
Abstract
Richter’s transformation (RT) is a rare complication arising in patients with chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) and is associated with an overall dismal outcome. The rarity of this entity poses many challenges in understanding its biology and outcomes seen and the optimal treatment approach. We utilized the SEER (Surveillance, Epidemiology and End Results) database to identify patients diagnosed with CLL/SLL between 2000 and 2016 and subsequently had a diagnosis of diffuse large B-cell lymphoma (DLBCL) or Hodgkin lymphoma (HL), thus capturing those who experienced an RT event. We compared the outcomes of those patients to those of patients in the database diagnosed with DLBCL without a preceding CLL/SLL diagnosis. We identified 530 patients who developed RT out of 74,116 patients diagnosed with CLL/SLL in the specified period. The median age at RT diagnosis was 66 years, and the median time from CLL/SLL diagnosis to RT development was roughly 4 years. Patients with RT had a dismal outcome with median overall survival of 10 months. We identified advanced Ann Arbor stage (III/IV) and prior treatment for CLL as predictors of worse outcome in patients with RT. Our study represents the largest dataset of patients with CLL/SLL and RT and adds to the existing literature indicating the poor outcomes for those patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The recent few years witnessed many strides in the treatment of patients with chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL).
However, significant gaps remain in risk factors, biology, prognosis, and optimal treatment approach for patients who develop Richter’s transformation (RT), a rare complication of CLL/SLL with a reported incidence of 4.8% at 10 years [1]. RT denotes disease transformation to an aggressive lymphoma, most commonly a diffuse large B-cell lymphoma (DLBCL), or less commonly a Hodgkin lymphoma (HL). Maurice Richter was the first to describe the pathologic appearance of a “generalized reticular cell sarcoma” in a patient with CLL in 1928 [2]. The terms Richter’s syndrome and Richter’s transformation (RT) are used interchangeably nowadays.
Available reports addressing RT are limited by their retrospective nature and generally the small number of patients included. Additionally, the rarity of RT meant that most retrospective studies spanned several years that saw several changes in the paradigms of treatment of CLL, HL, and DLBCL, making it very challenging to compare the results of those studies and discern prognostic/predictive factors for outcomes and the optimal treatment strategies in those patients. We sought to utilize the Surveillance, Epidemiology and End Results (SEER) database to evaluate outcomes with RT in a large cohort of patients.
Methods
Database
Covering approximately 35% of the US population; SEER is a program of the National Cancer Institute that collects and publishes cancer incidence and survival data from national population-based cancer registries (http://www.seer.cancer.gov). We used the SEER 2018 publication and identified three cohort of patients over the period 2000–2016 and labeled them as follows: Patients with CLL who did not develop RT (CLL WO RT), patients with CLL who developed RT (CLL W RT), and patients with DLBCL and no history of CLL in the SEER database (de novo DLBCL).
All data were collected and used following the SEER policies and procedures. All CLL/SLL cases during the period between January 2000 and December 2016 were identified in the SEER cancer registry, using the International Classification of Diseases ICD-O-3 code 9823/3. Patients who had subsequent diagnosis of DLBCL (ICD-O-3 code 9680/3) or HL (ICD-O-3 code 9650/3) at least 2 months after CLL/SLL diagnosis and within the study period were identified.
Data obtained included age, gender, race, history of treatment for CLL, overall survival (OS), lymphoma anatomical site, Ann Arbor stage, and the time period between CLL and RT diagnoses.
Definitions and statistical analysis
Overall survival was the primary endpoint of this study and was defined as the period from CLL/SLL diagnosis to death from any cause and was determined from the SEER records of survival time (total number of months) and vital status (dead or alive). The reversed Kaplan–Meier method was used to calculate median follow-up time. Secondary endpoints were comparison of baseline characteristics between the CLL/SLL cohort who developed RS and those who did not.
Statistical analyses were performed using PC SAS 9.4. χ-square tests and Fisher’s exact tests were used to compare differences between nominal variables, and the Mann–Whitney U test or the Kruskal–Wallis test was used for continuous variables. Kaplan–Meier method and log-rank test were used to analyze and create the survival curve. Finally, the Cox proportional hazards model was used to assess the influence of various prognostic factors on OS.
Results
We identified 74,116 patients diagnosed with CLL between 2000 and 2016. A total of 530 patients developed RT (0.72%). Of the 530 patients who developed RT, most patients experienced transformation to a DLBCL (85%), while 81 patients (15%) developed Hodgkin lymphoma (Table 1). Most of those transformation events were nodal (74%). The most commonly involved extra-nodal sites were the GI tract (25%), skeletal system (19%), and the brain/CNS (12%).
