
Abstract
LIGHT and herpes virus entry mediator (HVEM) comprise a ligand–receptor pair in the tumor necrosis factor superfamily. These molecules play an important role in regulating immunity, particularly in the intestinal mucosa. LIGHT also binds the lymphotoxin β receptor, and HVEM can act as a ligand for immunoglobulin family molecules, including B- and T-lymphocyte attenuator, which suppresses immune responses. Complexity in this pivotal system arises from several factors, including the non-monogamous pairing of ligands and receptors, and reverse signaling or the ability of some ligands to serve as receptors. As a result, recognition events in this fascinating network of interacting molecules can have pro- or anti-inflammatory consequences. Despite complexity, experiments we and others are carrying out are establishing rules for understanding when and in what cell types these molecules contribute to intestinal inflammation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The intestinal mucosa is constantly exposed to antigens derived from food and commensal bacteria, as well as from pathogenic organisms. Immune cells in the intestine are intimately associated with the intestinal epithelium and are constantly challenged by these antigens. The immune system of the host has evolved a number of regulatory mechanisms to discriminate between these different stimuli, ensuring protection against infection, while avoiding strong inflammatory responses to harmless antigens. Abnormal function of these regulatory mechanisms can lead to uncontrolled immune responses to gut antigens, causing in some cases severe diseases such as inflammatory bowel disease (IBD). IBD is a spectrum of immune-mediated conditions in which the target organ is the intestine. IBD is divided into two primary forms, Crohn’s disease (CD) and ulcerative colitis (UC) [1]. Although the etiology remains incompletely understood, increasing evidence suggests that IBD is the result of a dysregulated mucosal inflammatory response to the intestinal flora [2]. One of the most potent effector cytokines in the pathogenesis of IBD is tumor necrosis factor (TNF), and blockade of TNF with anti-TNF antibodies is an effective treatment for a subset of CD patients [3]. Several TNF-related molecules have also been associated with IBD pathogenesis in humans [4]. One of these TNF-related cytokines is LIGHT (homologous to lymphotoxin, shows inducible expression and competes with herpes simplex virus (HSV) glycoprotein D for herpes virus entry mediator, a receptor expressed by t lymphocytes (note letters in bold), also called TNF-SF14. In this review, we discuss recent findings about LIGHT, its receptors, particularly herpes virus entry mediator (HVEM), other molecules LIGHT and HVEM interact with, and their roles in mucosal immunology and IBD pathogenesis.
The TNF superfamily of cytokines
The TNF superfamily (TNF-SF) of cytokines provides essential communication signals that participate in the orchestration of inflammatory and immune responses. By targeting specific cellular receptors, these cytokines initiate signaling pathways that affect cell death, survival, and differentiation. Some of these molecules also regulate the development, organization, and homeostasis of lymphoid tissues. Members of the TNF family of ligands associate with one or more specific cell surface receptors that form a corresponding family of cognate receptors. More than 20 distinct ligand–receptor systems are part of the TNF-SF. The TNF-SF of ligands shares a common structure as type II trans-membrane proteins that assemble as compact trimers, which can be membrane bound or soluble. Members of the TNF receptor superfamily (TNFR-SF) are type I transmembrane glycoproteins defined by a cysteine-rich (CR) motif in the ligand-binding ectodomain. TNF and lymphotoxin (LT) are the prototypic members of the TNF-SF of cytokines, and they share structural and functional properties that are representative of the larger family [5]. TNF, LTα, LTβ, along with LIGHT, define a core of four closely related ligands that with their four cognate receptors, TNFR1, TNFR2, lymphotoxin β receptor (LTβR) and the HVEM, form an integrated signaling network necessary for efficient innate and adaptive immune responses. This group of molecules is referred to as the immediate TNF family, reflecting their relatively high degree of amino acid sequence similarity. They have an overlapping pattern of ligand–receptor binding, which constitutes a striking feature of this group of molecules. Although the shared utilization of ligands and receptors suggest functional redundancy, studies with gene-knockout mice have revealed unique and cooperative roles for the different ligand–receptor pairs [6].
Two distinct structural forms of LT have been characterized, LTα and LTβ, which form soluble or membrane-bound trimeric molecules that engage to different cellular receptors. After specific cleavage, soluble homotrimeric LTα (LTα3) binds to TNFR1 or TNFR2, sharing both of these receptors with TNF. Some evidence indicates that LTα also binds to the HVEM (or TNFR-SF14), although the binding to HVEM is relatively weak. The most abundant form of LT, however, is a membrane form consisting of two LTβ chains and one LTα molecules (LTαβ2). The LTβ subunit in the LTαβ2 heterotrimer changes the receptor binding specificity so that LTβ engages only another receptor, the LTβ receptor LTβR or TNFR-SF3), with high affinity [6] (Fig. 1).

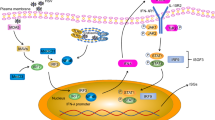
A schematic depiction of the known receptor–ligand interactions that involve HVEM, LIGHT, and their other binding partners or additional receptors is shown. Molecules are not drawn to scale. TNF proteins and their receptors tend to trimerize, as indicated, but the valency of the HVEM–BTLA interaction is monomeric. As noted in the text, DcR3 exists in humans but not in mice, the LTα3 interaction with HVEM is weak, and the in vivo significance of the trimolecular complex of LIGHT, BTLA, and HVEM has not been established
In the immediate TNF family, LIGHT is the fourth and most recently defined cytokine [7]. LIGHT binds to HVEM, and similar to LTαβ2, it also binds to the LTβR. In humans, there also is a soluble LIGHT-binding decoy receptor (DcR3) [8] (Fig. 1). Like many TNF superfamily ligands, LIGHT is predominantly a cell surface molecule, although evidence for a secreted form exists [9].
The pattern of expression of the immediate TNF family of ligands and receptors is summarized in Table 1. Activated T cells express LIGHT transiently, and LIGHT also is expressed by other cell types including immature dendritic cells (DC) and monocytes [10]. The two receptors for LIGHT have very different patterns of expression. The LTβR is expressed predominantly on stromal and epithelial cells [11–13], and although it is not expressed by lymphocytes, it is also found on some hematopoietic cells including DC and monocytes [6]. HVEM, by contrast, is expressed mostly by hematopoietic cells [14, 15], although there are reports of HVEM expression by other cell types including primary epithelial cells, breast cancer cell lines, and pancreatic β cells [16–18]. Most naive T cells constitutively express HVEM, but it becomes transiently downregulated following T-cell activation. This downregulation has been attributed to the ligation of HVEM with LIGHT [19]. In addition, HVEM expression can be detected on DC, macrophages, and monocytes [20].
BTLA: a new partner in the TNF/LT/LIGHT immediate family
Until recently, TNFR-SF members were thought to interact exclusively with TNF-SF ligands. However, HVEM is an exception, because it is able to bind to the herpes simplex virus type 1 glycoprotein D (HSV1 gD), which contains an immunoglobulin (Ig) homology domain [21]. More recently, interactions between HVEM and additional non-TNF-SF ligands were found. Unexpectedly, the immunoglobulin (Ig) homology domain-containing receptor known as the B- and T-lymphocyte attenuator (BTLA) binds to HVEM in both mice and humans [22, 23] (Fig. 1). BTLA is classified as a member of the B7 superfamily of co-stimulatory molecules. Although it shares relatively low sequence homology with the other proteins in this family [24], it is structurally most similar to the inhibitory B7 members, including the cytotoxic T-lymphocyte antigen-4 (CTLA-4 or CD152) and programmed death-1 (PD-1) [25]. BTLA is a type I transmembrane glycoprotein with a typical single IgV-like extracellular domain, a characteristic feature among all receptors for B7 family members, a transmembrane region and a cytoplasmic domain. Additionally, the BTLA intracellular region contains conserved tyrosine residues in two distinct sequence motifs. The tyrosine in the most membrane-proximal region is a potential binding site for growth-factor receptor-bound protein 2 (Grb2), whereas the other tyrosine residues are present in immune-receptor tyrosine-based inhibitory motif (ITIM) sequences [24]. Once engaged by HVEM, these ITIM motifs in the cytoplasmic tail of BTLA become tyrosine-phosphorylated, allowing the recruitment of the phosphatases SHP-1 and SHP-2 [24, 26]. BTLA is expressed mainly by lymphocytes and myeloid cells (Table 1), with particularly high expression by peripheral B cells. In addition, BTLA expression has also been detected in a subpopulation of CD11b+ macrophages and DX5+ natural killer (NK) cells [26, 27]. BTLA expression on T cells is upregulated upon T-cell receptor (TCR) stimulation, and it is selectively expressed by cells polarized to produce Th1 cytokines [26, 27].
