICAM1 (expressed most strongly in the myeloid-dominant RA synovial tissue) and CXCL13 (expressed most strongly in the lymphoid-dominant RA synovial tissue), that are elevated in RA sera but which do not correlate with each other with respect to their serum concentrations. Baseline samples were then assayed for these two proteins, from an RA therapeutic trial that compared two biologic agents, adalimumab (anti-TNF) and tocilizumab (anti-IL-6R). Patients with high sICAM1 and low CXCL13 were more likely to achieve a robust clinical response ACR50) with adalimumab, while those with low sICAM1 and high CXCL13 were much more likely to achieve an ACR50 with tocilizumab [103]. If replicated, these findings point towards selection of optimal biologic therapeutics for individual RA patients, based on circulating biomarkers that reflect the molecular composition of synovial tissue subtypes.
Conclusion
RA synovium may be an environment that is sufficiently unique to promote patterns of gene expression, inflammatory mediator production, cell surface protein expression, and cell–cell interactions that are distinct from those essential to normal host defenses and immune system function. For example, the hypoxic conditions in RA synovium lead to the local production by FLS of hypoxia-inducible factor-1alpha, which alters FLS gene expression in multiple ways relevant to the pathogenesis of RA [104], including augmentation of production of pro-inflammatory cytokines and of interactions of FLS with both T and B cells. The pathogenesis of RA does not reflect the actions of any one cell lineage, but rather the complex interactions between all cell populations in RA synovium, mediated by both direct cell–cell contact and by molecules that are secreted or shed by the various types of synovial cells. This network of multiple interactions is perhaps surprisingly non-redundant, since neutralization of a single inflammatory mediator, (out of dozens or hundreds that are present in the RA synovium), can be clinically effective. Further advances in the efficacy and safety of the treatment of RA are likely to arise from definition of pathogenic pathways that are less relevant to host defenses against infection or neoplasm, and from identification of clinically meaningful heterogeneity in the molecular pathogenesis of RA that will guide selection of therapeutics.
References
Pap T et al (2000) Fibroblast biology. Role of synovial fibroblasts in the pathogenesis of rheumatoid arthritis. Arthritis Res 2:361–367
Put S et al (2014) Molecular imaging of rheumatoid arthritis: emerging markers, tools, and techniques. Arthritis Res Ther 16:208
Valencia X et al (2004) Cadherin-11 provides specific cellular adhesion between fibroblast-like synoviocytes. J Exp Med 200:1673–1679
Noss EH et al (2015) Evidence for cadherin-11 cleavage in the synovium and partial characterization of its mechanism. Arthritis Res Ther 17:126
Pap T et al (2001) Invasiveness of synovial fibroblasts is regulated by p53 in the SCID mouse in vivo model of cartilage invasion. Arthritis Rheum 44:676–681
Lefevre S et al (2009) Synovial fibroblasts spread rheumatoid arthritis to unaffected joints. Nat Med 15:1414–1420
Mor A et al (2005) The fibroblast-like synovial cell in rheumatoid arthritis: a key player in inflammation and joint destruction. Clin Immunol 115:118–128
Crofford LJ et al (1994) Cyclooxygenase-1 and -2 expression in rheumatoid synovial tissues. Effects of interleukin-1 beta, phorbol ester, and corticosteroids. J Clin Invest 93:1095–1101
Yamada R et al (2000) Selective inhibition of cyclooxygenase-2 with antisense oligodeoxynucleotide restricts induction of rat adjuvant-induced arthritis. Biochem Biophys Res Commun 269:415–421
