
Abstract
C1 inhibitor (C1-INH) is a serine protease inhibitor (serpins) that inactivates several different proteases in the complement, contact, coagulation, and fibrinolytic systems. By its C-terminal part (serpin domain), characterized by three β-sheets and an exposed mobile reactive loop, C1-INH binds, and blocks the activity of its target proteases. The N-terminal end (nonserpin domain) confers to C1-INH the capacity to bind lipopolysaccharides and E-selectin. Owing to this moiety, C1-INH intervenes in regulation of the inflammatory reaction. The heterozygous deficiency of C1-INH results in hereditary angioedema (HAE). The clinical picture of HAE is characterized by bouts of local increase in vascular permeability. Depending on the affected site, patients suffer from disfiguring subcutaneous edema, abdominal pain, vomiting and/or diarrhoea for edema of the gastrointestinal mucosa, dysphagia, and dysphonia up to asphyxia for edema of the pharynx and larynx. Apart from its genetic deficiency, there are several pathological conditions such as ischemia–reperfusion, septic shock, capillary leak syndrome, and pancreatitis, in which C1-INH has been reported to either play a pathogenic role or be a potential therapeutic tool. These potential applications were identified long ago, but controlled studies have not been performed to confirm pilot experiences. Recombinant C1-INH, produced in transgenic animals, has recently been produced for treatment of HAE, and clinical trials are in progress. We can expect that the introduction of this new product, along with the existing plasma derivative, will renew interest in exploiting C1-INH as a therapeutic agent.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Functional and structural properties of C1 inhibitor
C1 inhibitor (C1-INH) is a serine protease inhibitor (serpins) that inactivates several different proteases: C1r, C1s, and MASPs in the complement system, Factor XII and kallikrein in the contact system, Factor XI and thrombin in the coagulation system, tPA and plasmin in the fibrinolytic system [1–14]. C1-INH binds and blocks the activity of its target proteases by the suicide mechanism, which is typical of the serpins [15]. Proteins of this family share an amino acid sequence homology, which confers a similar three-dimensional structure to the C-terminal part (serpin domain) characterized by three β-sheets and an exposed mobile reactive loop. The P1–P1′ amino acid bond in this loop behaves as a pseudo-substrate for the proteases that cleave the peptide bond and covalently bind the amino acid residue. After binding, C1-INH acts as a mousetrap swinging the inactivated protease from the upper to the lower pole with insertion of the reactive loop as a fourth strand in the β-sheet [16]. Mutations causing substitutions of an amino acid involved in this conformational change almost invariably result in loss of inhibitory activity.
The N-terminal end of C1-INH (non-serpin domain) has no homologies with other serpins and is not essential for protease-inhibitor complex formation [17]. It is heavily glycosylated with 3 N-linked and 7 O-linked carbohydrates and contains two disulfide bridges connecting the N-terminal domain to the serpin domain [18]. Mutations abolishing the disulfide bridges lead to a conformational change that eliminates the metastable properties of the serpin domain and allows multimerization [19]. In addition, recent studies brought important evidence that C1-INH has a novel biological role as an anti-inflammatory protein. This role does not depend on the presence of an intact reactive site, but on the carbohydrates linked to the amino acid residues of the non-serpin domain in the amino-terminal end of the molecule. Owing to this moiety, C1-INH is able to bind lipopolysaccharides (LPS) and E-selectin. Hence, via direct binding to LPS, C1-INH protects mice from lethal Gram-negative endotoxemia and inhibits LPS-triggered macrophage expression of TNF-alpha mRNA [20]. With this binding, C1-INH prevents the endothelial cell response to LPS, which causes altered cell morphology, intercellular gap formation, increased transendothelial permeability and eventually leads to the capillary leak syndrome that complicates Gram-negative sepsis and septic shock [21]. The capacity to bind selectins is mediated by the Sialyl Lewis epitope, a fucose-containing tetrasaccharide known to bind all three selectins [22], which is present in C1-INH [23]. By binding E-selectin present in endothelial cells, C1-INH can concentrate at sites of inflammation and can also regulate leukocyte adhesion and transmigration across the endothelial surface [24]. By combining protease-inhibitory functions and these newly described anti-inflammatory properties, C1-INH could play a major role in several pathological conditions: future studies will certainly delineate the actual in vivo relevance for human diseases.
In order to have a complete scenario of the potential consequences of changes in C1-INH structure and/or function, we must introduce the concept of conformational diseases and serpinopathies [25, 26]. These terms identify a growing number of diseases arising from the same general mechanism of abnormal unfolding and then aggregation of an underlying protein: the new conformation of the mutant protein, rather than the reduction of its specific function, is a fundamental pathological event. Alpha1 antitrypsin, the archetypal serpin, provided the first example of this disease mechanism. Its Z mutant undergoes polymerization and aggregation, and, as a consequence, it is not secreted and accumulated in the endoplasmic reticulum of the hepatocyte. Much of it is degraded, but the remainder aggregates to form insoluble intracellular inclusions. These inclusions are associated with hepatocellular damage, and 10% of newborn Z homozygotes develop liver disease, which often leads to fatal childhood cirrhosis [27, 28]. Another single monoacid substitution of alpha1 antitrypsin (alpha 1-antitrypsin Pittsburgh) modifies its protease specificity from elastase to thrombin and causes bleeding disorders [29, 30]. Conformational changes in antithrombin leading to polymerization have been shown to cause thrombosis [31]. Likewise, inappropriate folding of serpin is now investigated as the primary defect leading to degenerative encephalopathies and dementia [32]. The list of diseases in this group and the underlying responsible serpins are still undefined and will probably be growing in the near future.
Extrapolating from the above, one could predict several pathological consequences deriving from C1-INH defects and, on the other side, several potential applications of C1-INH as a therapeutic agent.
