
Abstract
Purpose
Through enhancement of 6-mercaptopurine (6MP) bioavailability and inhibition of purine de novo synthesis, high-dose methotrexate (HD-MTX) may increase incorporation into DNA of 6-thioguanine nucleotides, the cytotoxic metabolites of 6MP. Patients with intermediate activity of thiopurine methyltransferase (TPMTIA) have higher cytosol 6-thioguanine nucleotide levels. We investigated toxicity following HD-MTX during MTX/6MP maintenance therapy in relation to 6MP and TPMT.
Methods
Using linear mixed models, we explored myelo- and hepatotoxicity in relation to 6MP dosage and TPMT phenotype following 1,749 HD-MTX courses to 411 children with acute lymphoblastic leukemia on maintenance therapy.
Results
The degree of myelosuppression following HD-MTX was similar for patients with TPMTIA and patients with high TPMT activity (TPMTHA), when HD-MTX started with same blood counts and 6MP doses. However, since TPMTIA had lower blood counts at initiation of HD-MTX compared with TPMTHA patients (median WBC 2.8 vs. 3.3 × 109/L, P = 0.01; median ANC 1.4 vs. 1.7 × 109/L, P = 0.02), TPMTIA continued to have lower WBC and ANC levels compared with TPMTHA during all 28 days after HD-MTX [relative difference 9 % (95 % CI 2–17), P = 0.02 and 21 % (95 % CI 6–39), P = 0.005]. Still, the fractional decrease in WBC and ANC levels after HD-MTX did not differ between TPMTIA and TPMTHA patients (P = 0.47; P = 0.38). The degree of leukopenia, neutropenia, thrombocytopenia and rise in aminotransferases were all significantly related to 6MP dose (P < 0.001 for all analyses).
Conclusion
For both TPMTIA and TPMTHA patients, dose of 6MP prior to HD-MTX should be guided by pre-HD-MTX blood counts, but not by TPMT activity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Overall survival for children with acute lymphoblastic leukemia (ALL) has reached 85 % [1], and exploration of the balance between efficacy and side effects of antileukemic drugs has become a major research target. High-dose methotrexate (HD-MTX) is an important part of the therapy given to children with ALL to reduce the risk of both systemic and extramedullary relapse [2, 3]. HD-MTX refers to MTX doses between 0.5 and 8 g/m2 or even higher [2, 4] and is commonly given during consolidation therapy with or without concurrent oral 6-mercaptopurine (6MP) and as re-induction during maintenance therapy with daily oral 6MP and weekly oral MTX as the backbone [5, 6]. HD-MTX often causes significant bone marrow toxicity that carries a risk of infections and a need for transfusions [7]. This myelosuppression may lead to treatment interruptions and thus a reduction of the dose intensity, which may affect the cure rate [8–10].
MTX and 6MP act synergistically [11–14]. The degree of myelosuppression and duration of treatment interruptions following HD-MTX is related to the dose of concurrently administered oral 6MP [15, 16] and can be avoided by reductions of the dose of 6MP in the weeks before and after HD-MTX [17]. MTX may increase the bioavailability of 6MP through inhibition of xanthine oxidase, which catabolizes 6MP [11, 14]. Through inhibition of de novo purine synthesis, MTX may enhance the DNA incorporation of 6-thioguanine nucleotides (6TGN) that primarily exert the cytotoxic effect of 6MP [12, 13, 18]. The enzyme thiopurine methyltransferase (TPMT) competes with the formation of 6TGN, as it methylates 6MP and some of its metabolites creating less toxic compounds (Fig. 1) [19, 20]. Approximately 10 % of all white individuals are TPMT heterozygous and have intermediate TPMT activity (TPMTIA), and one in 300 individuals is TPMT deficient with extremely low or undetectable activity [21]. The interindividual variations in TPMT activity significantly influences the degree of methylation and intracellular 6TGN accumulation [20, 22] and thus modifies the effect of 6MP. In the present study, we explored the impact of TPMT activity on the risk of HD-MTX-related myelotoxicity, hepatotoxicity and treatment interruptions among 411 children (excluding those with trisomy 21) enrolled in the NOPHO ALL92 maintenance therapy study.