Pap T, et al. 2000. Fibroblast biology. Role of synovial fibroblasts in the pathogenesis of rheumatoid arthritis. In: Arthritis Res, p 361–367
Yamanishi Y, Firestein GS (2001) Pathogenesis of rheumatoid arthritis: the role of synoviocytes. [review] [119 refs]. Rheum Dis Clin N Am 27:355–371
Tolboom TC et al (2002) Invasive properties of fibroblast-like synoviocytes: correlation with growth characteristics and expression of MMP-1, MMP-3, and MMP-10. Ann Rheum Dis 61:975–980
Sabeh F et al (2010) Membrane-type I matrix metalloproteinase-dependent regulation of rheumatoid arthritis synoviocyte function. J Immunol 184:6396–6406
Qin S et al (2015) Immunolocalization of membrane-type 1 MMP in human rheumatoid synovium tissues. Int J Clin Exp Pathol 8:9286–9292
Koch AE et al (1991) Synovial tissue macrophage as a source of the chemotactic cytokine Il-8. J Immunol 147:2187–2195
Koch AE et al (1992) Interleukin-8 as a macrophage-derived mediator of angiogenesis. Science 258:1798–1801
Koch AE et al (1994) Epithelial neutrophil activating peptide-78: a novel chemotactic cytokine for neutrophils in arthritis. J Clin Invest 94:1012–1018
Koch AE et al (1992) Enhanced production of monocyte chemoattractant protein-1 in rheumatoid arthritis. J Clin Invest 90:772–779
Koch AE et al (1994) Macrophage inflammatory protein-1 alpha. A novel chemotactic cytokine for macrophages in rheumatoid arthritis J Clin Invest 93:921–928
Ruth JH et al (2001) Fractalkine, a novel chemokine in rheumatoid arthritis and in rat adjuvant-induced arthritis. Arthritis Rheum 44:1568–1581
Ruth JH et al (2003) Role of macrophage inflammatory protein-3alpha and its ligand CCR6 in rheumatoid arthritis. Lab Investig 83:579–588
Ruth JH et al (2006) CXCL16-mediated cell recruitment to rheumatoid arthritis synovial tissue and murine lymph nodes is dependent upon the MAPK pathway. Arthritis Rheum 54:765–778
Shahrara S et al (2005) Amelioration of rat adjuvant-induced arthritis by Met-RANTES. Arthritis Rheum 52:1907–1919
Shahrara S et al (2006) RANTES modulates TLR4-induced cytokine secretion in human peripheral blood monocytes. J Immunol 177:5077–5087
Kokkonen H et al (2010) Up-regulation of cytokines and chemokines predates the onset of rheumatoid arthritis. Arthritis Rheum 62:383–391
Ogura N et al (2005) Tumor necrosis factor-alpha increases chemokine gene expression and production in synovial fibroblasts from human temporomandibular joint. J Oral Pathol Med 34:357–363
Morgan R et al (2015) Expression and function of aminopeptidase N/CD13 produced by fibroblast-like synoviocytes in rheumatoid arthritis: role of CD13 in chemotaxis of cytokine-activated T cells independent of enzymatic activity. Arthritis Rheumatol 67:74–85
Morgan RL et al (2016) Localization, shedding, regulation and function of aminopeptidase N/CD13 on fibroblast like Synoviocytes. PLoS One 11:e0162008
Strieter RM et al (1995) The functional role of the ELR motif in CXC chemokine-mediated angiogenesis. J Biol Chem 270:27348–27357
Girbal-Neuhauser E et al (1999) The epitopes targeted by the rheumatoid arthritis-associated antifilaggrin autoantibodies are posttranslationally generated on various sites of (pro) filaggrin by deimination of arginine residues. J Immunol 162:585–594
Schellekens GA et al (1998) Citrulline is an essential constituent of antigenic determinants recognized by rheumatoid arthritis-specific autoantibodies. J Clin Invest 101:273–281
Mastronardi FG et al (2006) Increased citrullination of histone H3 in multiple sclerosis brain and animal models of demyelination: a role for tumor necrosis factor-induced peptidylarginine deiminase 4 translocation. J Neurosci 26:11387–11396