C1 inhibitor deficiency and angioedema
In 1963, Donaldson and Evans discovered that subjects with hereditary angioedema (HAE) genetically lacked C1-INH [33]. Angioedema is a self-limiting, transient, localized edema due to a reversible increase in vascular permeability. HAE patients present with functional plasma levels of C1-INH ranging from less than 10% up to 35% compared to normal subjects. This defect is associated to mutations in one of the two alleles of C1-INH gene which is located in chromosome 11q11.2–q13 [18, 34, 35]. More than 150 mutations in this gene, associated with HAE, have been described so far, and a register listing all published mutations is currently on the web (http://hae.biomembrane.hu/) [36]. The clinical phenotype of HAE, i.e. angioedema recurrence, is transmitted as an autosomal dominant trait [37]. The mutated allele and C1-INH deficiency segregate into families according to Mendelian law [38, 39]. From the above, one could conclude that HAE is a monogenic disease, but attempts to correlate specific mutations with phenotypic aspects such as age at onset, frequency or severity of symptoms were unsuccessful. In front of consistently similar C1-INH functional levels within the same patients throughout life and also among different patients, the clinical phenotype may be extremely varied. Frequency and severity of angioedema symptoms vary from patient to patient as well as within the same patient from time to time. This variability clearly does not segregate with the transmission of C1-INH deficiency. Therefore, in HAE, as in the majority of genetic diseases, mutations cannot predict the phenotype of a patient, indicating that genetic factors and the environment might be important modulating factors [40].
This conclusion does not lessen the importance of mutation screening in HAE families. Besides the obvious value of the genetic diagnosis, these experiments could provide new insight into the understanding of conformational diseases and serpinopathies. Moreover, identifying mutations in C1-INH gene may still shed some light on the clinical variability of disease expression. Clinical phenotypes primarily dependent on protein conformation more than on protein function have low penetrance and probably need a cofactor to become expressed. As we can learn from the molecular studies on the Z mutant of alpha1 antitrypsin, temperature and rate of protein synthesis influence its polymerization, and the pro-inflammatory effect of the polymers is exacerbated by the presence of endogenous mediators, such as inflammatory cytokines or external factors such as cigarette smoke [16, 27]. Therefore, we can expect that a clinical phenotype arising from the conformational change induced by specific mutations will present a wide range of variation depending on the individual history of exposure to infections, environmental factors, genetic background, etc. Hence, a better understanding of the size and impact of conformational diseases will come from combining careful analysis of specific genotype/phenotype correlates with the understanding of the structure/function correlates of specific mutations. C1-INH offers a valuable opportunity to reach this goal: there is the register of mutations already mentioned, and there is a register of HAE patients which already contains more than 1,000 entries with detailed clinical information (http://www.haeregister.org). Physicians, scientists, and also patients who are working on these registers in Europe are associated in an active and expanding network [41]. These resources represent the appropriate background to exploit molecular studies on C1-INH mutants to understand clinical variability.
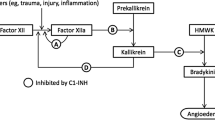
Identification of candidate factors governing the variability of angioedema recurrence in HAE patients is a main target for development of new therapeutic approaches. The episodic appearance of angioedema in C1-INH-deficient patients has been associated to further derangement of the defective control of activation of the contact/complement system, a direct result of the genetic defect [42–47]. The relative importance of these two systems in the pathogenesis of angioedema has been largely debated as detailed in recent reviews on this topic [48–50]. The controversial point has been the final mediator of the increased vascular permeability: a C2-derived peptide, in the “complementcentric theory” and bradykinin, in the “contactcentric theory”. Nowadays the contribution of the contact system and bradykinin is supported by a large mass of data coming from in vitro studies, analysis of patients, and analysis of C1-INH-deficient mice. We know now that contact system is activated during attacks [45, 46]. When depleted of enzymes inducing contact activation, C1-INH-deficient plasma loses its capacity to generate vascular permeability-enhancing activity [51]. During angioedema attacks, C1-INH-deficient patients have increased plasma levels of bradykinin, which is generated in the affected site [52, 53]. A C1-INH mutation, which selectively blocks the complement inhibitory activity but not the activity of C1-INH on the contact system, causes depletion of classical pathway complement components but does not result in angioedema [54]. C1-INH knockout mice, both homozygous and heterozygous, do not have obvious angioedema episodes but present an ongoing increased vascular permeability, which depends on C1-INH deficiency being corrected by intravenous C1-INH [55, 56]. Such an increased vascular permeability depends on contact system activation and is mediated via bradykinin receptor 2 (BK2R). These data lead to the conclusion that in C1-INH deficiency, angioedema takes place in sites where BK2R is stimulated by bradykinin released upon kallikrein generation. Highly specific and potent inhibitors of kallikrein and BK2R are now under investigation for their ability to revert angioedema in HAE patients [57–59]. Data from these studies should rapidly confirm, in a clinical setting, the appropriateness of the pathogenic view, which has been extrapolated from the laboratory.
As mentioned, the clinical picture of C1-INH deficiency is characterized by angioedema, i.e. bouts of local increase in vascular permeability. Depending on the affected site, patients suffer from disfiguring subcutaneous edema, abdominal pain, vomiting and/or diarrhoea for edema of the gastrointestinal mucosa, dysphagia, and dysphonia up to asphyxia for edema of the pharynx and larynx [60]. The episodes last from 2 to 5 days and usually render patients incapable of pursuing their normal activities. Half the patients become symptomatic during the first decade of life and less then 10% after the second. One third of untreated patients have more than one attack per month, while in another third, the disease tends to be mild with a few attacks per year. However, even patients with the mild form may experience life-threatening laryngeal edema [61]. Attacks can be spontaneous, but microtrauma and psychological stress are frequently recognized as triggering factors. Due to the disabling consequences of attacks and to the risk of fatality, treatment is a crucial issue in HAE. The classical approach comprises: (a) long-term prophylaxis, for patients with frequent severe attacks, (b) short-term prophylaxis, for patients exposed to potential triggers of laryngeal attacks (e.g. medical manipulation traumatizing the oropharynx), (c) treatment to revert an ongoing attack. Detailed reviews on the clinical approach to HAE and its treatment have recently been published, and therefore we will not further review this topic [41, 62–64].