Similar to HSV1 gD, the binding of BTLA to HVEM involves cysteine-rich-domain (CRD) 1, the most membrane-distal domain of HVEM [22]. LIGHT, by contrast, interacts with HVEM largely through CRD 2 and 3 [28]. Indeed, the binding of BTLA, but not LIGHT, to HVEM could be blocked by the addition of soluble HSV1 gD protein [23, 29]. Moreover, structural and functional studies of HVEM [30, 31] revealed that the binding site for LIGHT is topographically distinct from BTLA [22, 23, 29]. Based on the ability of these molecules to bind different CRDs on HVEM, and supported by co-crystal studies of the BTLA–HVEM interaction, it has been proposed that HVEM can interact simultaneously with BTLA and LIGHT [23, 29, 30] (Fig. 1). Experiments show that this is true, however, only for the soluble form of LIGHT, as membrane bound LIGHT non-competitively disrupts the HVEM–BTLA complex [29], suggesting it may act as a regulator of HVEM–BTLA inhibitory signaling [32]. Whereas these studies imply a role for a multimeric BTLA/HVEM/LIGHT complex, it remains to be determined if such a complex has an important function in vivo.
Recently, another Ig superfamily member, CD160, was identified as a third mammalian ligand able to bind HVEM [33] (Fig. 1). CD160 is a glycosylphosphatidylinositol (GPI)-anchored protein that contains a single IgV-like domain. It is expressed on the cell surface as a multimer [34]. CD160 is expressed mainly by T lymphocytes, natural killer T (NKT), and NK cells [34–37] (Table 1). Similar to BTLA and HSV1 gD, CD160 binds to the CRD 1 of HVEM [33]. A multimeric complex between CD160/HVEM/LIGHT also has been proposed, although in this case too, it is unknown if this structure exists in vivo [33]. Evidence from experiments performed with human CD4+ T cells suggested that the HVEM/CD160 interaction functions as a negative regulator of T-cell-mediated immune responses by interfering with TCR signaling pathways [33].
Signaling pathways in the immediate TNF family
The interaction of the immediate TNF family members, TNF, LT, and LIGHT, with their receptors constitutes a complex signaling network with multiple functions in the mucosal immune system. The concept of bidirectional signaling through TNF-TNFR family members has long been proposed, and a balance between forward and reverse signaling between ligands and receptors is tightly regulated in the immediate TNF family. As for the majority of the TNFR members, the LTβR and HVEM signaling pathways involve the recruitment of TNFR-associated factors (TRAF) and the subsequent activation of caspases and transcription factors (Fig. 2). Despite the general similarities between these two LIGHT receptors, the downstream signaling events triggered by LTβR and HVEM are distinct because of their differential expression patterns and their ability to associate with different signaling molecules [38]. LTβR signaling induces the activation of genes involved in pro-inflammatory responses, including MIP-1/2, adhesion molecules, and chemokines [39]. Furthermore, it has been recently described that LIGHT–LTβR signaling controls lipid homeostasis and is involved in dyslipidemia [40]. HVEM signaling controls the activation of genes important for T-cell proliferation, survival, and cytokine production [15]. In human monocytes and neutrophils, the LIGHT–HVEM interaction, but not LIGHT–LTβR, enhances antibacterial activity [41].

Signaling pathways downstream of HVEM, LIGHT, and their binding partners are shown. Hypothetical pathways are shown with a question mark. Signaling downstream of the LTβR activates the alternative NF-κB pathway that acts through the NF-κB inducing kinase (NIK)
Following LIGHT engagement, LTβR was shown to associate with, or form a signaling complex that includes TRAF2, TRAF3, and the inhibitor of κB (IκB)-kinase complex (IKKα/β; Fig. 2). Furthermore, the LIGHT–LTβR interaction mediates c-Jun N-terminal kinase (JNK), activator protein-1 (AP-1), and nuclear factor-κB (NF-κB) activation in a TRAF2-dependent fashion [42]. LIGHT–LTβR-mediated apoptosis seems to be dependent on TRAF3 but not TRAF2 [28] (Fig. 2). Engagement of LIGHT with HVEM also induces the recruitment of TRAFs, including TRAF2, and the activation of the NF-κB and JNK/AP-1 transcription factors, which are thought to contribute to T-cell survival in vivo [43, 44]. Similar to transmembrane TNF, there is evidence that the transmembrane form of LIGHT can mediate bidirectional signaling, which includes triggering of receptor trimerization (forward signaling), as well as acting as a receptor itself by transducing signals into LIGHT-expressing cells (reverse signaling) [45] (Fig. 2). Indeed, engagement of LIGHT by an immobilized DcR3:Ig fusion protein strongly co-stimulated T cells and enhanced cell proliferation, cytokine production, and cytotoxic activity in vitro [45, 46]. Furthermore, confocal microscopy analysis showed that LIGHT rapidly forms clusters and colocalizes with glycolipid-enriched rafts upon DcR3:Ig stimulation, and LIGHT cross-linking enhanced CD3-dependent p44/44 ERK activation [45]. The detailed mechanism as to how LIGHT-mediated signaling regulates downstream TCR signaling pathways is unknown. LIGHT has a relatively short intracellular domain, so the recruitment of signaling adaptors to LIGHT itself for signal transduction seems unlikely. Further investigation therefore will be required to determine the precise manner by which LIGHT reverse signaling regulates T-cell responses.
As mentioned above, BTLA is a co-inhibitory signaling molecule that binds to HVEM. It has been shown that upon TCR stimulation, BTLA was able to co-cluster with the CD3ζ chain at the immunological synapse, where in turn it could regulate molecules essential for T-cell signal transduction [47]. After engagement with HVEM, BTLA became tyrosine-phosphorylated and associated with both the TCR complex and the phosphatases SHP-1 and SHP-2, a characteristic shared by CTLA-4 and PD-1. BTLA also was found to associate with the B-cell receptor (BCR) and to recruit SHP-1 and SHP-2 in B cells, thereby attenuating B-cell activation by targeting the downstream signaling molecules Syk, BLNK, PLCγ2, as well as NF-κB [48]. Although at present the targets of SHP-1 and SHP-2 recruited to BTLA in T cells are unknown, by analogy with the events that occur in B lymphocytes, it is likely that they dephosphorylate signaling intermediates downstream of the TCR.
Interestingly, in recent work, we found that trans activation of HVEM signaling by BTLA induces TRAF2 recruitment and NF-κB activation, as do CD160 and HSV1 gD induced HVEM receptor trimerization [49] (Fig. 2). Furthermore, HVEM engagement by a BTLA:Ig fusion protein led to NF-κB activation and it promoted enhanced T-cell survival following TCR stimulation in vitro [49]. In light of these findings, the overall concept we have is that not only is there evidence that the LIGHT–HVEM axis may have bidirectional signaling, but the same is true for the BTLA–HVEM axis. Therefore, HVEM can exert both co-stimulatory and co-inhibitory functions depending on the nature of its ligands and its expression on different cell types.
LIGHT expression is related to IBD
LIGHT, HVEM, and BTLA each have been shown to play important roles in regulating diverse immune reactions. For example, LIGHT is involved in chronic allograft rejection, and HVEM is implicated in Concannavalin A-induced hepatitis and in experimental autoimmune encephalomyelitis (EAE), an animal model for multiple sclerosis. Mice deficient for BTLA spontaneously develop an autoimmune syndrome as they age. Given the critical importance of maintaining a balanced immune response in the intestinal mucosa, it is therefore not surprising that each of these molecules has been shown to have a role in the mucosal immune system as well.
Several lines of evidence point to LIGHT as an important mediator of mucosal inflammation and IBD pathogenesis [50–52]. The human LIGHT gene maps to chromosome 19p13.3, a region that has been implicated in the pathogenesis of Crohn’s disease [53]. Many other candidate genes map into this area, and specific evidence implicating LIGHT polymorphisms in IBD pathogenesis have not been reported yet. However, the ability of LIGHT to bind to HVEM on T cells and to enhance proliferation and cytokine production indicates that this cytokine can serve as a potent T cell co-stimulatory molecule [10, 46, 54, 55]. Moreover, high levels of LIGHT were detected in mature Th1 CD4+ T cells and mucosal T lymphocytes isolated from patients with IBD [50], and LIGHT expression was significantly increased in the intestine of active Crohn’s disease patients [52]. A role for LIGHT in IBD pathogenesis has also been suggested from data obtained from several experimental animal models for IBD, as outlined below.