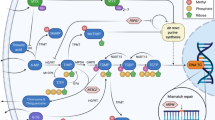
Simplified diagram of 6MP and MTX interaction.*Circumstantial evidence indicates that these metabolites are associated with myelotoxicity and hepatotoxicity, respectively. Ery-6TGN erythrocyte levels of 6-thioguanine nucleotides, FPGS folyl-polyglutamyl synthetase, HPRT hypoxanthine–guanine phosphoribosyltransferase, MTX methotrexate, MTXpg methotrexate polyglutamates, TPMT thiopurine methyltransferase, XO xanthine oxidase, 6MP 6-mercaptopurine
Methods
Study design
The present study is a retrospective analysis of data from the NOPHO ALL92 maintenance therapy study initially designed to analyze the impact of pharmacologic dosing oral 6MP and MTX by erythrocyte levels of 6TGN and MTX [23]. Patients were eligible for this study, if they: (1) were included in the NOPHO ALL92 maintenance therapy study [23]; (2) had their TPMT phenotype measured during maintenance therapy; (3) were treated at least once with HD-MTX 5.0 g/m2 (±10 %) during maintenance therapy; (4) had an interval between HD-MTX courses of at least 49 days; and (5) had at least one available measurement of blood counts or alanine aminotransferase (ALT) levels 1 week before or 4 weeks after HD-MTX. Patients included in the NOPHO ALL92 maintenance therapy study were (1) diagnosed with B cell precursor or T cell ALL in the Nordic countries (Denmark, Finland, Iceland, Norway and Sweden) between January 1992 and December 1996; (2) between 1.0 and 14.9 years of age at diagnosis; and (3) in first remission after induction and consolidation therapy. Five hundred and thirty eight patients with SR-, IR- or HR-ALL were entered into the NOPHO ALL92 maintenance therapy trial. As part of that study, all 28,580 data sets of blood counts as well as 6MP and MTX doses available for the 538 patients were prospectively registered [23, 24]. In the NOPHO ALL92 protocol, patients treated according to the high-risk arm received no HD-MTX during maintenance therapy and were not eligible for the present study. Thus, HD-MTX during maintenance therapy was only part of the treatment protocol for standard and intermediate risk (SR/IR-ALL) patients [15]. Of the 430 SR-/IR-ALL patients eligible for the present study, 11 patients who relapsed during maintenance therapy (all with high TPMT activity) and one patient who developed AML during maintenance therapy (TPMT heterozygous by genotyping) were excluded from the study. Furthermore, five patients with trisomy 21 (all with high TPMT activity) and two TPMT deficient patients (median TPMT activity 0.01 and 0.58 IU/mL) were excluded [25]. Since HD-MTX was given right at the start of maintenance therapy for IR-ALL without preceding oral 6MP therapy, this HD-MTX course was excluded from the analysis. If no blood counts or ALT measurement was registered on the day HD-MTX was given, the most recent measurement taken up to 1 week before counted as a measurement on the day HD-MTX was administered. The median number of white blood cell count (WBC), absolute neutrophil count (ANC), platelet count and ALT measures per HD-MTX course was 3 (50 % range 2–5), 3 (50 % range 1–5), 3 (50 % range 2–5) and 1 (50 % range 0–2), respectively. The total number of WBC, ANC, platelet counts and ALT measurements included in this study were 6,907, 5,869, 6,850 and 2,716, respectively. Measures taken at time-points with missing 6MP or MTX doses were excluded from the multiple linear mixed models. In total, the study population consisted of 411 SR/IR-ALL patients (411/430, 96 % of all eligible patients) with 1,749 HD-MTX courses.
Therapy
The risk group assignment was based on age and WBC at diagnosis (SR-ALL: age 2.0–9.9 years and WBC < 10 × 109/L; IR-ALL: age 1.0–1.9 years or age 10.0–14.9 years and/or WBC 10–49 × 109/L) and the absence of high-risk criteria (WBC 50 × 109/L, T cell disease, mediastinal or CNS or testicular or lymphomatous disease, t(4;11), t(9;22), a day 15 bone marrow with more than 25 % lymphoblasts, or a day 29 bone marrow with more than 5 % lymphoblasts) [26].