Yoshida K et al (2014) Citrullination of epithelial neutrophil-activating peptide 78/CXCL5 results in conversion from a non-monocyte-recruiting chemokine to a monocyte-recruiting chemokine. Arthritis Rheumatol 66:2716–2727
Ling S et al (2013) Citrullinated calreticulin potentiates rheumatoid arthritis shared epitope signaling. Arthritis Rheum 65:618–626
Isozaki T et al (2014) Inhibitor of DNA binding 1 as a secreted angiogenic transcription factor in rheumatoid arthritis. Arthritis Res Ther 16:R68
Edhayan G et al (2016) Inflammatory properties of inhibitor of DNA binding 1 secreted by synovial fibroblasts in rheumatoid arthritis. Arthritis Res Ther 18:87
Bourdonnay E et al (2015) Transcellular delivery of vesicular SOCS proteins from macrophages to epithelial cells blunts inflammatory signaling. J Exp Med 212:729–742
Lefevre S et al (2015) Role of synovial fibroblasts in rheumatoid arthritis. Curr Pharm Des 21:130–141
Muller-Ladner U et al (2007) Cells of the synovium in rheumatoid arthritis. Synovial fibroblasts Arthritis Res Ther 9:223
Bartok B, Firestein GS (2010) Fibroblast-like synoviocytes: key effector cells in rheumatoid arthritis. Immunol Rev 233:233–255
Mulherin D et al (1996) Synovial tissue macrophage populations and articular damage in rheumatoid arthritis. Arthritis Rheum 39:115–124
Davignon JL et al (2013) Targeting monocytes/macrophages in the treatment of rheumatoid arthritis. Rheumatology (Oxford) 52:590–598
McInnes IB, Schett G (2007) Cytokines in the pathogenesis of rheumatoid arthritis. Nat Rev Immunol 7:429–442
Athanasou NA (1995) Synovial macrophages. Ann Rheum Dis 54:392–394
Epelman S et al (2014) Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 40:91–104
Merad M et al (2002) Langerhans cells renew in the skin throughout life under steady-state conditions. Nat Immunol 3:1135–1141
Bain CC et al (2014) Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat Immunol 15:929–937
Tamoutounour S et al (2013) Origins and functional specialization of macrophages and of conventional and monocyte-derived dendritic cells in mouse skin. Immunity 39:925–938
Ingersoll MA et al (2011) Monocyte trafficking in acute and chronic inflammation. Trends Immunol 32:470–477
Geissmann F et al (2003) Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 19:71–82
Si Y et al (2010) CCR2 mediates hematopoietic stem and progenitor cell trafficking to sites of inflammation in mice. J Clin Invest 120:1192–1203
Boring L et al (1997) Impaired monocyte migration and reduced type 1 (Th1) cytokine responses in C-C chemokine receptor 2 knockout mice. J Clin Invest 100:2552–2561
Serbina NV, Pamer EG (2006) Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat Immunol 7:311–317
Hamilton JA (2008) Colony-stimulating factors in inflammation and autoimmunity. Nat Rev Immunol 8:533–544
Croxford AL et al (2015) The cytokine GM-CSF drives the inflammatory signature of CCR2+ monocytes and licenses autoimmunity. Immunity 43:502–514
Serbina NV et al (2008) Monocyte-mediated defense against microbial pathogens. Annu Rev Immunol 26:421–452
Barbalat R et al (2009) Toll-like receptor 2 on inflammatory monocytes induces type I interferon in response to viral but not bacterial ligands. Nat Immunol 10:1200–1207
Audoy-Remus J et al (2008) Rod-shaped monocytes patrol the brain vasculature and give rise to perivascular macrophages under the influence of proinflammatory cytokines and angiopoietin-2. J Neurosci 28:10187–10199
Ziegler-Heitbrock L et al (2010) Nomenclature of monocytes and dendritic cells in blood. Blood 116:e74–e80
Cros J et al (2010) Human CD14dim monocytes patrol and sense nucleic acids and viruses via TLR7 and TLR8 receptors. Immunity 33:375–386