LIGHT-transgenic mice spontaneously develop intestinal inflammation
Studies from our group [56] and from Wang et al. [57] demonstrated that transgenic mice constitutively expressing LIGHT on T cells spontaneously develop multi-organ inflammation, with particularly severe inflammation in the intestine. Overall, the phenotype of these LIGHT-transgenic animals is reminiscent of that from transgenic mice that overexpress TNF [58–61], suggesting that constitutive or increased LIGHT expression, predominantly if not exclusively by T cells, is highly pro-inflammatory. LIGHT expression in the transgenic animals generated by our group and by Wang et al. was driven for two different T-cell-specific promoters, the CD2 promoter and the proximal lck promoter, respectively. The spectrum and severity of autoimmune syndromes in the LIGHT-transgenic mice were somewhat different, which might reflect the different promoters used, differences in the amount expressed, the use of heterologous human LIGHT in one case [56], or differences in the mouse colonies. However, despite the use of different promoters, both LIGHT-transgenic mice showed multi-organ inflammation, demonstrating that unregulated LIGHT expression by T cells leads to severe inflammation and loss of peripheral tolerance. In both types of LIGHT-transgenic mice, the small and large intestine had signs of chronic inflammation with substantial mononuclear cell infiltrates, loss of goblets cells, distortion and hyperplasia of crypts, and villus atrophy [56, 57]. In the LIGHT-transgenic mice generated by our group, LIGHT expression was detected mainly in T lymphocytes, including thymocytes and peripheral T cells. A fraction of T cells from peripheral lymph nodes (PLN) and mesenteric lymph nodes (MLN) were highly positive for human LIGHT (hLIGHT) expression. The percentage of T cells expressing hLIGHT in the spleen of transgenic mice, however, was greatly reduced and not significantly different from the endogenous LIGHT expressed by control splenocytes. The greatest percentage of cells expressing high levels of hLIGHT was observed in the intraepithelial lymphocytes (IEL) and lamina propria lymphocytes from the small and large intestine of the transgenic mice [56]. Analysis of different T-cell subsets in the intestine of these animals revealed decreased CD8αα-expressing (IEL), including TCRγδ+ cells and TCRαβ+ cells that express CD8αα exclusively, and increased populations of conventional, activated, or memory-type TCRαβ+ CD4+ and CD8αβ+ T cells. This type of population shift, toward the more conventional, peripheral T cells, and away from the IEL-specific CD8αα expressing lymphocytes, is characteristic of a number of inflammatory models. In the transgenic animals generated by Wang et al. [57], mouse LIGHT expression on T cells was higher in the spleen and PLN when compared to wild-type (WT) mice, although in this study the percentage of LIGHT-expressing cells in the gut of the transgenic mice was not assessed [57, 62]. Furthermore, activated T cells in the MLN, but not in the PLN [4], expressed higher amounts of the α4β7 integrin, which is involved in mucosal homing [63]. Interestingly, the expression of the vascular addressin MAdCAM-1, the binding partner for the α4β7 integrin, was dramatically enhanced in the colon of LIGHT-transgenic mice [62]. High levels of α4β7 expression allow T cells to efficiently attach to MAdCAM-1 that is expressed by intestinal microvessels [64]. Therefore, by upregulating gut homing molecules on T cells and counter-adhesion receptors on intestinal endothelial cells, LIGHT promoted the accumulation of activated T cells in the intestine of the transgenic mice, contributing to severe inflammation. Although the manner by which LIGHT promotes intestinal inflammation remains to be determined, there is evidence that LIGHT-mediated IBD pathogenesis in mice involves its interaction with both LTβR and HVEM [52].
Involvement of LT and the LTβR in intestinal inflammation
The LTβR is part of a complex communication system linking lymphocytes and surrounding parenchymal and stromal cells. Multiple phenotypes affecting the structure and function of the immune system are associated with the disruption of the LTαβ2/LTβR system (reviewed in [6] and [65]). During embryonic development, engagement of the LTβR is critical for the formation of lymphoid tissues. LTα−/− and LTβR−/− mice lack Peyer’s patches (PP), MLN, and PLN, and they have disrupted splenic microarchitecture [66–68]. LTβ−/− mice, however, lack PLN and PP but retain MLN [69], suggesting that in these mice, a different LTβR ligand may contribute to the development of these gut-related lymphoid structures. It is possible that LIGHT could exert such a lymphoid-structural effect through the LTβR signaling pathway. In agreement with this, the simultaneous ablation of LIGHT and LTβ led to the absence of MLN, recapitulating the phenotype observed in LTβR−/− mice [70]. More evidence for a role for LIGHT in organogenesis was obtained in complementation experiments, where transgenic LIGHT expression in LTα−/− mice restored some of the splenic architecture deficiencies in these animals [71].
Besides its role in the organogenesis of lymphoid structures, engagement of the LTβR by LIGHT is associated with inflammatory responses in the intestine. The first evidence for a role for the LIGHT/LTβR interaction in intestinal inflammation came from studies performed by Mackay and colleagues [72]. This was accomplished using two different mouse colitis models, consisting of either the transfer of CD4+ CD45RBhigh T cells into immune-deficient hosts, or the transfer of WT bone marrow into T-cell-deficient Tgε26 recipients. The authors demonstrated that treatment of the recipient mice with a soluble LTβR decoy, in the form of an LTβR–Ig fusion protein, blocked disease development in either model. In a mouse model in which colitis is induced by the intrarectal administration of trinitrobenzene sulfonic acid, disease was also attenuated by the chimeric LTβR decoy protein [73]. Similar observations were obtained by Stofer et al. in mice with chronic colitis induced by dextran sodium sulfate (DSS) [74], a model of pathogenesis that is not strictly dependent on the action of B or T lymphocytes. In this model, where inflammation is caused by epithelial damage and propagated by innate immunity, animals treated with the LTβR–Ig had reduced inflammatory cytokines in the intestine and lower expression of MAdCAM-1. Although these data from the different colitis models suggest that the LTβR is critical for colitis pathogenesis, it remains unknown which LTβR ligand participates in the inflammatory process. The LTβR decoy could prevent colitis by blocking any or more of several types of interactions, including those of LIGHT and/or LT with their respective receptors. Because LT and LIGHT are induced transiently on activated T cells under Th1 conditions, the LTβR decoy will selectively bind to these activated lymphocytes. Therefore, it also remains possible that the LTβR:Ig fusion protein leads to the selective elimination of the pathogenic T cells by the reticuloendothelial system or by complement. Although such a depletion has not been reported, the control frequently used for these experiments, the use of an “empty” Ig Fc, does not adequately control for this possibility.
Opposing effects of LTβR signals in acute and chronic disease models
In acute colitis models induced by chemical treatment or bacterial infection, it is paradoxical that interruption of LTβR signaling increased rather than decreased disease symptoms. For example, a recent report by Jungbeck et al. showed that LTβR–Ig treatment exacerbated acute DSS-induced colitis and secretion of high levels of pro-inflammatory cytokines, including TNF, IL6 and IFNγ, by LN cells from the treated mice [75]. Furthermore, exacerbated acute DSS-induced colitis was also observed in LTβR−/−, LTαβ−/−, and T-cell-specific LTβ-deficient (T-LTβ−/−) mice [75]. Surprisingly, B-cell-specific LTβ-deficient (B-LTβ−/−) mice had an opposite phenotype with reduced signs of intestinal inflammation [75]. A common feature of the LTβR−/−, LTβ−/−, and LTα−/− mice is the absence of secondary lymphoid structures, while B-LTβ−/− mice have relatively normal LN and PP, although their splenic microarchitecture is abnormal [76]. The absence of PP and MLN may lead to disruption of the “dialogues” between T and B lymphocytes with regulatory T cells and/or regulatory mucosal DC, ultimately resulting in the differentiation of abnormal effector T cells. Previous reports also showed exacerbated acute DSS-induced disease not only in mice lacking PP and LN due to gene disruption, but also in mice treated in utero with an LTβR:Ig [65, 77]. Furthermore, acute colitis induced by the enteropathogenic bacteria Citrobacter rodentium, was also enhanced in LTβR−/−, LTβ−/−, and LTα−/− mice, and in LTβR:Ig-treated mice. In this infection-induced acute colitis model, mice with disrupted LT–LTβR interactions had increased disease-related mortality, more severe weight loss, intestinal bacterial abscesses, and a higher burden of bacteria in the spleen and liver [78].
While exacerbated colitis in LTβR−/−, LTβ−/− and LTα−/− animals may be related to the absence of secondary lymphoid structures, this cannot explain the divergent outcomes comparing B-LTβ−/− and T-LTβ−/− mice. It remains possible therefore that an anti-inflammatory interaction between LTαβ and LTβR prevents exacerbated colitis in the acute models, perhaps with counterbalancing anti-inflammatory and pro-inflammatory functions for LTαβ when expressed by T or B lymphocytes. In different experimental settings, the relative expression of LIGHT and LTαβ, or LTαβ by different cell types, may vary. It could be speculated that in the chronic colitis, LTβR:Ig treatment prevents disease development by targeting principally LIGHT and B-cell-expressed LTαβ, while it may preferentially target T-cell-expressed LTαβ in the acute model. The contrasting effects of the LTβR:Ig treatment in the acute and chronic DSS colitis models are intriguing, and further investigation remains to be done to determine the precise mechanisms by which blockade of the LT/LIGHT/LTβR pathway can lead to reduced or exacerbated colitis in these experimental situations.