As induction therapy, all patients received prednisolone (60 mg/m2/day on days 1–36, then tapered), weekly vincristine (VCR, 2.0 mg/m2 times six), doxorubicin (40 mg/m2 times three), Erwinia asparaginase (30,000 IU/m2 daily on days 37–46) and intrathecal (i.t.) MTX on four occasions. Consolidation therapy for SR-ALL included three courses of HD-MTX given at 2-week intervals without concurrent oral 6MP. For IR-ALL patients, consolidation therapy consisted of: (1) from day 50; daily oral 6MP (75 mg/m2 for two times 2 weeks), cyclophosphamide (1 g/m2 times two), four series of low-dose cytarabine (each with i.v. doses of 75 mg/m2 on four consecutive days), and i.t. MTX (times two); (2) from day 106; daily oral 6MP for 8 weeks (25 mg/m2) with four courses of HD-MTX given at 2-week intervals; (3) from day 169; 4 weeks of re-induction with dexamethasone (10 mg/m2/day divided into three doses for 3 weeks, then tapered), weekly VCR (2.0 mg/m2/day times four), weekly daunorubicin (30 mg/m2/day times four) and asparaginase four times (30,000 IU/m2 at 3–4 days of intervals); and (4) from day 197; daily oral thioguanine (60 mg/m2 for 2 weeks), cyclophosphamide (1 g/m2 times one), two series of low-dose cytarabine (each with i.v. doses of 75 mg/m2 on four consecutive days) and i.t. MTX (times one).
MTX/6MP maintenance therapy was initiated at treatment weeks 13 (SR-ALL) or 32 (IR-ALL) and continued until 2.0 (IR-ALL) or 2.5 years (SR-ALL) after diagnosis. The starting dose of 6MP was 75 mg/m2/day, and starting dose of MTX was 20 mg/m2/week. The doses of oral 6MP and oral MTX were to be targeted to a WBC of 1.5–3.5 × 109/L and reduced to 50 % at a WBC < 1.5 × 109/L and interrupted at a WBC < 1.0 × 109/L and/or a platelet count <100 × 109/L. During the first year of maintenance therapy, patients with SR- or IR-ALL received alternate pulses at 4-week intervals of (1) VCR (2.0 mg/m2 once) and prednisolone (60 mg/m2/day for 1 week), and (2) HD-MTX until five courses of HD-MTX had been given.
High-dose methotrexate
According to the NOPHO ALL92 protocol, 5.0 g of MTX/m2 was given i.v. over 24 h with i.t. MTX in age-related doses (10–12 mg) [26]. Starting 36 h from the beginning of the HD-MTX infusion, leucovorin rescue was given at a dose of 15 mg/m2 i.v. every 6 h until the serum MTX was below 0.2 μmol/L. If serum MTX was >1 µmol/L at 42 h, the leucovorin doses were increased according to the serum MTX concentrations [6].
Thiopurine methyltransferase
The four hundred and eleven patients had their erythrocyte TPMT activity measured 1–6 times during maintenance therapy. For patients with more than one TPMT activity measurement, an arithmetic mean TPMT activity was calculated. All TPMT phenotype assays were performed at least 8 weeks after the most recent blood transfusion. The 49 patients with a TPMT activity below 14 IU/mL (antimode of distribution) were classified as TPMTIA (median TPMT activity 10.7, range 6.2–13.9 IU/mL) [24]. The remaining 362 patients were classified as TPMT high activity (TPMTHA) (median TPMT activity 18.7, range 14.2–27.6 IU/mL). The TPMT phenotype was not revealed to the physicians while the patients were on therapy.
Statistics
The Fisher exact test was used to compare the distribution of categorical variables across subgroups, and the Mann–Whitney test was used to compare the distributions of quantitative variables. The duration of event-free survival (EFS) was defined as the time from diagnosis until the date of relapse, death, the diagnosis of a secondary cancer (whichever first) or the last registered follow-up for event-free survivors. The Kaplan–Meier method was applied for estimation of remission duration [27], and subgroups were compared with the log-rank test [28]. Comparing groups with respect to outcomes defined as summary measures for each HD-MTX course, tests taking the repeated outcomes for each patient into account were applied: For binary outcomes, proportions were compared by logistic regression based on Generalized Estimating Equations [29]. For quantitative outcomes, the tests were based on a Monte Carlo permutation [30] of the Kruskal–Wallis test.
The analyses of the degree of toxicity were based on measurements of WBC, ANC, platelet count and ALT taken from the time of administration of HD-MTX and the following 28 days. If no measurement was registered on the day HD-MTX was given, the most recent measurement taken up to 1 week before counted as a measurement on the day HD-MTX was administered. The intraindividual variation was large, and the patients were not monitored regularly, making it impossible to determine the exact time for nadir/peak and the corresponding degree of myelosuppression and hepatotoxicity for each patient based on the available measurements. To analyze whether the time to nadir/time to peak and the degree of myelo- and hepatotoxicity depended on the 6MP dose and TPMT activity, we formulated multivariable hierarchical linear mixed models describing the level of each outcome for each time-point up to 28 days after HD-MTX.