Three new compounds are now being tested in clinical trials to assess their capacity to revert acute attacks in HAE patients. The first one, Dx88 (Dyax Corp., Cambridge, MA), is a novel recombinant protein with potent and specific inhibitory activity for plasma kallikrein. One randomized placebo-controlled and two open label studies have been performed so far with a total of 230 doses administered intravenously to more than 130 patients [59]. The second is a recombinant human C1-INH isolated from the milk of transgenic rabbits (Pharming Technologies BV, Leiden). Nine acute attacks have been treated in a phase II open label study; a randomized, controlled study is ongoing [65]. The third is Icatibant (Jerini AG, Berlin), a potent, specific bradykinin B2 receptor antagonist. Twenty attacks have been treated so far in an open label study, 12 by intravenous infusion and 8 by subcutaneous injection [66]. Randomized, controlled studies are ongoing with the subcutaneous formulation. For all three compounds, the available results appear to be favorable.
Ongoing clinical trials for treatment of acute attacks in HAE patients are not restricted to new compounds. Plasma-derived C1-INH represents an established treatment for acute attacks in HAE patients [67–70] and has been approved for this purpose in most European countries for 20 years or more. However, this product is not available in the United States. For this reason, ZLB Behring (Marburg) and Lev Pharmaceutical Inc. (New York, NY) have just started recruiting HAE patients for clinical trials with their plasma-derived C1-INHs.
C1 inhibitor and other diseases
Based on the wide range of biological activities of C1-INH, one could expect several clinical abnormalities to result from its deficiency. Instead, patients lacking C1-INH appear perfectly normal except for angioedema; also C1-INH K.O. mice, both heterozygous and homozygous, do not present obvious phenotypic abnormalities [55]. Nevertheless, there are several pathological conditions in which C1-INH has been reported to either play a pathogenic role or be a potential therapeutic tool. These conditions will be briefly analyzed below.
Ischemia–reperfusion injury
This term refers to the pathological events that follow restoration of blood flow to ischemic tissues. Ischemia rapidly damages metabolically active tissues, and reperfusion paradoxically initiates a cascade of pathologies that leads to additional cell or tissue injury. It can occur spontaneously during a pathological event, but its clinical relevance has grown tremendously with expansion of techniques aimed at restoring patency of occluded arteries and of organ transplantation. Complement activation and neutrophil stimulation are two major components in events leading to tissue injury [71]. During reperfusion, complement can be activated by exposure to intracellular components such as mitochondrial membranes or intermediate filaments. Two elements of the activated complement contribute directly or indirectly to damage: anaphylatoxins (C3a and C5a) and the membrane attack complex [72]. The use of specific inhibitors may find wide clinical application because there are no effective drug therapies currently available to treat I/R injuries [73]. In a model of I/R, organs of transgenic animals overexpressing C1-INH are protected from endothelial cell damage [74]. C1-INH has been tested as potential therapy for I/R injury either in models such as the ischemic muscle [75, 76] or in specific diseases.
I/R injury of the myocardium that follows coronary artery occlusion has been investigated in animal studies, in which it has been shown that C1-INH has the capacity to reduce the myocardial ischemia–reperfusion injury [77–80]. The effect of intravenous C1-INH following reperfusion therapy for acute myocardial infarction has also been tested in pilot human studies. Three patients received C1-INH as rescue therapy for severe reperfusion injury after coronary surgery, with rapid hemodynamic stabilization [81]. In one study, specifically designed to assess the effects of intravenous C1-INH following reperfusion therapy in 22 patients with acute myocardial infarction, this treatment was proved to be safe and effective in reducing complement activation [82]. Due to the small size of the population and the absence of a control group, it was not possible to reach a statistically significant conclusion on clinical effectiveness.
Brain damage following ischemia has recently become a major area of investigation with the possibility to exploit these studies for treatment of stroke, one of the major causes of death and disability worldwide, and for which there is still no effective therapeutic tool. Different animal models of ischemic brain have been studied. Important evidence for a neuroprotective activity of C1-INH has been provided by the group of Bergamaschini using a murine model of transient ischemia induced by introducing a nylon microfilament into the middle cerebral artery [83–85]. Analogous positive conclusions were also reached with other animal models of brain ischemia and of brain damage deriving from hypothermic circulatory arrest [86–88]. We expect that these results will be confirmed in controlled human studies.
I/R injury mediated by complement activation also occurs during organ transplantation. Such an activation and related pathological consequences can be prevented by C1-INH. C1-INH has been circulated into the organ before implant [89, 90] or added to the preserving solution that protects the organ during transport [91]. Exposure of organs to be transplanted to high concentrations of C1-INH significantly reduces such complications due to the capacity of C1-INH to bind endothelial cells maintaining intact functional capacity [92].
Septic shock
Severe sepsis and septic shock are inflammatory diseases triggered by bacterial infection. Despite antibiotics and supportive care, mortality remains extremely high varying from 20 to 50%. Complement and contact systems are activated in sepsis and probably involved in its pathogenesis [93, 94]. Specifically in plasma from septic patients, there is a relative deficiency of C1-INH due to its proteolytic inactivation by neutrophil elastase. In addition to these studies, the rationale for supplying C1-INH to patients with sepsis is further supported by animal models of this condition showing the protective effect of this protein [95–98]. When these studies have been transferred to humans, the positive effect of C1-INH has been confirmed either in anecdotal reports or in pilot studies [99–101]. However, none of these studies has been designed with appropriate control groups to reach a definitive and statistically proven conclusion.
Other diseases
In addition to the above conditions, diverse reports suggest additional clinical indications where C1-INH infusion could be beneficial. Experimental pancreatitis can be prevented by conditioning animals with human C1-INH. These findings have been confirmed in several different animal models [102–106] but are difficult to transfer to humans where treatment cannot obviously anticipate the development of the disease. Only few experiences have been reported in humans and in very specific situations. One is the report of two children who improved upon C1-INH after developing an acute pancreatitis following allogeneic hematopoietic stem cell transplantation [107]. The other is a pilot study performed by our group, in which we demonstrated that administration of C1-INH reduces hyperamylasemia consequent to endoscopic cholangio-pancreatography, a procedure that can be complicated by overt clinical pancreatitis [108].