LIGHT–LTβR interactions in mucosal immunity and colitis pathogenesis
A direct demonstration of a role for the LIGHT/LTβR interaction in promoting intestinal inflammation comes from experiments performed by Wang et al. [52]. As mentioned earlier, LIGHT-transgenic mice spontaneously developed severe intestinal inflammation. When these transgenic animals were crossed with LTβR−/− mice, however, the resulting LIGHT-transgenic/LTβR−/− mice did not develop intestinal inflammation [52]. Furthermore, severe colitis induced by the adoptive transfer of LIGHT-transgenic MLN cells into lymphopenic hosts did not occur in hosts lacking LTβR expression on stromal cells [52]. LTβR−/− mice have severe defects in MLN and PP development, and therefore the absence of these lymphoid structures could affect the function of colitogenic T cells in disease development. Thus, a developmental defect in LTβR−/− mice rather that a defect in the LIGHT/LTβR signaling pathway in pathogenic cells could lead to the absence of disease in LIGHT-transgenic/LTβR−/− animals. However, LTα−/− mice that had severe defects in LN development, similar to those observed in LTβR−/− mice, developed colitis following the transfer of LIGHT-transgenic MLN cells [52]. These data demonstrate that the absence of disease in LTβR−/− mice is not related to defects in the development of MLN and PP but to the disruption of inflammatory signals triggered by the binding of LIGHT to LTβR.
Although the precise manner by which the LIGHT/LTβR interaction promotes intestinal inflammation remains unknown, it has been proposed that the binding of LIGHT to the LTβR induces the transformation of the gut mucosa to promote the formation of pathological lymphoid structures [52]. As mentioned above, the expression of α4β7 in T cells and MAdCAM-1 in colonic endothelial cells was dramatically enhanced in LIGHT-transgenic mice [4, 62]; moreover, in the chronic DSS colitis model, animals treated with the LTβR:Ig had reduced inflammatory cytokines in the gut and lower expression of MAdCAM-1 [74]. The ability of LIGHT to induce expression of chemokines and adhesion molecules may lead to the transformation of the local chemokine milieu, allowing for the formation of expanded isolated lymphoid follicles and colonic patches, as well as the recruitment of inflammatory leukocytes into the gut mucosa. DC content in the colonic patches in the inflamed gut also was reduced by LTβR:Ig treatment [73]. By imprinting T cells for gut tropism, mucosal DC can promote the recruitment of inflammatory leukocytes to the intestine. Therefore, the LIGHT/LTβR interaction may allow for the permanence in the gut-associated lymphoid tissue of mucosal DC that are required for the differentiation of gut-homing effector T cells. In addition, the LIGHT/LTβR interaction may also promote the recruitment to the intestine of DC that, once in the inflamed tissue, may produce pro-inflammatory cytokines.
Another plausible mechanism by which the LIGHT/LTβR interaction promotes intestinal inflammation comes from recent reports involving LIGHT in the regulation of the epithelial barrier function [17, 79]. LIGHT engagement of the LTβR, but not HVEM, induced the activation of intestinal epithelial myosin II regulatory light chain kinase and it stimulated occludin endocytosis through a caveolar pathway, both of which contribute to the loss of barrier function [17]. This effect of LIGHT required IFNγ-dependent upregulation of LTβR expression on intestinal epithelial cells. Therefore, similar to TNF, LIGHT can act synergistically with IFNγ to cause barrier dysfunction. LIGHT is principally expressed by activated T cells, and it has been shown that LIGHT enhanced IFNγ release from T cells isolated from IBD patients [50, 52]. Therefore, LIGHT may promote inflammation acting at two different levels; initially by regulating production of IFNγ that, in turn, will upregulate LTβR and TNFR2 expression on epithelial cells.
The LIGHT/LTβR interaction also has been associated with dysregulated production of IgA in the intestine [56, 57, 62, 80]. IgA is produced abundantly in mammals and it plays an important role in the defense against microorganisms at mucosal surfaces. This Ig isotype is synthesized by IgA-committed B cells in both PP and intestinal lamina propria [62]. It has been shown that constitutive expression of LIGHT on T cells induced a selective increase in the number of lamina propria B cells along with increases in serum IgA [56, 57]. Furthermore, LIGHT transgene expression by T cells led to glomerular IgA deposition in the kidneys of the transgenic animals, indicating that dysregulated LIGHT expression contributes to IgA nephropathy [62]. Interestingly, IBD patients with active inflammation in the intestine present with elevated serum IgA and significantly increased numbers of IgA-producing cells in the gut [62]. A role for the LTβR in the production of IgA has been described, as LTβR−/− mice display extremely low levels of baseline IgA in serum and fecal extracts [80]. These data suggest that LIGHT expressed by T cells may interact with LTβR on gut stromal cells, promoting recruitment of IgA-committed precursors and IgA production.
LIGHT–HVEM interactions in colitis pathogenesis
Although LIGHT can promote inflammation by signaling through the LTβR, the LIGHT/HVEM interaction can promote T-cell-mediated inflammatory responses. Indeed, engagement of HVEM by LIGHT delivers co-stimulatory signals leading to T-cell activation [15, 54, 55, 81] and enhanced production of pro-inflammatory cytokines [14, 56, 57, 62, 82].
It has been described that engagement of HVEM by LIGHT induced activation of human mucosal T cells, contributing to IBD pathogenesis [50]. In human T cells, LIGHT expression was detected in memory CD45RO CD4+ T lymphocytes and in IFNγ-producing CD4+ T cells [50]. Kinetic analyses of LIGHT expression revealed a rapid induction in lamina propria T cells compared to a slow upregulation in T cells from lymph nodes or peripheral blood. Stimulation of HVEM by recombinant LIGHT induced IFNγ production by lamina propria T cells, and blocking LIGHT with a specific anti-human LIGHT antibody inhibited CD2-dependent induction of IFNγ expression [50]. Therefore, the LIGHT/HVEM pathway acts synergistically with the gut-associated CD2 stimulation pathway to enhance IFNγ production by mucosal T cells.
As mentioned above, the adoptive transfer of LIGHT-transgenic MLN cells into Rag −/− mice induced a consistent, rapid, and synchronized intestinal inflammation [52]. Using this MLN cell transfer model, Wang et al. demonstrated that the transfer of LIGHT-transgenic/Hvem −/− MLN cells failed to induce full development of disease in Rag −/− recipients [52]. Furthermore, when LIGHT-transgenic/Hvem −/− T cells were compared with LIGHT-transgenic HVEM wild-type T lymphocytes, the HVEM-deficient cells showed a significant reduction in the upregulation of expression of activation markers [52]. These results indicate that HVEM expression by T cells is necessary for optimal LIGHT-mediated activation and expansion. Therefore, T-cell-derived LIGHT interacting with HVEM expressed by the same T lymphocyte, or by another T cell, may be required for the full differentiation of gut-homing effector T cells. This is consistent with other evidence suggesting a potential for T cell–T cell interactions between LIGHT and HVEM [50, 83].
In the CD4+CD45RBhigh T-cell transfer model of colitis, HVEM expression on T cells also contributed to intestinal inflammation, but only to a limited extent [16]. Although Rag −/− recipient mice transferred with Hvem −/− T cells unquestionably became ill, the animals had lower mononuclear cell infiltrates in the proximal and distal colons. In addition, these recipients had a higher number of goblet cells in the distal colon, indicating that inflammation in these animals was milder than that observed in animals transferred with WT T cells. We found similar results when Rag −/− mice were transferred with Light −/− CD4+CD45RBhigh T cells. Therefore, while T-cell expression of LIGHT and HVEM are not required for colitis pathogenesis, the increased disease when the donor T cells can express both HVEM and LIGHT is consistent with a co-stimulatory or inflammation-promoting role for LIGHT and HVEM when both molecules are expressed by T lymphocytes.
Besides a role for the LIGHT/HVEM interaction in T-cell activation and T-cell-mediated inflammation, LIGHT and HVEM also are involved with the activation of innate immune cells [41, 84–86]. Indeed, LIGHT activated NK cell proliferation and cytokine production in a predominantly HVEM-dependent manner [86]. Furthermore, in an in vitro model of DC maturation, LIGHT induced the maturation of human DC in synergism with CD40 ligation [85]. Specifically, LIGHT induced expression of MHC class II, CD80, and CD86 and reduced macropinocytosis and increased cytokine synthesis. Additionally, it has been shown that LIGHT can bind to HVEM expressed on human monocytes and neutrophils [41]. HVEM, but not LTβR, is highly expressed on monocytes and neutrophils isolated from peripheral blood, and LIGHT can activate these cells and enhance effector functions of monocytes and neutrophils. Engagement of HVEM on these cells increased the bactericidal activity against Listeria monocytogenes and Staphylococcus aureus. Furthermore, LIGHT–HVEM signaling in monocytes and neutrophils enhanced the production of IL-8, TNF, nitric oxide, and reactive oxygen species [41]. In summary, this evidence indicates that the LIGHT–HVEM interaction can activate subsets of innate immune cells.