The median age of the subgroup of CLL WO RT was 70 years, males constituted 60% of this population, and 90% were white. Regarding treatment status, 84% of these patients had not received chemotherapy for CLL or had unknown treatment status, while 1% were reported to have received radiation therapy. A significant improvement in median overall survival was noted for patients diagnosed with CLL between 2011 and 2016 compared to those diagnosed between 2000 and 2010 (Fig. 1, Table 2) with median OS not reached versus 98 months (HR 1.2, 95% CI 1.15–1.23), p < 0.0001.

KM curves for OS of CLL/SLL patients
The median time from the diagnosis of CLL to the development of RT was almost 4 years, with no significant difference in the time interval based on histology of the transformation event comparing DLBCL and HL (47 months versus 49 months, P = 0.22) and the median age at the time of RT was 66 years.
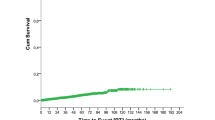
The median overall survival from the RT event diagnosis among the entire population that developed RT was 10 months (Fig. 2), with a significantly better median overall survival for those who were treatment naïve for CLL as opposed to those with prior treatment for CLL (12 months versus 7 months, P = 0.02). We found that 72% of patients received chemotherapy for RT, resulting in a significantly improved outcome compared to the remainder of patients who did not receive treatment (or had unknown status for treatment with chemotherapy), with median overall survival of 15 months versus 2 months (HR 2.2, 95% CI 1.74–2.79, p < 0.0001), and an advanced Ann Arbor stage (3/4 versus 1/2) was associated with worse overall survival on multivariate analysis with HR of 1.58 (95% CI 1.23–2.03, P = 0.0003).

KM curve for OS of de novo DLBCL versus DLBCL arising from RT
Fourteen percent of the entire cohort of RT patients received radiation as part of their treatment.
When stratified by the histology of the transformation event (Fig. 3), no significant difference in median OS was noted between DLBCL and Hodgkin lymphoma (10 months versus 14 months, P = 0.22).

KM curves for RT stratified by histology (DLBCL versus HL)
Within the SEER database, we found a total of 97,415 patients who were diagnosed with DLBCL over the study specified period. The median age at the time of diagnosis was 68 years, with male predominance (55%). The median overall survival for all patients was 62 months. Factors associated with a significantly worse OS on multivariate analysis included age > 60 years, male gender, and advanced Ann Arbor stage. Black patients had a significantly worse outcome compared to white patients with HR of 1.24 (95% CI 1.20–1.29, p < 0.0001) (Table 3).
Discussion
To our knowledge, our study represents the largest dataset of patients with RT published to date. Several limitations exist due the nature of the SEER registry and lack of central review of the pathology slides. As no reassurance exists to the quality of pathological review used to label each patient included in the dataset, we aimed to minimize the risk of incorporating patients who were incorrectly diagnosed with HL or DLBCL without a true culprit transformation event by incorporating a delay of 2 months between the diagnosis of CLL and RT in the registry. We thus excluded patients who were likely to have presented with symptomatic disease leading to a diagnosis of one of the three entities (CLL/SLL, HL, or DLBCL).
The results from our review of this large dataset are comparable to many of the previous reports. However, while some of the observations were consistent among several reports of RT, significant differences are also noted.
Our study found that the median overall survival from the RT event diagnosis among the entire population that developed RT was 10 months. This is in line with previous US reports such as the large MD Anderson series by Tsimberidou et al. [3] (n = 148, median OS 8 months), and the Mayo clinic series by Parikh et al. [1] (n = 120, median OS 0.9 years). The similarity in results between the two series is notable given the difference in the timeframe covered (1975–2005 and 1995–2013, respectively).
Interestingly, significantly better outcomes were seen in the study by Ben-Dali et al. [4] which used data from the Danish National CLL registry, providing coverage of nearly 99% of patients diagnosed with CLL between 2008 and 2016, with a median OS after development of RT (n = 113) in this patient population of 3.1 years for DLBCL and 2.9 years for HL.
The reason for this remarkably better outcome is unclear. Only 12 patients received a hematopoietic stem cell transplant (HSCT) in this cohort (5 autologous and 7 allogeneic, with median OS of 4.1 years and 4.3 years, respectively). The results from our large dataset make it likely that a role for referral bias contributing to the poor prognosis noted in the previous single-center US studies is minimal, if any.
A clonal relationship to the underlying CLL has been reported to be present in more than 75% of cases of RT to DLBCL [5]. Those with a clonally unrelated DLBCL have a significantly lower prevalence of TP53 disruption and better outcomes compared to those with clonally related DLBCL (median 62.5 months versus 14.2 months) [6].