Random effects were included at two levels: One random effect for the patient and one random effect for each HD-MTX treatment within each patient (nested random effects) [31]. The level of the log-transformed outcome as a function of the number of days (time) since administration of HD-MTX was modeled by the use of restricted cubic splines [32]. The courses were allowed to depend on 6MP (divided into the three groups 0–50, 50–75 and >75 mg/m2/day) and TPMT activity (dichotomized as TPMTIA and TPMTHA groups). For each outcome, it was evaluated whether the courses following HD-MTX depended on TPMT groups. If not, the courses were only allowed to depend on the 6MP dose. To illustrate the differences in courses following HD-MTX according to the 6MP and TPMT groups, plots were made conditioning on a specific value of the outcome assumed to be measured prior to the administration of HD-MTX (corresponding to day 0 in the analyses). The analyses were adjusted for sex, risk group, TPMT group, age at diagnosis, number of years on maintenance therapy, the most recent MTX dose (quantitative, units of 10 mg/m2/week) and the most recent 6MP dose (0 = <50, 1 = 50–75, 2 = >75 mg/m2/day). The estimates are reported in Online Resource 1.
The linear mixed models were used to estimate time to nadir for WBC, ANC and platelet count and time to peak for ALT for each 6MP group. For the models for which the courses depended also on the TPMT status, nadir values were estimated for each combination of 6MP and TPMT activity groups. For each estimated nadir peak value, the corresponding percent-wise change was estimated. Confidence intervals for time to nadir/time to peak and the percentage change in outcome at nadir or peak were determined by the bootstrap percentile method [33], resampling patients rather than individual measurements to account for repeated measurements. Two-sided P values <0.05 were regarded as significant. Statistical analyses were performed with the SAS statistical software (SAS Institute Inc., Cary, NC, USA), R version 2.15.2 and the SPSS 19.0 software package (SPSS Inc., Chicago, IL, USA).
Results
Myelotoxicity and hepatotoxicity
The clinical characteristics and treatment assignments of the 220 boys and 191 girls are given in Table 1. TPMTIA patients had lower blood counts at the initiation of HD-MTX compared with TPMTHA patients (median WBC 2.8 vs. 3.3 × 109/L, P = 0.01; median ANC 1.4 vs. 1.7 × 109/L, P = 0.02). As a group, the TPMTIA patients continued to have lower WBC and ANC levels compared with TPMTHA patients during the entire 28 days after HD-MTX [relative difference 9 % (95 % CI 2–17; P = 0.02) lower WBC levels and 21 % (95 % CI 6–39; P = 0.005) lower ANC levels, respectively]. However, the fractional decrease in WBC and ANC levels after HD-MTX among TPMTIA patients was the same as for TPMTHA patients (P = 0.47; P = 0.38, respectively). Thus, the WBC and ANC nadir value following HD-MTX was the same for patients with TPMTIA or TPMTHA starting a HD-MTX course with the same blood count value and the same 6MP dose. Furthermore, the time to nadir was the same for TPMTIA and TPMTHA patients. Although the TPMT activity was significantly associated with the course of platelet counts following HD-MTX (P = 0.006), the difference in the estimated platelet nadir between TPMTIA and TPMTHA was clinically irrelevant [e.g., 78 × 109/L (95 % CI 68–93) vs. 93 × 109/L (95 % CI 85–100) for patients receiving a 6MP dose of 50–75 mg/m2/day] (Fig. 2). The estimated time to platelet count nadir was 8.4 days (TPMTHA) to 8.5 days (TPMTIA) for patients receiving a 6MP dose of 50–75 mg/m2/day.

Course of platelet count following HD-MTX in relation to TPMT activity and coadministered 6MP dose. The course is conditional on an observed platelet count of 250 × 109/L at the day of administration of HD-MTX. The plot depicts patients receiving a 6MP dose of 50–75 mg/m2/day. P value for test of no effect of TPMT on courses of platelet count after HD-MTX: P = 0.006. P value for test of no effect of 6MP dose on courses of platelet count after HD-MTX: P < 0.001
The estimated courses of WBC, ANC, platelet count and ALT after HD-MTX depended on the 6MP dose (P < 0.001 for all analyses) (Figs. 3, 4). Depending on the 6MP dose, the estimated time to nadir ranged from 10.0 days (6MP dose >75 mg/m2/day) to 12.8 days (6MP dose <50 mg/m2/day) for WBC, 13.2 days (6MP dose >75 mg/m2/day) to 16.2 days (6MP dose <50 mg/m2/day) for ANC and 7.4 days (6MP dose <50 and >75 mg/m2/day) to 8.1 days (6MP dose 50–75 mg/m2/day) for ALT peak. The estimated parameters of these models are shown in Online Resource 1.