Reduction in C1-INH plasma levels and complement activation have been involved in the pathogenesis of reactions to radiographic contrast media (RCM). The pathogenic mechanism causing this reaction is still not defined. Signs of activation of complement and contact systems have been detected during RCM infusion. These findings, along with the evidence for reduced plasma levels of protease inhibitors, and specifically of C1-INH, led to the hypothesis that reduced control of these systems could underlie RCM reactions [109–115]. This hypothesis has never been definitively proven, and supporting data have been hampered, at least partially, by hemodilution accompanying infusion of RCM with elevated osmotic activity that alters plasma protein concentration [116].
A final field of intervention for C1-INH is vascular leakage. It may occur spontaneously as in idiopathic capillary leak syndrome (Clarkson disease), a lethal disease characterized by recurrent fluid extravasations leading to hypovolemic shock underlain by marked hemoconcentration (hematocrit as high as 80%) [117]. However, a clinical picture dominated by fluid extravasation can complicate diseases such as septic shock or burns, or specific therapeutic interventions such as treatment with recombinant interleukin 2, bone marrow transplantation, and cardiopulmonary bypass. A role for C1-INH in these conditions is supported by evidence of activation of its target proteases in animal models and by anecdotal experience in humans [118–130].
In conclusion, we can see a broad area of intervention for C1-INH as a therapeutic tool. Some of these potential applications were identified long time ago, but we have not yet seen any controlled study supporting anecdotal reports or pilot experiences. In the last few years, a recombinant C1-INH, produced in transgenic animals, has been proposed for treatment of acute attacks in patients with HAE, and clinical trials with this product are, at present, in progress in North America and Europe [65]. We can expect that the introduction of this new product, along with the existing plasma derivative, will renew interest in exploiting C1-INH as a therapeutic agent in conditions other than genetic deficiency.
References
Pensky J, Levy LR, Lepow IH (1961) Partial purification of a serum inhibitor of C1 esterase. J Biol Chem 236:1674–1679
Ratnoff OD, Pensky J, Donaldson VH, Amir J (1972) The inhibitory properties of plasma against activated plasma thromboplastin antecedent (factor XIa) in hereditary angioneurotic edema. J Lab Clin Med 80:803–809
Harpel PC, Cooper NR (1975) Studies on human plasma C1 inactivator–enzyme interactions. I. Mechanisms of interaction with C1s, plasmin, and trypsin. J Clin Invest 55:593–604
Sim RB, Arlaud GJ, Colomb MG (1979) C1 inhibitor-dependent dissociation of human complement component C1 bound to immune complexes. Biochem J 179:449–457
Ziccardi RJ (1981) Activation of the early components of the classical complement pathway under physiologic conditions. J Immunol 126:1769–1773
Schapira M, Scott CF, Colman RW (1982) Contribution of plasma protease inhibitors to the inactivation of kallikrein in plasma. J Clin Invest 69:462–468
van der Graaf F, Koedam JA, Bouma BN (1983) Inactivation of kallikrein in human plasma. J Clin Invest 71:149–158
de Agostini A, Lijnen HR, Pixley RA, Colman RW, Schapira M (1984) Inactivation of factor XII active fragment in normal plasma. Predominant role of C-1-inhibitor. J Clin Invest 73:1542–1549
Harpel PC, Lewin MF, Kaplan AP (1985) Distribution of plasma kallikrein between C-1 inactivator and alpha 2-macroglobulin in plasma utilizing a new assay for alpha 2-macroglobulin–kallikrein complexes. J Biol Chem 260:4257–4263
Pixley RA, Schapira M, Colman RW (1985) The regulation of human factor XIIa by plasma proteinase inhibitors. J Biol Chem 260:1723–1729
Booth NA, Walker E, Maughan R, Bennett B (1987) Plasminogen activator in normal subjects after exercise and venous occlusion: t-PA circulates as complexes with C1-inhibitor and PAI-1. Blood 69:1600–1604
Matsushita M, Thiel S, Jensenius JC, Terai I, Fujita T (2000) Proteolytic activities of two types of mannose-binding lectin-associated serine protease. J Immunol 165:2637–2642
Jiang H, Wagner E, Zhang H, Frank MM (2001) Complement 1 inhibitor is a regulator of the alternative complement pathway. J Exp Med 194:1609–1616
Cugno M, Bos I, Lubbers Y, Hack CE, Agostoni A (2001) In vitro interaction of C1-inhibitor with thrombin. Blood Coagul Fibrinolysis 12:253–260
Patston PA, Gettins P, Beechem J, Schapira M (1991) Mechanism of serpin action: evidence that C1 inhibitor functions as a suicide substrate. Biochemistry 30:8876–8882
Lomas DA, Belorgey D, Mallya M, Miranda E, Kinghorn KJ, Sharp LK et al (2005) Molecular mousetraps and the serpinopathies(1). Biochem Soc Trans 33:321–330
Coutinho M, Aulak KS, Davis AE III (1994) Functional analysis of the serpin domain of C1 inhibitor. J Immunol 153:3648–3654
Bock SC, Skriver K, Nielsen E, Thogersen HC, Wiman B, Donaldson VH et al (1986) Human C1 inhibitor: primary structure, cDNA cloning, and chromosomal localization. Biochemistry 25:4292–4301
Bos IG, Lubbers YT, Roem D, Abrahams JP, Hack CE, Eldering E (2003) The functional integrity of the serpin domain of C1-inhibitor depends on the unique N-terminal domain, as revealed by a pathological mutant. J Biol Chem 278:29463–29470