HVEM–BTLA interactions attenuate intestinal inflammation
A growing literature indicates that HVEM, in addition to its activity as a TNF receptor, can trigger inhibitory signals by acting as a ligand that binds to BTLA. Studies have shown that HVEM-deficient as well as BTLA-deficient T cells are hyper-responsive to TCR-induced stimulation in vitro [24, 26, 87]. Additionally, both HVEM and BTLA-deficient mice exhibit a higher susceptibility to the induction of EAE [24, 87], while the loss of BTLA dramatically accelerated partially MHC-mismatched cardiac allograft rejection [88] and prolonged airway inflammation [89]. Furthermore, the BTLA–HVEM interaction limited T-cell activity in vivo, negatively regulating the homeostatic expansion of a subset of CD8+ memory T cells [90]. A recent report also suggested an inhibitory role for HVEM expressed on regulatory T cells in the suppression of proliferation and function of BTLA-expressing effector T cells [91]. Despite its generally inhibitory role, BTLA has recently been reported to play a role in promoting the survival of activated T cells in a mouse model of graft versus host disease [92] and the survival of T cells in the lung in a mouse allergic asthma model [93]. Whereas most of the evidence is consistent with the hypothesis that HVEM acts a negative regulator of T-cell responses by triggering BTLA-derived inhibitory signals, the actual contribution of HVEM and BTLA to T-cell-mediated immune responses in vivo, and to the pathogenesis of T-cell-mediated inflammatory conditions such as IBD, has not been fully investigated.
During the past years, we have extensively investigated the role of HVEM and its ligands in the development of colitis induced by the transfer of CD4+CD45RBhigh T cells into Rag −/− mice. This experimental system is well suited to the analysis of complex networks of receptor–ligand interactions, because disease development is synchronous and the genotypes of the donor T cells and the immune deficient host can be independently varied. Although as noted the absence of HVEM in the donor CD4+CD45RBhigh T lymphocytes slightly reduced the severity of colitis development, an opposite effect was observed for HVEM expression by cells in the RAG-deficient hosts. Strikingly, we found that the transfer of WT CD4+CD45RBhigh T cells into Hvem −/− Rag −/− mice led to a dramatic acceleration of intestinal inflammation and colitis [16]. Clinical signs in Hvem-deficient Rag −/− animals correlated with an extremely fast weight loss and elevated histological scores in the colon. Colitis acceleration in Hvem −/− Rag −/− recipient mice was characterized by a rapid accumulation of activated T cells producing Th1 cytokines and IL-17 in the large intestine of these recipients. Therefore, colitis acceleration in Hvem −/− Rag −/− mice was probably induced by an enhanced pro-inflammatory response mediated by CD4+ T cells in the large intestine. When either Light −/− or Hvem −/− CD4+CD45RBhigh T cells were transferred into Hvem −/− Rag −/− animals, the disease-accelerating effects of the absence of HVEM in the Rag −/− hosts were dominant over the more subtle disease-delaying effects when these molecules were not expressed by the gene-deficient donor T cells. Therefore, the predominant effect of HVEM in this colitis model system is anti-inflammatory, and it is related to its expression by cells other than lymphocytes present in the Rag −/− hosts.
Mapping the cell types involved in HVEM–BTLA interactions
Because BTLA is an inhibitory receptor, it was a good candidate for mediating the anti-inflammatory effect of HVEM expression in the Rag −/− recipients. Consistent with previous reports [27], we detected BTLA expression on naive and activated CD4+ T lymphocytes. Likewise, we found BTLA expression on CD4+ T cells isolated from spleen, MLN, and lamina propria of Rag −/− recipients of CD4+CD45RBhigh T cells. Therefore, we speculated that the interaction between Rag −/− host HVEM and T-cell BTLA could alter the acceleration of colitis. Consistent with this hypothesis, treatment of Hvem −/− Rag −/− recipients of CD4+CD45RBhigh T cells with an anti-BTLA mAb with agonistic properties was able to reverse the accelerated colitis, but only when the T cells expressed BTLA. Hvem −/− Rag −/− mice transferred with Btla −/− T cells developed a fast disease that was not affected by treatment with the agonistic anti-BTLA mAb. While our data clearly demonstrate that a major function of the anti-BTLA mAb is to attenuate T-cell responses by reacting with T lymphocytes, BTLA expression by host cells is also relevant. This was demonstrated by the results from transfer of WT T cells into BTLA-deficient Rag −/− recipients, which also led to accelerated colitis. Therefore, T-cell expression of BTLA is necessary but not sufficient for preventing accelerated disease, and there must be a non-redundant function for BTLA expression by non-lymphoid cells of the innate immune system in preventing colitis acceleration. BTLA expression has been detected on myeloid cells, including CD11c+ DC and a subpopulation of CD11b+ macrophages, and also by DX5+ NK cells [26, 27]. Therefore, engagement of BTLA on one or more of these innate immune cell types could deliver inhibitory signals, thereby attenuating their functions and colitis pathogenesis. Contrary to the situation in Hvem −/− Rag −/− recipients, in the transfer of WT CD4+CD45RBhigh T cells to Btla −/− Rag −/− animals, there is no defect in the signaling of host-expressed HVEM to T cell BTLA, and therefore a somewhat different mechanism, perhaps more dependent on alterations in the behavior of innate host cells, could be operating to accelerate disease in the Btla −/− Rag −/− recipients. Conditional ablation of BTLA expression in different cell types in Rag −/− mice with a floxed Btla allele ultimately will be required to identify the cell type(s) that must express this protein to prevent disease acceleration.
Contrary to our expectations, the transfer of Btla −/− CD4+CD45RBhigh T cells into Rag −/− mice did not replicate the severe disease phenotype in the Hvem −/− Rag −/− recipients, despite the finding that the agonistic anti-BTLA mAb must in part engage BTLA expressed by T cells. Surprisingly, we found that there was a significant reduction in the number of T cells in different lymphoid organs and in the large intestine following transfer of Btla −/− CD4+ T cells when compared to transfer of WT CD4+ T cells. In accordance with this finding, it has recently been shown that under conditions of chronic stimulation, Btla −/− T cells did not survive as well [92, 93]. Therefore, decreased T-cell survival could partially explain the absence of a dramatic colitis acceleration following transfer of Btla −/− T cells into Rag −/− mice. It should be noted that although BTLA deficiency in donor T cells negatively affected T-cell accumulation, the transfer of Btla −/− T cells into Hvem −/− Rag −/− mice led to accelerated colitis. We speculate that accelerated disease can be induced even by reduced numbers of effector T cells when BTLA signaling in innate immune cells also is absent. In Rag −/− mice transferred with Btla −/− T cells, HVEM can still induce inhibitory signals by interacting with BTLA expressed by host cells. However, when the stromal cells cannot deliver the critical anti-inflammatory HVEM-mediated signals to either the transferred T cells or to Rag −/− host cells, as in the case in Hvem −/− Rag −/− recipients, then accelerated disease occurs, even following transfer of Btla −/− CD4+ T cells that do not survive as well as wild CD4+ T cells.
To identify the crucial host cell type that must express HVEM to prevent accelerated disease, we created bone marrow chimeric Rag −/− recipients, by using Hvem −/− Rag −/− bone marrow to reconstitute irradiated Rag −/− mice (Hvem −/− Rag −/− → Rag −/−) or by constructing the reverse chimeras (Rag −/− → Hvem −/− Rag −/−). After reconstitution, the chimeras were used as hosts for CD4+CD45RBhigh T cells. In this way, we showed that HVEM expression by an irradiation-resistant host cell is required to prevent accelerated and severe colitis. Interestingly, we also found that colonic epithelial cells (CEC) constitutively express high levels of HVEM [16]. Not only do intestinal epithelial cells provide a point of contact for enteric antigens, but they play a direct role in mucosal immunity, particularly by regulating T-cell responses to enteric antigens [94]. Thus, expression of HVEM by host CEC could be essential for engagement of BTLA on T cells to avoid an exaggerated T-cell activation. It remains possible, however, that some other radio-resistant, HVEM-expressing cell type is responsible for preventing exacerbated T-cell responses leading to accelerated colitis. There are several possible candidates, including a recently identified, radio-resistant, CD70-expressing, tissue-specific APC, which was required for the proliferation of mucosal T cells after oral infection [95]. Similarly, engagement by HVEM of BTLA on mucosal DC or macrophages, cell types that participate in the inflammatory responses leading to colitis development in different mouse colitis models [96, 97], could regulate innate cell activation and cytokine production. Whereas most of the studies that investigate the role of BTLA binding to its ligand have emphasized the expression of these molecules by B and T lymphocytes, and their roles in directly regulating lymphocyte responses, the effect of engaging BTLA expressed on myeloid lineages has not been well characterized.