a Course of WBC following HD-MTX in relation to coadministered 6MP dose. The course is conditional on an observed WBC value of 2.5 × 109/L at the day of administration of HD-MTX. P value for test of no difference between curves: P < 0.001. b Course of ANC following HD-MTX in relation to coadministered 6MP dose. The course is conditional on an observed ANC value of 1.5 × 109/L at the day of administration of HD-MTX. P value for test of no difference between curves: P < 0.001

Course of ALT following HD-MTX in relation to coadministered 6MP dose. The course is conditional on an observed ALT value of 75 IU/L at the day of administration of HD-MTX. P value for test of no difference between curves: P < 0.001
Treatment interruptions
Among the 1,657 courses with available data on 6MP dosing 0–28 days after HD-MTX, 6MP maintenance therapy was discontinued more often following HD-MTX among TPMTIA patients compared with the TPMTHA patients [45 % (79 of 174 courses) vs. 29 % (425 of 1,483 courses), P = 0.002]. The higher number of treatment interruptions among TPMTIA compared with TPMTHA patients may be explained by their lower blood counts at the start of HD-MTX. However, once interrupted, the duration of treatment interruptions did not differ between TPMTIA and TPMTHA patients (median: 10 vs. 9 days, P = 0.25). Among the 1,716 courses with available data on 6MP dosing within 8 weeks prior to and on the day of HD-MTX, more HD-MTX courses were initiated while 6MP was discontinued among TPMTIA patients compared with the TPMTHA patients [9 % (18 of 192 courses) vs. 4 % (55 of 1,524 courses), P = 0.001].
Discussion
HD-MTX has been given to patients with ALL since the 1960s to reduce the risk of relapse [15]. However, post-HD-MTX toxicity is common and associated with increased risk of infections and interruption of maintenance treatment, which has been linked to a reduced cure rate [9, 10, 17]. Anticancer treatment programmes generally combine drugs with non-overlapping toxicity, and accordingly significant drug–drug interactions with respect to MTX-induced toxicity are not common. An important exception is the combination with oral 6MP in both low- and HD-MTX therapy [34].
The association between complete TPMT deficiency and increased toxicity of treatment with 6MP in ALL patients is well established as these individuals may develop life-threatening myelotoxicity from normal doses of 6MP [25, 35–39]. However, the clinical relevance in TPMTIA patients is less clear. In this study, we found the 6MP dose to be the most important predictor of myelo- and hepatotoxicity after HD-MTX. Compared to low-intensity treatment of 6MP, increased 6MP dose was associated with an increase in hematological and hepatic toxicity. However, the myelo- and hepatotoxicity following HD-MTX did not depend on TPMT status. Toxicity was the same for patients with TPMTIA and TPMTHA starting a HD-MTX course on the same 6MP dose with the same blood count or ALT value. Previous studies have found intermediate activity of TPMT to be both related [39] and unrelated [37] to increased toxicity measured as 6MP dose reduction and 6MP withdrawal rate. However, neither of these studies specifically explored the time-points of HD-MTX administration, but rather the entire ALL maintenance therapy phase.
Benefits of 6MP dose reduction in relation to TPMT status are complex. We have previously shown that TPMT activity is related to the risk of secondary cancer in the NOPHO ALL92 study [40], and subsequently that reducing 6MP starting doses at initiation of maintenance therapy for patients with TPMT heterozygosity may reduce the risk of second malignant neoplasm (SMN), but also increase relapse rates [41], probably due to the overall lower 6MP dose intensity obtained for TPMT low activity patients throughout maintenance therapy phase. An increased risk of SMN was not associated with TPMT genotype in a recent large German study [42]. The present study indicates that these differences between the Nordic and German studies do not reflect the administration of HD-MTX during maintenance therapy in NOPHO ALL92. In the current NOPHO ALL2008 protocol, there are no guidelines for 6MP dose adjustments during HD-MTX according to TPMT status.
This is the largest material analyzed regarding the influence of TPMT activity on toxicity after HD-MTX. However, it has a few limitations. First, we were not able to determine the nadirs for each patient, since some patients did not have a sufficient number of measurements. However, with the population-based approach, we attempted to overcome the intraindividual variability of measurements and estimate nadir values. Second, patients with delayed MTX clearance have long been known to be at increased risk for myelotoxicity, but we did not include serum MTX values in the analyses of post-HD-MTX toxicity, since the target was to explore the impact of parameters that were available when HD-MTX was initiated. Third, the TPMT activity was determined in erythrocytes rather than in bone marrow or liver cells, but TPMT genotype is strongly correlated with TPMT activity in several tissues [43–45].