Liu D, Cai S, Gu X, Scafidi J, Wu X, Davis AE III (2003) C1 inhibitor prevents endotoxin shock via a direct interaction with lipopolysaccharide. J Immunol 171:2594–2601
Liu D, Zhang D, Scafidi J, Wu X, Cramer CC, Davis AE III (2005) C1 inhibitor prevents Gram-negative bacterial lipopolysaccharide-induced vascular permeability. Blood 105:2350–2355
Vestweber D, Blanks JE (1999) Mechanisms that regulate the function of the selectins and their ligands. Physiol Rev 79:181–213
Cai S, Davis AE III (2003) Complement regulatory protein C1 inhibitor binds to selectins and interferes with endothelial-leukocyte adhesion. J Immunol 171:4786–4791
Cai S, Dole VS, Bergmeier W, Scafidi J, Feng H, Wagner DD et al (2005) A direct role for C1 inhibitor in regulation of leukocyte adhesion. J Immunol 174:6462–6466
Lomas DA, Carrell RW (2002) Serpinopathies and the conformational dementias. Nat Rev Genet 3:759–768
Carrell RW, Lomas DA (1997) Conformational disease. Lancet 350:134–138
Lomas DA, Evans DL, Finch JT, Carrell RW (1992) The mechanism of Z alpha 1-antitrypsin accumulation in the liver. Nature 357:605–607
Carrell RW, Whisstock J, Lomas DA (1994) Conformational changes in serpins and the mechanism of alpha 1-antitrypsin deficiency [published erratum appears in Am J Respir Crit Care Med 1995 Mar;151(3 Pt 1):926]. Am J Respir Crit Care Med 150:S171–S175
Owen MC, Brennan SO, Lewis JH, Carrell RW (1983) Mutation of antitrypsin to antithrombin. Alpha 1-antitrypsin Pittsburgh (358 Met leads to Arg), a fatal bleeding disorder. N Engl J Med 309:694–698
Peterson FC, Gettins PG (2001) Insight into the mechanism of serpin-proteinase inhibition from 2D [1H–15N] NMR studies of the 69 kDa alpha 1-proteinase inhibitor Pittsburgh–trypsin covalent complex. Biochemistry 40:6284–6292
Millar DS, Wacey AI, Ribando J, Melissari E, Laursen B, Woods P et al (1994) Three novel missense mutations in the antithrombin III (AT3) gene causing recurrent venous thrombosis. Hum Genet 94:509–512
Lomas DA, Lourbakos A, Cumming SA, Belorgey D (2002) Hypersensitive mousetraps, alpha1-antitrypsin deficiency and dementia. Biochem Soc Trans 30:89–92
Donaldson VH, Evans RR (1963) A Biochemical abnormality in hereditary angioneurotic edema: absence of serum inhibitor of C′ 1-esterase. Am J Sci 31:37–44
Davis AE III, Whitehead AS, Harrison RA, Dauphinais A, Bruns GA, Cicardi M et al (1986) Human inhibitor of the first component of complement, C1: characterization of cDNA clones and localization of the gene to chromosome 11. Proc Natl Acad Sci U S A 83:3161–3165
Tosi M, Duponchel C, Bourgarel P, Colomb M, Meo T (1986) Molecular cloning of human C1 inhibitor: sequence homologies with alpha 1-antitrypsin and other members of the serpins superfamily. Gene 42:265–272
Kalmar L, Hegedus T, Farkas H, Nagy M, Tordai A (2005) HAEdb: a novel interactive, locus-specific mutation database for the C1 inhibitor gene. Hum Mutat 25:1–5
Crowder JR, Crowder TR (1917) Five generations of angioneurotic edema. Arch Inter Med 20:840–852
Cicardi M, Igarashi T, Kim MS, Frangi D, Agostoni A, Davis AE III (1987) Restriction fragment length polymorphism of the C1 inhibitor gene in hereditary angioneurotic edema. J Clin Invest 80:1640–1643
Stoppa-Lyonnet D, Tosi M, Laurent J, Sobel A, Lagrue G, Meo T (1987) Altered C1 inhibitor genes in type I hereditary angioedema. N Engl J Med 317:1–6
Badano JL, Katsanis N (2002) Beyond Mendel: an evolving view of human genetic disease transmission. Nat Rev Genet 3:779–789
Agostoni A, Aygoren-Pursun E, Binkley KE, Blanch A, Bork K, Bouillet L et al (2004) Hereditary and acquired angioedema: problems and progress: proceedings of the third C1 esterase inhibitor deficiency workshop and beyond. J Allergy Clin Immunol 114:S51–S131
Klemperer MR, Donaldson VH, Rosen FS (1968) Effect of C′1 esterase on vascular permeability in man: studies in normal and complement-deficient individuals and in patients with hereditary angioneurotic edema. J Clin Invest 47:604–611
Donaldson VH, Ratnoff OD, Dias Da Silva W, Rosen FS (1969) Permeability-increasing activity in hereditary angioneurotic edema plasma. II. Mechanism of formation and partial characterization. J Clin Invest 48:642–653
Donaldson VH, Rosen FS, Bing DH (1977) Role of the second component of complement (C2) and plasmin in kinin release in hereditary angioneurotic edema (H.A.N.E.) plasma. Trans Assoc Am Physicians 90:174–183
Schapira M, Silver LD, Scott CF, Schmaier AH, Prograis LJ Jr, Curd JG et al (1983) Prekallikrein activation and high-molecular-weight kininogen consumption in hereditary angioedema. N Engl J Med 308:1050–1053
Curd JG, Prograis LJ Jr, Cochrane CG (1980) Detection of active kallikrein in induced blister fluids of hereditary angioedema patients. J Exp Med 152:742–747
Fields T, Ghebrehiwet B, Kaplan AP (1983) Kinin formation in hereditary angioedema plasma: evidence against kinin derivation from C2 and in support of “spontaneous” formation of bradykinin. J Allergy Clin Immunol 72:54–60
Agostoni A, Aygoren-Pursun E, Binkley KE, Blanch A, Bork K, Bouillet L et al (2004) Hereditary and acquired angioedema: problems and progress: proceedings of the third C1 esterase inhibitor deficiency workshop and beyond. J Allergy Clin Immunol 114:S51–S131
Davis AE (2003) The pathogenesis of hereditary angioedema. Transfus Apher Sci 29:195–203