Can BTLA binding to HVEM lead to HVEM-mediated signal transduction events opposite from the suppressive effects mediated by BTLA signaling? There was some prior evidence suggesting this could be the case [22], but recently we have shown unambiguously that BTLA delivers reverse signals to HVEM [49]. Indeed, BTLA binding to HVEM strongly induced HVEM trimerization and activation of the NF-κB protein RelA in a TRAF2-dependent fashion [49]. Because the NF-κB pathway is related to cell survival, these results suggest that in certain situations BTLA can act as a cell survival factor by regulating HVEM-dependent activation of RelA. In view of these findings, we speculate that colitis acceleration in Btla −/− Rag −/− and Hvem −/− Rag −/− mice could be caused, at least in part, by a reduction in NF-κB signaling and survival of innate regulatory cells, such as mucosal CD103+DC or CD11b+ macrophages. Several groups have recently demonstrated that mucosal DC have unique regulatory functions, including the capacity to produce anti-inflammatory mediators such us IL-10, TGF-β, and/or retinoic acid (RA) [40, 98, 99]. Mucosal DC are especially capable of releasing RA [100] and efficiently converting naive CD4+ T cells into Foxp3+ regulatory T cells in a TGF-β-dependent fashion [40]. A subset of LP CD11b+ macrophages also have been described as potent regulatory APC that are able to convert naive T lymphocytes into Foxp3+ regulatory T cells in an IL-10-, RA- and TGF-β-dependent manner [101]. Interestingly, a recent report showed that BTLA and HVEM expression on splenic DC is required for the maintenance of the normal homeostasis of a subset of CD11c+ CD8α− cells [20]. It remains unknown, however, if this regulatory function of BTLA and HVEM is also required for mucosal DC homeostasis. Because HVEM is highly expressed on mouse intestinal epithelial cells, one could propose that reduced HVEM-dependent NF-κB signaling in epithelial cells could lead to survival defects that ultimately could affect the normal homeostasis and function of the epithelium. Nevertheless, epithelial permeability was not altered in Hvem −/− mice, and these mice were not more susceptible to DSS colitis [16]. Therefore, it seems that the absence of HVEM expression does not by itself alter the physiological function of intestinal epithelial cells, but it might when epithelial HVEM deficiency is combined with T-cell activation and pro-inflammatory cytokine production. Therefore, while epithelial cell expression of HVEM is intriguing, it remains to be proven that HVEM expression by IEC is required to prevent colitis acceleration.
Conclusions
The mucosal immune system faces a special problem in maintaining homeostasis in the face of constant antigenic challenge. Many molecules play a role in maintaining this homeostasis, but prominent among them is a TNF superfamily member LIGHT, and HVEM, one of its two receptors. Receptor–ligand interactions in the TNF superfamily are often non-monogamous, and HVEM and LIGHT have other partners. Together, HVEM and LIGHT therefore are located at the center of a network of receptor–ligand interactions that play an important role in regulating immune responses in the intestine. LIGHT–HVEM binding, and LIGHT interaction with its other receptor, the LTβR, are mostly involved in pro-inflammatory signaling. The involvement of LIGHT in mucosal immune regulation is more complex, however, than a single-minded role as a ligand inducing pro-inflammatory responses. In addition to acting as a ligand, LIGHT may be capable of acting as a receptor that transmits reverse signals. Furthermore, there is evidence, although still incomplete, that LIGHT-induced signaling can play an anti-inflammatory role, through the LTβR in acute colitis models, or as a soluble molecule that may enhance HVEM interactions with BTLA.
HVEM, like LIGHT, probably can serve dual roles as a ligand as well as acting as TNF superfamily receptor by binding to LIGHT. Its unique attribute as a TNF receptor is its ability to bind to Ig superfamily molecules encoded by viral as well as mammalian genomes. The interactions with the Ig family member BTLA are the best characterized in terms of effects on immune regulation. HVEM clearly can either enhance or prevent inflammation, depending on the receptor and cell type it interacts with. Whereas its expression on T lymphocytes is mainly pro-inflammatory by acting as a receptor for LIGHT, HVEM expression on non-lymphoid cells seems to be principally anti-inflammatory via interactions with BTLA on T cells and non-lymphoid cells. Importantly, the inhibitory effect of HVEM is dominant over its inflammatory function in the context of the colitis model we have studied.
Future work will pinpoint the important cell types involved in mediating the crucial molecular interactions between LIGHT, HVEM, and BTLA that enhance or prevent inflammation in the intestine. They also will sort out the complications related to cis versus trans signaling and reverse signaling by ligands that also act as receptors. This sorting out is a pre-requisite for the ultimate goal of studying the crucial signal transduction events acting downstream of these ligand–receptor pairs. Ultimately, these types of interactions must be investigated using human cells, in order to determine their relevance for IBD patients. The results from studies on LIGHT using human mucosal T lymphocytes encourage our belief that the molecular interactions described in this article will help in understanding at least some forms of IBD in humans.
References
Bouma G, Strober W (2003) The immunological and genetic basis of inflammatory bowel disease. Nat Rev Immunol 3(7):521–533
Sartor RB (2003) Innate immunity in the pathogenesis and therapy of IBD. J Gastroenterol 38(Suppl 15):43–47
Targan SR (2000) Biology of inflammation in Crohn’s disease: mechanisms of action of anti-TNF-a therapy. Can J Gastroenterol 14(Suppl C):13C–16C
Wang J, Fu YX (2005) Tumor necrosis factor family members and inflammatory bowel disease. Immunol Rev 204:144–155
Ware CF (2005) Network communications: lymphotoxins, LIGHT, and TNF. Annu Rev Immunol 23:787–819
Schneider K, Potter KG, Ware CF (2004) Lymphotoxin and LIGHT signaling pathways and target genes. Immunol Rev 202:49–66
Mauri D (1998) LIGHT, a new member of the TNF superfamily, and lymphotoxin α are ligands for herpesvirus entry mediator. Immunity 8:21–30
Locksley RM, Killeen N, Lenardo MJ (2001) The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell 104(4):487–501
Granger SW, Butrovich KD, Houshmand P, Edwards WR, Ware CF (2001) Genomic characterization of LIGHT reveals linkage to an immune response locus on chromosome 19p13.3 and distinct isoforms generated by alternate splicing or proteolysis. J Immunol 167(9):5122–5128
Gommerman JL, Browning JL (2003) Lymphotoxin/light, lymphoid microenvironments and autoimmune disease. Nat Rev Immunol 3(8):642–655
Endres R, Alimzhanov MB, Plitz T et al (1999) Mature follicular dendritic cell networks depend on expression of lymphotoxin beta receptor by radioresistant stromal cells and of lymphotoxin beta and tumor necrosis factor by B cells. J Exp Med 189(1):159–168
Browning JL, French LE (2002) Visualization of lymphotoxin-beta and lymphotoxin-beta receptor expression in mouse embryos. J Immunol 168(10):5079–5087
Murphy M, Walter BN, Pike-Nobile L et al (1998) Expression of the lymphotoxin beta receptor on follicular stromal cells in human lymphoid tissues. Cell Death Differ 5(6):497–505
Harrop JA, Reddy M, Dede K et al (1998) Antibodies to TR2 (herpesvirus entry mediator), a new member of the TNF receptor superfamily, block T cell proliferation, expression of activation markers, and production of cytokines. J Immunol 161(4):1786–1794
Harrop JA, McDonnell PC, Brigham-Burke M et al (1998) Herpesvirus entry mediator ligand (HVEM-L), a novel ligand for HVEM/TR2, stimulates proliferation of T cells and inhibits HT29 cell growth. J Biol Chem 273(42):27548–27556
Steinberg MW, Turovskaya O, Shaikh RB et al (2008) A crucial role for HVEM and BTLA in preventing intestinal inflammation. J Exp Med 205(6):1463–1476