In conclusion, TPMTIA and TPMTHA groups had the same degree of myelo- and hepatotoxicity following HD-MTX when they received the same 6MP dose and started a HD-MTX course with the same blood count or ALT value. TPMT deficient patients were excluded from this study, but previous research has shown that TPMT deficiency causes severe myelotoxicity after HD-MTX [25]. TPMT intermediate activity does not predict myelo- and hepatotoxicity following HD-MTX with concurrent oral 6MP when compared to TPMTHA. TPMTIA should not due to their TPMT activity have their 6MP dose reduced prior to HD-MTX. Dosage of 6MP prior to HD-MTX should be guided by pre-HD-MTX blood counts for both TPMTIA and TPMTHA patients.
References
Schrappe M, Nachman J, Hunger S, Schmiegelow K, Conter V, Masera G, Pieters R, Pui CH (2010) Educational symposium on long-term results of large prospective clinical trials for childhood acute lymphoblastic leukemia (1985–2000). Leukemia 24:253–254
Clarke M, Gaynon P, Hann I, Harrison G, Masera G, Peto R, Richards S (2003) CNS-directed therapy for childhood acute lymphoblastic leukemia: Childhood ALL Collaborative Group overview of 43 randomized trials. J Clin Oncol 21:1798–1809
Pui CH, Thiel E (2009) Central nervous system disease in hematologic malignancies: historical perspective and practical applications. Semin Oncol 36:S2–S16
Nathan PC, Whitcomb T, Wolters PL, Steinberg SM, Balis FM, Brouwers P, Hunsberger S, Feusner J, Sather H, Miser J, Odom LF, Poplack D, Reaman G, Bleyer WA (2006) Very high-dose methotrexate (33.6 g/m2) as central nervous system preventive therapy for childhood acute lymphoblastic leukemia: results of National Cancer Institute/Children’s Cancer Group trials CCG-191P, CCG-134P and CCG-144P. Leuk Lymphoma 47:2488–2504
Pui CH, Evans WE (1998) Acute lymphoblastic leukemia. N Engl J Med 339:605–615
Skarby TV, Anderson H, Heldrup J, Kanerva JA, Seidel H, Schmiegelow K (2006) High leucovorin doses during high-dose methotrexate treatment may reduce the cure rate in childhood acute lymphoblastic leukemia. Leukemia 20:1955–1962
Rask C, Albertioni F, Bentzen SM, Schroeder H, Peterson C (1998) Clinical and pharmacokinetic risk factors for high-dose methotrexate-induced toxicity in children with acute lymphoblastic leukemia—a logistic regression analysis. Acta Oncol 37:277–284
Peeters M, Koren G, Jakubovicz D, Zipursky A (1988) Physician compliance and relapse rates of acute lymphoblastic leukemia in children. Clin Pharmacol Ther 43:228–232
Relling MV, Hancock ML, Boyett JM, Pui CH, Evans WE (1999) Prognostic importance of 6-mercaptopurine dose intensity in acute lymphoblastic leukemia. Blood 93:2817–2823
Schmiegelow K (1991) Prognostic significance of methotrexate and 6-mercaptopurine dosage during maintenance chemotherapy for childhood acute lymphoblastic leukemia. Pediatr Hematol Oncol 8:301–312
Balis FM, Holcenberg JS, Zimm S, Tubergen D, Collins JM, Murphy RF, Gilchrist GS, Hammond D, Poplack DG (1987) The effect of methotrexate on the bioavailability of oral 6-mercaptopurine. Clin Pharmacol Ther 41:384–387
Bokkerink JP, Bakker MA, Hulscher TW, De Abreu RA, Schretlen ED (1988) Purine de novo synthesis as the basis of synergism of methotrexate and 6-mercaptopurine in human malignant lymphoblasts of different lineages. Biochem Pharmacol 37:2321–2327
Giverhaug T, Loennechen T, Aarbakke J (1999) The interaction of 6-mercaptopurine (6-MP) and methotrexate (MTX). Gen Pharmacol 33:341–346
Innocenti F, Danesi R, Di PA, Loru B, Favre C, Nardi M, Bocci G, Nardini D, Macchia P, Del TM (1996) Clinical and experimental pharmacokinetic interaction between 6-mercaptopurine and methotrexate. Cancer Chemother Pharmacol 37:409–414
Schmiegelow K, Bretton-Meyer U (2001) 6-mercaptopurine dosage and pharmacokinetics influence the degree of bone marrow toxicity following high-dose methotrexate in children with acute lymphoblastic leukemia. Leukemia 15:74–79