Davis AE III (2005) The pathophysiology of hereditary angioedema. Clin Immunol 114:3–9
Shoemaker LR, Schurman SJ, Donaldson VH, Davis AE III (1994) Hereditary angioneurotic oedema: characterization of plasma kinin and vascular permeability-enhancing activities. Clin Exp Immunol 95:22–28
Nussberger J, Cugno M, Amstutz C, Cicardi M, Pellacani A, Agostoni A (1998) Plasma bradykinin in angio-oedema. Lancet 351:1693–1697
Nussberger J, Cugno M, Cicardi M, Agostoni A (1999) Local bradykinin generation in hereditary angioedema. J Allergy Clin Immunol 104:1321–1322
Zahedi R, Wisnieski J, Davis AE III (1997) Role of the P2 residue of complement 1 inhibitor (Ala443) in determination of target protease specificity: inhibition of complement and contact system proteases. J Immunol 159:983–988
Han ED, MacFarlane RC, Mulligan AN, Scafidi J, Davis AE III (2002) Increased vascular permeability in C1 inhibitor-deficient mice mediated by the bradykinin type 2 receptor. J Clin Invest 109:1057–1063
Han Lee ED, Pappalardo E, Scafidi J, Davis AE III (2003) Approaches toward reversal of increased vascular permeability in C1 inhibitor deficient mice. Immunol Lett 89:155–160
Icatibant: HOE 140, JE 049, JE049. Drugs R D (2004) 5:343–348
Williams A, Baird LG (2003) DX-88 and HAE: a developmental perspective. Transfus Apher Sci 29:255–258
Cicardi M, Morrison J, Baird L, Williams A (2005) Plasma kallikrein inhibition: a viable method of treating laryngeal edema secondary to hereditary angioedema (HAE). J Allergy Clin Immunol 115:S203
Agostoni A, Cicardi M (1992) Hereditary and acquired C1-inhibitor deficiency: biological and clinical characteristics in 235 patients. Medicine (Baltimore) 71:206–215
Bork K, Ressel N (2003) Sudden upper airway obstruction in patients with hereditary angioedema. Transfus Apher Sci 29:235–238
Gompels MM, Lock RJ, Abinun M, Bethune CA, Davies G, Grattan C et al (2005) C1 inhibitor deficiency: consensus document. Clin Exp Immunol 139:379–394
Bowen T, Cicardi M, Farkas H, Bork K, Kreuz W, Zingale L et al (2004) Canadian 2003 international consensus algorithm for the diagnosis, therapy, and management of hereditary angioedema. J Allergy Clin Immunol 114:629–637
Cicardi M, Zingale L (2003) How do we treat patients with hereditary angioedema. Transfus Apher Sci 29:221–227
Nuijens J, Verdonk R, Van Doorn M, Burggraaf K, Cohen A, Choi G et al (2005) Clinical studies of recombinant human C1 inhibitor in subjects with hereditary angioedema. J Allergy Clin Immunol 115:S202
Bork K, Frank J, Kreuz W, Dong L, Rosenkranz B, Knolle J. Novel approach to treatment of hereditary angioedema with Icatibant, a bradykinin receptor antagonist. IV C1 inhibitor deficiency workshop, Budapest 29 April–1 May 2005; Abstract Book:19
Agostoni A, Bergamaschini L, Martignoni G, Cicardi M, Marasini B (1980) Treatment of acute attacks of hereditary angioedema with C1-inhibitor concentrate. Ann Allergy 44:299–301
Gadek JE, Hosea SW, Gelfand JA, Santaella M, Wickerhauser M, Triantaphyllopoulos DC et al (1980) Replacement therapy in hereditary angioedema: successful treatment of acute episodes of angioedema with partly purified C1 inhibitor. N Engl J Med 302:542–546
Waytes AT, Rosen FS, Frank MM (1996) Treatment of hereditary angioedema with a vapor-heated C1 inhibitor concentrate. N Engl J Med 334:1630–1634
Bork K, Barnstedt SE (2001) Treatment of 193 episodes of laryngeal edema with C1 inhibitor concentrate in patients with hereditary angioedema. Arch Intern Med 161:714–718
Riedemann NC, Ward PA (2003) Complement in ischemia reperfusion injury. Am J Pathol 162:363–367
Monsinjon T, Richard V, Fontaine M (2001) Complement and its implications in cardiac ischemia/reperfusion: strategies to inhibit complement. Fundam Clin Pharmacol 15:293–306
Arumugam TV, Shiels IA, Woodruff TM, Granger DN, Taylor SM (2004) The role of the complement system in ischemia–reperfusion injury. Shock 21:401–409
Inderbitzin D, Beldi G, Avital I, Vinci G, Candinas D (2004) Local and remote ischemia–reperfusion injury is mitigated in mice overexpressing human C1 inhibitor. Eur Surg Res 36:142–147
Toomayan GA, Chen LE, Jiang HX, Qi WN, Seaber AV, Frank MM et al (2003) C1-esterase inhibitor and a novel peptide inhibitor improve contractile function in reperfused skeletal muscle. Microsurgery 23:561–567
Nielsen EW, Mollnes TE, Harlan JM, Winn RK (2002) C1-inhibitor reduces the ischaemia–reperfusion injury of skeletal muscles in mice after aortic cross-clamping. Scand J Immunol 56:588–592
Buerke M, Murohara T, Lefer AM (1995) Cardioprotective effects of a C1 esterase inhibitor in myocardial ischemia and reperfusion [see comments]. Circulation 91:393–402
Horstick G, Heimann A, Gotze O, Hafner G, Berg O, Boehmer P et al (1997) Intracoronary application of C1 esterase inhibitor improves cardiac function and reduces myocardial necrosis in an experimental model of ischemia and reperfusion. Circulation 95:701–708
Buerke M, Prufer D, Dahm M, Oelert H, Meyer J, Darius H (1998) Blocking of classical complement pathway inhibits endothelial adhesion molecule expression and preserves ischemic myocardium from reperfusion injury. J Pharmacol Exp Ther 286:429–438
Horstick G, Berg O, Heimann A, Gotze O, Loos M, Hafner G et al (2001) Application of C1-esterase inhibitor during reperfusion of ischemic myocardium: dose-related beneficial versus detrimental effects. Circulation 104:3125–3131