Schwarz BT, Wang F, Shen L et al (2007) LIGHT signals directly to intestinal epithelia to cause barrier dysfunction via cytoskeletal and endocytic mechanisms. Gastroenterology 132(7):2383–2394
Pakala SV, Ilic A, Chen L, Sarvetnick N (2001) TNF-alpha receptor 1 (p55) on islets is necessary for the expression of LIGHT on diabetogenic T cells. Clin Immunol 100(2):198–207
Morel Y, Schiano de Colella JM, Harrop J et al (2000) Reciprocal expression of the TNF family receptor herpes virus entry mediator and its ligand LIGHT on activated T cells: LIGHT down-regulates its own receptor. J Immunol 165(8):4397–4404
De Trez C, Schneider K, Potter K et al (2008) The inhibitory HVEM–BTLA pathway counter regulates lymphotoxin receptor signaling to achieve homeostasis of dendritic cells. J Immunol 180(1):238–248
Montgomery RI, Warner MS, Lum BJ, Spear PG (1996) Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell 87(3):427–436
Sedy JR, Gavrieli M, Potter KG et al (2005) B and T lymphocyte attenuator regulates T cell activation through interaction with herpesvirus entry mediator. Nat Immunol 6(1):90–98
Gonzalez LC, Loyet KM, Calemine-Fenaux J et al (2005) A coreceptor interaction between the CD28 and TNF receptor family members B and T lymphocyte attenuator and herpesvirus entry mediator. Proc Natl Acad Sci USA 102(4):1116–1121
Watanabe N, Gavrieli M, Sedy JR et al (2003) BTLA is a lymphocyte inhibitory receptor with similarities to CTLA-4 and PD-1. Nat Immunol 4(7):670–679
Carter LL, Carreno BM (2003) Cytotoxic T-lymphocyte antigen-4 and programmed death-1 function as negative regulators of lymphocyte activation. Immunol Res 28(1):49–59
Han P, Goularte OD, Rufner K, Wilkinson B, Kaye J (2004) An inhibitory Ig superfamily protein expressed by lymphocytes and APCs is also an early marker of thymocyte positive selection. J Immunol 172(10):5931–5939
Hurchla MA, Sedy JR, Gavrieli M et al (2005) B and T lymphocyte attenuator exhibits structural and expression polymorphisms and is highly induced in anergic CD4+ T cells. J Immunol 174(6):3377–3385
Rooney IA, Butrovich KD, Glass AA et al (2000) The lymphotoxin-beta receptor is necessary and sufficient for LIGHT-mediated apoptosis of tumor cells. J Biol Chem 275(19):14307–14315
Cheung TC, Humphreys IR, Potter KG et al (2005) Evolutionarily divergent herpesviruses modulate T cell activation by targeting the herpesvirus entry mediator cosignaling pathway. Proc Natl Acad Sci USA 102(37):13218–13223
Compaan DM, Gonzalez LC, Tom I et al (2005) Attenuating lymphocyte activity: the crystal structure of the BTLA–HVEM complex. J Biol Chem 280(47):39553–39561
Nelson CA, Fremont MD, Sedy JR et al (2008) Structural determinants of herpesvirus entry mediator recognition by murine B and T lymphocyte attenuator. J Immunol 180(2):940–947
Ware CF (2008) The TNF superfamily—2008. Cytokine Growth Factor Rev 19(3–4):183–186
Cai G, Anumanthan A, Brown JA et al (2008) CD160 inhibits activation of human CD4+ T cells through interaction with herpesvirus entry mediator. Nat Immunol 9(2):176–185
Anumanthan A, Bensussan A, Boumsell L et al (1998) Cloning of BY55, a novel Ig superfamily member expressed on NK cells, CTL, and intestinal intraepithelial lymphocytes. J Immunol 161(6):2780–2790
Bensussan A, Gluckman E, el Marsafy S et al (1994) BY55 monoclonal antibody delineates within human cord blood and bone marrow lymphocytes distinct cell subsets mediating cytotoxic activity. Proc Natl Acad Sci USA 91(19):9136–9140
Maiza H, Leca G, Mansur IG et al (1993) A novel 80-kD cell surface structure identifies human circulating lymphocytes with natural killer activity. J Exp Med 178(3):1121–1126
Maeda M, Carpenito C, Russell RC et al (2005) Murine CD160, Ig-like receptor on NK cells and NKT cells, recognizes classical and nonclassical MHC class I and regulates NK cell activation. J Immunol 175(7):4426–4432
Zhai Y (1998) LIGHT, a novel ligand for lymphotoxin β receptor and TR2/HVEM induces apoptosis and suppresses in vivo tumor formation via gene transfer. J Clin Invest 102(6):1142–1151
Dejardin E, Droin NM, Delhase M et al (2002) The lymphotoxin-beta receptor induces different patterns of gene expression via two NF-kappaB pathways. Immunity 17(4):525
Coombes JL, Siddiqui KR, Arancibia-Carcamo CV et al (2007) A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med 204(8):1757–1764
Heo SK, Ju SA, Lee SC et al (2006) LIGHT enhances the bactericidal activity of human monocytes and neutrophils via HVEM. J Leukoc Biol 79(2):330–338
Kim WJ, Kang YJ, Koh EM et al (2005) LIGHT is involved in the pathogenesis of rheumatoid arthritis by inducing the expression of pro-inflammatory cytokines and MMP-9 in macrophages. Immunology 114(2):272–279
Hsu H, Solovyev I, Colombero A et al (1997) ATAR, a novel tumor necrosis factor receptor family member, signals through TRAF2 and TRAF5. J Biol Chem 272(21):13471–13474
Marsters SA, Ayres TM, Skubatch M et al (1997) Herpesvirus entry mediator, a member of the tumor necrosis factor receptor (TNFR) family, interacts with members of the TNFR-associated factor family and activates the transcription factors NF-kappaB and AP-1. J Biol Chem 272(22):14029–14032
Shi G, Luo H, Wan X et al (2002) Mouse T cells receive costimulatory signals from LIGHT, a TNF family member. Blood 100(9):3279–3286
Wan X, Zhang J, Luo H et al (2002) A TNF family member LIGHT transduces costimulatory signals into human T cells. J Immunol 169(12):6813–6821
Wu TH, Zhen Y, Zeng C, Yi HF, Zhao Y (2007) B and T lymphocyte attenuator interacts with CD3zeta and inhibits tyrosine phosphorylation of TCRzeta complex during T-cell activation. Immunol Cell Biol 85(8):590–595
Vendel AC, Calemine-Fenaux J, Izrael-Tomasevic A et al (2009) B and T lymphocyte attenuator regulates B cell receptor signaling by targeting Syk and BLNK. J Immunol 182(3):1509–1517
Cheung TC, Steinberg MW, Oborne LM et al (2009) Unconventional ligand activation of herpesvirus entry mediator signals cell survival. Proc Natl Acad Sci USA 106(15):6244–6249
Cohavy O, Zhou J, Granger SW, Ware CF, Targan SR (2004) LIGHT expression by mucosal T cells may regulate IFN-gamma expression in the intestine. J Immunol 173(1):251–258
Cohavy O, Zhou J, Ware CF, Targan SR (2005) LIGHT is constitutively expressed on T and NK cells in the human gut and can be induced by CD2-mediated signaling. J Immunol 174(2):646–653
Wang J, Anders RA, Wang Y et al (2005) The critical role of LIGHT in promoting intestinal inflammation and Crohn's disease. J Immunol 174(12):8173–8182
Maloy KJ, Salaun L, Cahill R et al (2003) CD4+CD25+ T(R) cells suppress innate immune pathology through cytokine-dependent mechanisms. J Exp Med 197(1):111–119
Tamada K, Shimozaki K, Chapoval AI et al (2000) LIGHT, a TNF-like molecule, costimulates T cell proliferation and is required for dendritic cell-mediated allogeneic T cell response. J Immunol 164(8):4105–4110
Tamada K, Shimozaki K, Chapoval AI et al (2000) Modulation of T-cell-mediated immunity in tumor and graft-versus-host disease models through the LIGHT co-stimulatory pathway. Nat Med 6(3):283–289
Shaikh RB, Santee S, Granger SW et al (2001) Constitutive expression of LIGHT on T cells leads to lymphocyte activation, inflammation, and tissue destruction. J Immunol 167(11):6330–6337
Wang J, Lo JC, Foster A et al (2001) The regulation of T cell homeostasis and autoimmunity by T cell-derived LIGHT. J Clin Invest 108(12):1771–1780
Probert LKJ, Corbella P, Cazlaris H, Patsavoudi E, Stephens S, Kaslaris E, Kioussis D, Kollias G (1993) Wasting, ischemia, and lymphoid abnormalities in mice expressin T cell-targeted human tumor necrosis factor transgenes. J Immunol 151(4):1894–1906
Crew MD, Effros RB, Walford RL et al (1998) Transgenic mice expressing a truncated Peromyscus leucopus TNF-alpha gene manifest an arthritis resembling ankylosing spondylitis. J Interferon Cytokine Res 18(4):219–225
Kontoyiannis D, Pasparakis M, Pizarro TT, Cominelli F, Kollias G (1999) Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Immunity 10(3):387–398
Kontoyiannis D, Boulougouris G, Manoloukos M et al (2002) Genetic dissection of the cellular pathways and signaling mechanisms in modeled tumor necrosis factor-induced Crohn's-like inflammatory bowel disease. J Exp Med 196(12):1563–1574