van Kooten Niekerk PB, Schmiegelow K, Schroeder H (2008) Influence of methylene tetrahydrofolate reductase polymorphisms and coadministration of antimetabolites on toxicity after high dose methotrexate. Eur J Haematol 81:391–398
Nygaard U, Schmiegelow K (2003) Dose reduction of coadministered 6-mercaptopurine decreases myelotoxicity following high-dose methotrexate in childhood leukemia. Leukemia 17:1344–1348
Karran P, Attard N (2008) Thiopurines in current medical practice: molecular mechanisms and contributions to therapy-related cancer. Nat Rev Cancer 8:24–36
Duley JA, Florin TH (2005) Thiopurine therapies: problems, complexities, and progress with monitoring thioguanine nucleotides. Ther Drug Monit 27:647–654
Weinshilboum RM, Otterness DM, Szumlanski CL (1999) Methylation pharmacogenetics: catechol O-methyltransferase, thiopurine methyltransferase, and histamine N-methyltransferase. Annu Rev Pharmacol Toxicol 39:19–52
Wang L, Weinshilboum R (2006) Thiopurine S-methyltransferase pharmacogenetics: insights, challenges and future directions. Oncogene 25:1629–1638
Ebbesen MS, Nersting J, Jacobsen JH, Frandsen TL, Vettenranta K, Abramsson J, Wesenberg F, Schmiegelow K (2013) Incorporation of 6-thioguanine nucleotides into DNA during maintenance therapy of childhood acute lymphoblastic leukemia—the influence of thiopurine methyltransferase genotypes. J Clin Pharmacol 53:670–674
Schmiegelow K, Bjork O, Glomstein A, Gustafsson G, Keiding N, Kristinsson J, Makipernaa A, Rosthoj S, Szumlanski C, Sorensen TM, Weinshilboum R (2003) Intensification of mercaptopurine/methotrexate maintenance chemotherapy may increase the risk of relapse for some children with acute lymphoblastic leukemia. J Clin Oncol 21:1332–1339
Schmiegelow K, Forestier E, Kristinsson J, Soderhall S, Vettenranta K, Weinshilboum R, Wesenberg F (2009) Thiopurine methyltransferase activity is related to the risk of relapse of childhood acute lymphoblastic leukemia: results from the NOPHO ALL-92 study. Leukemia 23:557–564
Andersen JB, Szumlanski C, Weinshilboum RM, Schmiegelow K (1998) Pharmacokinetics, dose adjustments, and 6-mercaptopurine/methotrexate drug interactions in two patients with thiopurine methyltransferase deficiency. Acta Paediatr 87:108–111
Gustafsson G, Schmiegelow K, Forestier E, Clausen N, Glomstein A, Jonmundsson G, Mellander L, Makipernaa A, Nygaard R, Saarinen-Pihkala UM (2000) Improving outcome through two decades in childhood ALL in the Nordic countries: the impact of high-dose methotrexate in the reduction of CNS irradiation. Nordic Society of Pediatric Haematology and Oncology (NOPHO). Leukemia 14:2267–2275
Kaplan EJ, Meier P (1958) Non-parametric estimation from incomplete observations. J Am Stat Assoc 53:457–481
Mantel N (1966) Evaluation of survival data and two new rank order statistics arising in its consideration. Cancer Chemother 50:163–170
Liang KY, Zeger SL (1986) Longitudinal data analysis using generalized linear models. Biometrika 73:13–22
Pesarin F, Salmaso L (2010) Permutation tests for complex data: theory, applications and software. Wiley, Chicester
Pinheiro JC, Bates DM (2000) Mixed-effect models in S and S-Plus. Springer, New York
Harrell FE (2001) Regression modeling strategies: with applications to linear models, logistic regression and survival analysis. Springer, New York
Efron B (1981) Nonparametric estimates of standard error: the jackknife, the bootstrap and other methods. Biometrika 68:589–599
Schmiegelow K (2009) Advances in individual prediction of methotrexate toxicity: a review. Br J Haematol 146:489–503
Evans WE, Horner M, Chu YQ, Kalwinsky D, Roberts WM (1991) Altered mercaptopurine metabolism, toxic effects, and dosage requirement in a thiopurine methyltransferase-deficient child with acute lymphocytic leukemia. J Pediatr 119:985–989