Bauernschmitt R, Bohrer H, Hagl S (1998) Rescue therapy with C1-esterase inhibitor concentrate after emergency coronary surgery for failed PTCA. Intensive Care Med 24:635–638
de Zwaan C, Kleine AH, Diris JH, Glatz JF, Wellens HJ, Strengers PF et al (2002) Continuous 48-h C1-inhibitor treatment, following reperfusion therapy, in patients with acute myocardial infarction. Eur Heart J 23:1670–1677
De Simoni MG, Rossi E, Storini C, Pizzimenti S, Echart C, Bergamaschini L (2004) The powerful neuroprotective action of C1-inhibitor on brain ischemia–reperfusion injury does not require C1q. Am J Pathol 164:1857–1863
De Simoni MG, Storini C, Barba M, Catapano L, Arabia AM, Rossi E et al (2003) Neuroprotection by complement (C1) inhibitor in mouse transient brain ischemia. J Cereb Blood Flow Metab 23:232–239
Storini C, Rossi E, Marrella V, Distaso M, Veerhuis R, Vergani C et al (2005) C1-inhibitor protects against brain ischemia–reperfusion injury via inhibition of cell recruitment and inflammation. Neurobiol Dis 19:10–17
Heimann A, Takeshima T, Horstick G, Kempski O (1999) C1-esterase inhibitor reduces infarct volume after cortical vein occlusion. Brain Res 838:210–213
Akita N, Nakase H, Kaido T, Kanemoto Y, Sakaki T (2003) Protective effect of C1 esterase inhibitor on reperfusion injury in the rat middle cerebral artery occlusion model. Neurosurgery 52:395–400 (discussion-1)
Heikkinen J, Koskenkari J, Kaakinen T, Dahlbacka S, Kiviluoma K, Salomaki T et al (2004) Apotransferrin, C1-esterase inhibitor, and alpha 1-acid glycoprotein for cerebral protection during experimental hypothermic circulatory arrest. Scand Cardiovasc J 38:178–186
Lehmann TG, Heger M, Munch S, Kirschfink M, Klar E (2000) In vivo microscopy reveals that complement inhibition by C1-esterase inhibitor reduces ischemia/reperfusion injury in the liver. Transpl Int 13:S547–S550
Scherer M, Demertzis S, Langer F, Moritz A, Schafers HJ (2002) C1-esterase inhibitor reduces reperfusion injury after lung transplantation. Ann Thorac Surg 73:233–238 (discussion 8–9)
Bergamaschini L, Gatti S, Caccamo L, Prato P, Latham L, Trezza P et al (2001) C1 inhibitor potentiates the protective effect of organ preservation solution on endothelial cells during cold storage. Transplant Proc 33:939–941
Bergamaschini L, Gobbo G, Gatti S, Caccamo L, Prato P, Maggioni M et al (2001) Endothelial targeting with C1-inhibitor reduces complement activation in vitro and during ex vivo reperfusion of pig liver. Clin Exp Immunol 126:412–420
Kalter ES, Daha MR, ten Cate JW, Verhoef J, Bouma BN (1985) Activation and inhibition of Hageman factor-dependent pathways and the complement system in uncomplicated bacteremia or bacterial shock. J Infect Dis 151:1019–1027
Nuijens JH, Huijbregts CC, Eerenberg-Belmer AJ, Abbink JJ, Strack van Schijndel RJ, Felt-Bersma RJ et al (1988) Quantification of plasma factor XIIa-Cl(−)-inhibitor and kallikrein-Cl(−)-inhibitor complexes in sepsis. Blood 72:1841–1848
de Boer JP, Creasey AA, Chang A, Roem D, Eerenberg AJ, Hack CE et al (1993) Activation of the complement system in baboons challenged with live Escherichia coli: correlation with mortality and evidence for a biphasic activation pattern. Infect Immun 61:4293–4301
Jansen PM, Eisele B, de Jong IW, Chang A, Delvos U, Taylor FB Jr et al (1992) Effect of C1 inhibitor on inflammatory and physiologic response patterns in primates suffering from lethal septic shock. J Immunol 160:475–484
Jansen PM, Pixley RA, Brouwer M, de Jong IW, Chang AC, Hack CE et al (1996) Inhibition of factor XII in septic baboons attenuates the activation of complement and fibrinolytic systems and reduces the release of interleukin-6 and neutrophil elastase. Blood 87:2337–2344
Fischer MB, Prodeus AP, Nicholson-Weller A, Ma M, Murrow J, Reid RR et al (1997) Increased susceptibility to endotoxin shock in complement C3- and C4-deficient mice is corrected by C1 inhibitor replacement. J Immunol 159:976–982
Hack CE, Voerman HJ, Eisele B, Keinecke HO, Nuijens JH, Eerenberg AJ et al (1992) C1-esterase inhibitor substitution in sepsis. Lancet 339:378
Marx G, Nashan B, Cobas Meyer M, Vangerow B, Schlitt HJ, Ziesing S et al (1999) Septic shock after liver transplantation for Caroli’s disease: clinical improvement after treatment with C1-esterase inhibitor. Intensive Care Med 25:1017–1020
Caliezi C, Zeerleder S, Redondo M, Regli B, Rothen HU, Zurcher-Zenklusen R et al (2002) C1-inhibitor in patients with severe sepsis and septic shock: beneficial effect on renal dysfunction. Crit Care Med 30:1722–1728
Kruse P, Hage E, Lasson A (1999) Proteases and protease inhibitors in taurocholate-induced acute pancreatitis in rats. Int J Pancreatol 25:113–121
Yamaguchi H, Weidenbach H, Luhrs H, Lerch MM, Dickneite G, Adler G (1997) Combined treatment with C1 esterase inhibitor and antithrombin III improves survival in severe acute experimental pancreatitis. Gut 40:531–535
Niederau C, Brinsa R, Niederau M, Luthen R, Strohmeyer G, Ferrell LD (1995) Effects of C1-esterase inhibitor in three models of acute pancreatitis. Int J Pancreatol 17:189–196
Vesentini S, Benetti L, Bassi C, Bonora A, Campedelli A, Zamboni G et al (1993) Effects of choline-esterase inhibitor in experimental acute pancreatitis in rats. Preliminary results. Int J Pancreatol 13:217–220