Wang J, Anders RA, Wu Q et al (2004) Dysregulated LIGHT expression on T cells mediates intestinal inflammation and contributes to IgA nephropathy. J Clin Invest 113(6):826–835
Johansson-Lindbom B, Svensson M, Wurbel MA et al (2003) Selective generation of gut tropic T cells in gut-associated lymphoid tissue (GALT): requirement for GALT dendritic cells and adjuvant. J Exp Med 198(6):963–969
Berlin C, Berg EL, Briskin MJ et al (1993) Alpha 4 beta 7 integrin mediates lymphocyte binding to the mucosal vascular addressin MAdCAM-1. Cell 74(1):185–185
Spahn TW, Kucharzik T (2004) Modulating the intestinal immune system: the role of lymphotoxin and GALT organs. Gut 53(3):456–465
Banks TA, Rouse BT, Kerley MK et al (1995) Lymphotoxin-alpha-deficient mice. Effects on secondary lymphoid organ development and humoral immune responsiveness. J Immunol 155(4):1685–1693
De Togni P, Goellner J, Ruddle NH et al (1994) Abnormal development of peripheral lymphoid organs in mice deficient in lymphotoxin. Science 264(5159):703–707
Futterer A, Mink K, Luz A, Kosco-Vilbois MH, Pfeffer K (1998) The lymphotoxin beta receptor controls organogenesis and affinity maturation in peripheral lymphoid tissues. Immunity 9(1):59–70
Koni PA, Sacca R, Lawton P et al (1997) Distinct roles in lymphoid organogenesis for lymphotoxins alpha and beta revealed in lymphotoxin beta-deficient mice. Immunity 6(4):491–500
Scheu S, Alferink J, Potzel T et al (2002) Targeted disruption of LIGHT causes defects in costimulatory T cell activation and reveals cooperation with lymphotoxin beta in mesenteric lymph node genesis. J Exp Med 195(12):1613–1624
Wang J, Foster A, Chin R et al (2002) The complementation of lymphotoxin deficiency with LIGHT, a newly discovered TNF family member, for the restoration of secondary lymphoid structure and function. Eur J Immunol 32(7):1969–1979
Mackay F, Browning JL, Lawton P et al (1998) Both the lymphotoxin and tumor necrosis factor pathways are involved in experimental murine models of colitis. Gastroenterology 115(6):1464–1475
Dohi T, Rennert PD, Fujihashi K et al (2001) Elimination of colonic patches with lymphotoxin beta receptor-Ig prevents Th2 cell-type colitis. J Immunol 167(5):2781–2790
Stopfer P, Obermeier F, Dunger N et al (2004) Blocking lymphotoxin-beta receptor activation diminishes inflammation via reduced mucosal addressin cell adhesion molecule-1 (MAdCAM-1) expression and leucocyte margination in chronic DSS-induced colitis. Clin Exp Immunol 136(1):21–29
Jungbeck M, Stopfer P, Bataille F et al (2008) Blocking lymphotoxin beta receptor signalling exacerbates acute DSS-induced intestinal inflammation—opposite functions for surface lymphotoxin expressed by T and B lymphocytes. Mol Immunol 45(1):34–41
Tumanov A, Kuprash D, Lagarkova M et al (2002) Distinct role of surface lymphotoxin expressed by B cells in the organization of secondary lymphoid tissues. Immunity 17(3):239–250
Spahn TW, Herbst H, Rennert PD et al (2002) Induction of colitis in mice deficient of Peyer's patches and mesenteric lymph nodes is associated with increased disease severity and formation of colonic lymphoid patches. Am J Pathol 161(6):2273–2282
Spahn TW, Maaser C, Eckmann L et al (2004) The lymphotoxin-beta receptor is critical for control of murine Citrobacter rodentium-induced colitis. Gastroenterology 127(5):1463–1473
Clayburgh DR, Musch MW, Leitges M, Fu YX, Turner JR (2006) Coordinated epithelial NHE3 inhibition and barrier dysfunction are required for TNF-mediated diarrhea in vivo. J Clin Invest 116(10):2682–2694
Kang HS, Chin RK, Wang Y et al (2002) Signaling via LTbetaR on the lamina propria stromal cells of the gut is required for IgA production. Nat Immunol 3(6):576–582
Yu P, Lee Y, Liu W et al (2004) Priming of naive T cells inside tumors leads to eradication of established tumors. Nat Immunol 5(2):141–149
Granger SW, Rickert S (2003) LIGHT–HVEM signaling and the regulation of T cell-mediated immunity. Cytokine Growth Factor Rev 14(3–4):289–296
Xu Y, Flies AS, Flies DB et al (2007) Selective targeting of the LIGHT–HVEM co-stimulatory system for the treatment of graft-versus-host disease. Blood 109:4097–4101
Morel Y, Truneh A, Costello RT, Olive D (2003) LIGHT, a new TNF superfamily member, is essential for memory T helper cell-mediated activation of dendritic cells. Eur J Immunol 33(11):3213–3219
Morel Y, Truneh A, Sweet RW, Olive D, Costello RT (2001) The TNF superfamily members LIGHT and CD154 (CD40 ligand) costimulate induction of dendritic cell maturation and elicit specific CTL activity. J Immunol 167(5):2479–2486
Fan Z, Yu P, Wang Y et al (2006) NK-cell activation by LIGHT triggers tumor-specific CD8+ T-cell immunity to reject established tumors. Blood 107(4):1342–1351
Wang Y, Subudhi SK, Anders RA et al (2005) The role of herpesvirus entry mediator as a negative regulator of T cell-mediated responses. J Clin Invest 115(3):711–717
Tao R, Wang L, Han R et al (2005) Differential effects of B and T lymphocyte attenuator and programmed death-1 on acceptance of partially versus fully MHC-mismatched cardiac allografts. J Immunol 175(9):5774–5782
Deppong C, Juehne TI, Hurchla M et al (2006) Cutting edge: B and T lymphocyte attenuator and programmed death receptor-1 inhibitory receptors are required for termination of acute allergic airway inflammation. J Immunol 176(7):3909–3913
Krieg C, Boyman O, Fu YX, Kaye J (2007) B and T lymphocyte attenuator regulates CD8(+) T cell-intrinsic homeostasis and memory cell generation. Nat Immunol 8:162–171
Tao R, Wang L, Murphy KM, Fraser CC, Hancock WW (2008) Regulatory T cell expression of herpesvirus entry mediator suppresses the function of B and T lymphocyte attenuator-positive effector T cells. J Immunol 180(10):6649–6655
Hurchla MA, Sedy JR, Murphy KM (2007) Unexpected role of B and T lymphocyte attenuator in sustaining cell survival during chronic allostimulation. J Immunol 178(10):6073–6082
Deppong C, Degnan JM, Murphy TL, Murphy KM, Green JM (2008) B and T lymphocyte attenuator regulates T cell survival in the lung. J Immunol 181(5):2973–2979
Shao L, Serrano D, Mayer L (2001) The role of epithelial cells in immune regulation in the gut. Semin Immunol 13(3):163–176
Laouar A, Haridas V, Vargas D et al (2005) CD70+ antigen-presenting cells control the proliferation and differentiation of T cells in the intestinal mucosa. Nat Immunol 6(7):698–706
Uhlig HH, McKenzie BS, Hue S et al (2006) Differential activity of IL-12 and IL-23 in mucosal and systemic innate immune pathology. Immunity 25(2):309–318
Corazza N, Eichenberger S, Eugster HP, Mueller C (1999) Nonlymphocyte-derived tumor necrosis factor is required for induction of colitis in recombination activating gene (RAG) 2(−/−) mice upon transfer of CD4(+) CD45RB(hi) T cells. J Exp Med 190(10):1479–1492
Mucida D, Park Y, Kim G et al (2007) Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science 317(5835):256–260
Sun CM, Hall JA, Blank RB et al (2007) Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J Exp Med 204(8):1775–1785
Mucida D, Park Y, Cheroutre H (2009) From the diet to the nucleus: vitamin A and TGF-beta join efforts at the mucosal interface of the intestine. Semin Immunol 21(1):14–21
Denning TL, Wang YC, Patel SR, Williams IR, Pulendran B (2007) Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17-producing T cell responses. Nat Immunol 8(10):1086–1094
Acknowledgments
This work was supported by grants from the National Institutes of Health AI61516 to M.K. and AI06789001 to C.F.W.; by a NIDDK postdoctoral fellowship (DK082249) to J-W. S; by a Research Fellowship Award from the Crohn’s & Colitis Foundation of America to M.W.S. and by the University of California, San Diego, Digestive Diseases Research Development Center (DK 080506). The authors have no conflicting financial interests. This is manuscript number 1143 from the La Jolla Institute for Allergy and Immunology.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Steinberg, M.W., Shui, JW., Ware, C.F. et al. Regulating the mucosal immune system: the contrasting roles of LIGHT, HVEM, and their various partners. Semin Immunopathol 31, 207–221 (2009). https://doi.org/10.1007/s00281-009-0157-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-009-0157-4