Lennard L, Gibson BE, Nicole T, Lilleyman JS (1993) Congenital thiopurine methyltransferase deficiency and 6-mercaptopurine toxicity during treatment for acute lymphoblastic leukaemia. Arch Dis Child 69:577–579
McLeod HL, Coulthard S, Thomas AE, Pritchard SC, King DJ, Richards SM, Eden OB, Hall AG, Gibson BE (1999) Analysis of thiopurine methyltransferase variant alleles in childhood acute lymphoblastic leukaemia. Br J Haematol 105:696–700
McLeod HL, Krynetski EY, Relling MV, Evans WE (2000) Genetic polymorphism of thiopurine methyltransferase and its clinical relevance for childhood acute lymphoblastic leukemia. Leukemia 14:567–572
Relling MV, Hancock ML, Rivera GK, Sandlund JT, Ribeiro RC, Krynetski EY, Pui CH, Evans WE (1999) Mercaptopurine therapy intolerance and heterozygosity at the thiopurine S-methyltransferase gene locus. J Natl Cancer Inst 91:2001–2008
Schmiegelow K, Al-Modhwahi I, Andersen MK, Behrendtz M, Forestier E, Hasle H, Heyman M, Kristinsson J, Nersting J, Nygaard R, Svendsen AL, Vettenranta K, Weinshilboum R (2009) Methotrexate/6-mercaptopurine maintenance therapy influences the risk of a second malignant neoplasm after childhood acute lymphoblastic leukemia: results from the NOPHO ALL-92 study. Blood 113:6077–6084
Levinsen M, Rotevatn EO, Rosthoj S, Nersting J, Abrahamsson J, Appell ML, Bergan S, Bechensteen AG, Harila-Saari A, Heyman M, Jonsson OG, Maxild JB, Niemi M, Soderhall S, Schmiegelow K (2014) Pharmacogenetically based dosing of thiopurines in childhood acute lymphoblastic leukemia: influence on cure rates and risk of second cancer. Pediatr Blood Cancer 61:797–802
Stanulla M, Schaeffeler E, Moricke A, Coulthard SA, Cario G, Schrauder A, Kaatsch P, Dordelmann M, Welte K, Zimmermann M, Reiter A, Eichelbaum M, Riehm H, Schrappe M, Schwab M (2009) Thiopurine methyltransferase genetics is not a major risk factor for secondary malignant neoplasms after treatment of childhood acute lymphoblastic leukemia on Berlin–Frankfurt–Munster protocols. Blood 114:1314–1318
Coulthard SA, Howell C, Robson J, Hall AG (1998) The relationship between thiopurine methyltransferase activity and genotype in blasts from patients with acute leukemia. Blood 92:2856–2862
Lennard L (2014) Implementation of TPMT testing. Br J Clin Pharmacol 77:704–714
Schaeffeler E, Fischer C, Brockmeier D, Wernet D, Moerike K, Eichelbaum M, Zanger UM, Schwab M (2004) Comprehensive analysis of thiopurine S-methyltransferase phenotype–genotype correlation in a large population of German-Caucasians and identification of novel TPMT variants. Pharmacogenetics 14:407–417
Acknowledgments
We acknowledge the critical input of Dr. P. D. Cole. The commitment and skillful technical assistance of Michael Timm are greatly appreciated. The study was funded by The Otto Christensens Fund, The Danish Childhood Cancer foundation, The Carl and Ellen Hertz Foundation, The Children’s Cancer Foundation of Sweden, The Danish Cancer Society, The JPC Foundation, The Lundbeck Foundation, The Minister Erna Hamilton Foundation, The Nordic Cancer Union and the US National Institutes of Health Grants R01 GM28157 and U19 GM61388.
Conflict of interest
None.
Ethical standard
The ALL92 protocol was approved by the Ethical Committee of Copenhagen as well as by the local ethical committees, and participants gave informed consent according to the Helsinki Declaration.
Author information
Authors and Affiliations
Corresponding author
Additional information
For the Nordic Society of Paediatric Haematology and Oncology (NOPHO).
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Levinsen, M., Rosthøj, S., Nygaard, U. et al. Myelotoxicity after high-dose methotrexate in childhood acute leukemia is influenced by 6-mercaptopurine dosing but not by intermediate thiopurine methyltransferase activity. Cancer Chemother Pharmacol 75, 59–66 (2015). https://doi.org/10.1007/s00280-014-2613-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-014-2613-7