Ruud TE, Aasen AO, Pillgram-Larsen J, Stadaas JO (1986) Effects on peritoneal proteolysis and hemodynamics of prophylactic infusion with C1 inhibitor in experimental acute pancreatitis. Scand J Gastroenterol 21:1018–1024
Schneider DT, Nurnberger W, Stannigel H, Bonig H, Gobel U (1999) Adjuvant treatment of severe acute pancreatitis with C1 esterase inhibitor concentrate after haematopoietic stem cell transplantation. Gut 45:733–736
Testoni PA, Cicardi M, Bergamaschini L, Guzzoni S, Cugno M, Buizza M et al (1995) Infusion of C1-inhibitor plasma concentrate prevents hyperamylasemia induced by endoscopic sphincterotomy. Gastrointest Endosc 42:301–305
Mikkonen R, Aronen HJ, Kivisaari L, Piilonen A, Syrjala M (1997) Plasma levels of prekallikrein, alpha-2-macroglobulin and C1-esterase inhibitor in patients with urticarial reaction to contrast media. Acta Radiol 38:466–473
Hoffmeister HM, Heller W (1996) Radiographic contrast media and the coagulation and complement systems [see comments]. Invest Radiol 31:591–595
Aronen HJ, Kivisaari L, Torstila I, Paavonen T, Meri S, Karonen SL et al (1992) Level of plasma prekallikrein and its inhibitors in reactors and nonreactors during intravenous enhancement with contrast media. Acta Radiol 33:374–378
Hagan JJ, Yost FJ, Nickoloff EL (1987) Prekallikrein activation, C1 esterase inhibitor, and factor XII as predictors of adverse reaction to contrast media. A prospective study. Invest Radiol 22:490–494
Mathews KP, Pan PM, Amendola MA, Lewis FH (1986) Plasma protease inhibitor and anaphylatoxin inactivator levels in chronic urticaria/angioedema and in patients experiencing anaphylactoid reactions to radiographic contrast media. Int Arch Allergy Appl Immunol 79:220–223
Johansen HT, Hoem NO, Veggeland T, Briseid K (1986) Assay of kallikrein inhibitors and levels of acetone-activated kallikrein in plasma specimens from reactors to dextran or to contrast media. Int J Tissue React 8:185–192
Lasser EC, Lang JH, Lyon SG, Hamblin AE (1979) Complement and contrast material reactors. J Allergy Clin Immunol 64:105–112
Gardinali M, Cicardi M, Frangi D, Franzinelli M, Gattoni F, Uslenghi C et al (1986) In vivo study of the complement system during infusion of radiographic contrast media. J Allergy Clin Immunol 77:690–692
Clarkson B, Thompson D, Horwith M, Luckey EH (1960) Cyclical edema and shock due to increased capillary permeability. Am J Med 29:193–216
Kirschfink M, Nurnberger W (1999) C1 inhibitor in anti-inflammatory therapy: from animal experiment to clinical application. Mol Immunol 36:225–232
Hazelzet JA, de Groot R, van Mierlo G, Joosten KF, van der Voort E, Eerenberg A et al (1998) Complement activation in relation to capillary leakage in children with septic shock and purpura. Infect Immun 66:5350–5356
Stiller B, Sonntag J, Dahnert I, Alexi-Meskishvili V, Hetzer R, Fischer T et al (2001) Capillary leak syndrome in children who undergo cardiopulmonary bypass: clinical outcome in comparison with complement activation and C1 inhibitor. Intensive Care Med 27:193–200
Radke A, Mottaghy K, Goldmann C, Khorram-Sefat R, Kovacs B, Janssen A et al (2000) C1 inhibitor prevents capillary leakage after thermal trauma. Crit Care Med 28:3224–3232
Seghaye MC, Grabitz RG, Duchateau J, Busse S, Dabritz S, Koch D et al (1996) Inflammatory reaction and capillary leak syndrome related to cardiopulmonary bypass in neonates undergoing cardiac operations. J Thorac Cardiovasc Surg 112:687–697
Heller A, Kunz M, Samakas A, Haase M, Kirschfink M, Koch T (2000) The complement regulators C1 inhibitor and soluble complement receptor 1 attenuate acute lung injury in rabbits. Shock 13:285–290
Schmidt S, Hertfelder HJ, von Spiegel T, Hering R, Harzheim M, Lassmann H et al (1999) Lethal capillary leak syndrome after a single administration of interferon beta-1b. Neurology 53:220–222
Nurnberger W, Heying R, Burdach S, Gobel U (1997) C1 esterase inhibitor concentrate for capillary leakage syndrome following bone marrow transplantation. Ann Hematol 75:95–101
Salat C, Holler E, Schleuning M, Eisele B, Reinhardt B, Kolb H et al (1995) Levels of the terminal complement complex, C3a-desArg and C1-inhibitor in adult patients with capillary leak syndrome following bone marrow transplantation. Ann Hematol 71:271–274
Hack CE, Ogilvie AC, Eisele B, Jansen PM, Wagstaff J, Thijs LG (1994) Initial studies on the administration of C1-esterase inhibitor to patients with septic shock or with a vascular leak syndrome induced by interleukin-2 therapy. Prog Clin Biol Res 388:335–357
Nurnberger W, Michelmann I, Petrik K, Holthausen S, Willers R, Lauermann G et al (1993) Activity of C1 esterase inhibitor in patients with vascular leak syndrome after bone marrow transplantation. Ann Hematol 67:17–21
Hack CE, Ogilvie AC, Eisele B, Eerenberg AJ, Wagstaff J, Thijs LG (1993) C1-inhibitor substitution therapy in septic shock and in the vascular leak syndrome induced by high doses of interleukin-2. Intensive Care Med 19:S19–S28
Nurnberger W, Gobel U, Stannigel H, Eisele B, Janssen A, Delvos U (1992) C1-inhibitor concentrate for sepsis-related capillary leak syndrome. Lancet 339:990
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Cicardi, M., Zingale, L., Zanichelli, A. et al. C1 inhibitor: molecular and clinical aspects. Springer Semin Immun 27, 286–298 (2005). https://doi.org/10.1007/s00281-005-0001-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-005-0001-4