Wong KL et al (2011) Gene expression profiling reveals the defining features of the classical, intermediate, and nonclassical human monocyte subsets. Blood 118:e16–e31
Rossol M et al (2012) The CD14(bright) CD16+ monocyte subset is expanded in rheumatoid arthritis and promotes expansion of the Th17 cell population. Arthritis Rheum 64:671–677
Yoon BR et al (2014) Functional phenotype of synovial monocytes modulating inflammatory T-cell responses in rheumatoid arthritis (RA). PLoS One 9:e109775
Coulthard LR et al (2012) Differential effects of infliximab on absolute circulating blood leucocyte counts of innate immune cells in early and late rheumatoid arthritis patients. Clin Exp Immunol 170:36–46
Cauli A et al (1997) Interleukin-1, interleukin-1 receptor antagonist and macrophage populations in rheumatoid arthritis synovial membrane. Br J Rheumatol 36:935–940
Koller M et al (1999) Expression of adhesion molecules on synovial fluid and peripheral blood monocytes in patients with inflammatory joint disease and osteoarthritis. Ann Rheum Dis 58:709–712
Kinne RW et al (2000) Macrophages in rheumatoid arthritis. Arthritis Res 2:189–202
van Amelsfort JM et al (2007) Proinflammatory mediator-induced reversal of CD4+,CD25+ regulatory T cell-mediated suppression in rheumatoid arthritis. Arthritis Rheum 56:732–742
Evans HG et al (2009) In vivo activated monocytes from the site of inflammation in humans specifically promote Th17 responses. Proc Natl Acad Sci U S A 106:6232–6237
Yeadon C, Karsh J (1983) Lymphapheresis in rheumatoid arthritis. The clinical and laboratory effects of a limited course of cell depletion. Clin Exp Rheumatol 1:119–124
Hahn G et al (1993) Modulation of monocyte activation in patients with rheumatoid arthritis by leukapheresis therapy. J Clin Invest 91:862–870
Ramprasad MP et al (1996) Cell surface expression of mouse macrosialin and human CD68 and their role as macrophage receptors for oxidized low density lipoprotein. Proc Natl Acad Sci U S A 93:14833–14838
Tak PP et al (1997) Analysis of the synovial cell infiltrate in early rheumatoid synovial tissue in relation to local disease activity. Arthritis Rheum 40:217–225
Bresnihan B et al (2007) Synovial macrophages as a biomarker of response to therapeutic intervention in rheumatoid arthritis: standardization and consistency across centers. J Rheumatol 34:620–622
Strobl H et al (1995) Flow cytometric analysis of intracellular CD68 molecule expression in normal and malignant haemopoiesis. Br J Haematol 90:774–782
Haringman JJ et al (2005) Synovial tissue macrophages: a sensitive biomarker for response to treatment in patients with rheumatoid arthritis. Ann Rheum Dis 64:834–838
Van Gorp H et al (2010) Scavenger receptor CD163, a Jack-of-all-trades and potential target for cell-directed therapy. Mol Immunol 47:1650–1660
Fonseca JE et al (2002) Macrophage subpopulations in rheumatoid synovium: reduced CD163 expression in CD4+ T lymphocyte-rich microenvironments. Arthritis Rheum 46:1210–1216
Bleesing J et al (2007) The diagnostic significance of soluble CD163 and soluble interleukin-2 receptor alpha-chain in macrophage activation syndrome and untreated new-onset systemic juvenile idiopathic arthritis. Arthritis Rheum 56:965–971
Mantovani A et al (2004) The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol 25:677–686
Lawrence T, Natoli G (2011) Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol 11:750–761
Mosser DM, Edwards JP (2008) Exploring the full spectrum of macrophage activation. Nat Rev Immunol 8:958–969
Gordon S, Martinez FO (2010) Alternative activation of macrophages: mechanism and functions. Immunity 32:593–604
Mantovani A et al (2005) Macrophage polarization comes of age. Immunity 23:344–346
Murray PJ et al (2014) Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 41:14–20
Xue J et al (2014) Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 40:274–288
Campbell IK et al (2000) The colony-stimulating factors and collagen-induced arthritis: exacerbation of disease by M-CSF and G-CSF and requirement for endogenous M-CSF. J Leukoc Biol 68:144–150
Cook AD et al (2001) Blockade of collagen-induced arthritis post-onset by antibody to granulocyte-macrophage colony-stimulating factor (GM-CSF): requirement for GM-CSF in the effector phase of disease. Arthritis Res 3:293–298
Cornish AL et al (2009) G-CSF and GM-CSF as therapeutic targets in rheumatoid arthritis. Nat Rev Rheumatol 5:554–559
Duby AD et al (1989) Clonal heterogeneity of synovial fluid T lymphocytes from patients with rheumatoid arthritis. Proc Natl Acad Sci U S A 86:6206–6210
Brennan FM et al (2008) Resting CD4+ effector memory T cells are precursors of bystander-activated effectors: a surrogate model of rheumatoid arthritis synovial T-cell function. Arthritis Res Ther 10:R36
Brennan FM et al (2002) Evidence that rheumatoid arthritis synovial T cells are similar to cytokine-activated T cells: involvement of phosphatidylinositol 3-kinase and nuclear factor kappaB pathways in tumor necrosis factor alpha production in rheumatoid arthritis. Arthritis Rheum 46:31–41
Tran CN et al (2007) Molecular interactions between T cells and fibroblast-like synoviocytes: role of membrane tumor necrosis factor-alpha on cytokine-activated T cells. Am J Pathol 171:1588–1598
Tran CN et al (2008) Interactions of T cells with fibroblast-like synoviocytes: role of the B7 family costimulatory ligand B7-H3. J Immunol 180:2989–2998
Tsai C et al (1996) Responsiveness of human T lymphocytes to bacterial superantigens presented by cultured rheumatoid arthritis synoviocytes. Arthritis Rheum 39:125–136
Tran CN et al (2007) Presentation of arthritogenic peptide to antigen-specific T cells by fibroblast-like synoviocytes. Arthritis Rheum 56:1497–1506
Roberts CA et al (2015) The interplay between monocytes/macrophages and CD4(+) T cell subsets in rheumatoid arthritis. Front Immunol 6:571
Rao DA et al (2017) Pathologically expanded peripheral T helper cell subset drives B cells in rheumatoid arthritis. Nature 542:110–114
Lebre MC, et al. 2016. Synovial IL-21/TNF-producing CD4+ T cells induce joint destruction in rheumatoid arthritis by inducing matrix metalloproteinase production by fibroblast-like synoviocytes. J Leukoc Biol
Xing R et al (2016) Interleukin-21 induces migration and invasion of fibroblast-like synoviocytes from patients with rheumatoid arthritis. Clin Exp Immunol 184:147–158
Cooles FA et al (2013) Treg cells in rheumatoid arthritis: an update. Curr Rheumatol Rep 15:352
Weyand CM, Goronzy JJ (2003) Ectopic germinal center formation in rheumatoid synovitis. Ann N Y Acad Sci 987:140–149
Dennis G Jr et al (2014) Synovial phenotypes in rheumatoid arthritis correlate with response to biologic therapeutics. Arthritis Res Ther 16:R90
Hu F et al (2016) Hypoxia-inducible factor-1alpha perpetuates synovial fibroblast interactions with T cells and B cells in rheumatoid arthritis. Eur J Immunol 46:742–751
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is a contribution to the special issue on Immunopathology of Rheumatoid Arthritis - Guest Editors: Cem Gabay and Paul Hasler
Rights and permissions
About this article

Cite this article
Asif Amin, M., Fox, D.A. & Ruth, J.H. Synovial cellular and molecular markers in rheumatoid arthritis. Semin Immunopathol 39, 385–393 (2017). https://doi.org/10.1007/s00281-017-0631-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-017-0631-3