While data on clonal relationship of the RT in our study are not available from the SEER database, the poor median overall survival supports the existing evidence that the clonally unrelated (de novo) DLBCL that is associated with better prognosis represents a minority of cases in RT. Other limitations of our study related to the nature of the SEER database are the lack of specific data about CLL genetic markers and specific CLL prior therapies.
Similar to previous reports [4], our analysis found a significantly better outcome for those who were treatment naïve for CLL as opposed to those who were previously treated for CLL (12 months versus 7 months, P = 0.02). The association of prior treatment for CLL with a poor median overall survival for RT is still noted in the era of novel agents. [7, 8]
This is likely related to biological differences in DLBCL in the setting of RT or the inherently different population selected for treatment with novel agents as opposed to chemoimmunotherapy, as exemplified by the frequent findings of TP53 disruption in RT developing while on venetoclax or ibrutinib therapy [9, 10], and 17p deletion has been reported as an independent prognostic factor for development of RT [11]. The findings of worse outcome for RT during treatment with novel agents may also be reflective of outcomes in a heavily pretreated population.
By assessing the outcomes for patients diagnosed with DLBCL without a preceding CLL/SLL diagnosis in the same timeframe (Fig. 2), we were able to identify a comparison group from the same dataset to properly compare outcomes of de novo DLBCL to those arising from RT. The median overall survival for de novo DLBCL was 62 months.
The significant improvement in median overall survival noted for patients diagnosed between 2011 and 2016 compared to those diagnosed between 2000 and 2010 (median OS not reached versus 98 months) likely reflects improvement in supportive care and salvage therapy options. Our study also found a significantly worse outcome for black patients compared to white patients (HR 1.238, p < 0.0001). Prior reports have demonstrated such disparities in outcomes [12], as well as younger age and a more advanced stage at presentation for black patients. [13]
Factors reported to affect the outcome of patients with RT-DLBCL included platelet count < 100 × 109 /L and presence of TP53 alterations [14]. Our study identified advanced Ann Arbor stage and prior therapy for CLL as factors associated with worse overall survival in patients with RT.
Our analysis shows no significant difference in the time from CLL diagnosis to the RT event whether it was to a DLBCL or a HL (47 months versus 49 months, P = 0.2163).
Transformation to a HL is a rare event. Our study identified 81 patients who developed RT to HL, corresponding to an incidence of 0.1% with a time from CLL/SLL to RT of 49 months that is in line with previous reports, where median time to transformation to HL was between 37.2 and 55 months. [11, 15]
The reported outcomes for HL arising from RT are variable, with some reporting good outcomes such as that seen in the pooled analysis of German CLL study group (GCLLSG) front line treatment trials [11], which reported a median OS of 82.6 months (n = 8), contrary to a series of 18 patients from MD Anderson that had a median OS of 0.8 years. [15]
For the HL variant of RT, advanced age was reported as the strongest predictor for shorter overall survival, and the clonal relationship did not affect the outcome in a large series that included 77 patients [16].
Our findings of a median OS for HL-RT of 14 months when considered with the previous reports suggest that the outcomes are generally worse compared to de novo Hodgkin lymphoma, though some patients may have favorable outcomes.
The optimal treatment approach for patients with RT is unclear. Majority of patients with RT to DLBCL are treated with R-CHOP like regimens. [4, 11]
Given the poor prognosis, the ASBMT (now the ASTCT) in their 2016 guideline for use of allogeneic HSCT in CLL provided strong recommendation for allogeneic stem cell transplantation (alloSCT) in RT patients who achieve an objective response to anthracycline-based chemotherapy. [17]
A retrospective, survey-based analysis from the European Group for Blood and Marrow Transplantation (EBMT) identified 59 patients who received HSCT (34 autologous transplants, 25 allogeneic transplants) with 36% of patients refractory to chemotherapy at the time of stem cell transplant. Outcomes for post-remission HSCT in DLBCL type RT showed a 3-year survival of 36% for allogeneic transplant and 59% after autologous SCT. [18] For patients who received an allogeneic transplant, the disease status at the time of transplant significantly impacted survival (3-year survival of 41% for those transplanted in complete response or partial response compared to 17% for those transplanted with progressive disease).
With evolving treatment options for CLL, our understanding of the biology and pathogenesis of RT continues to grow. Particularly poor outcomes have been reported for patients developing RT on ibrutinib therapy (median OS of 2 months and 3.5 months in two series). [7, 19]
More interestingly, Parikh et al. [20] recently described three patients who were diagnosed with RT incidentally during temporary interruption of ibrutinib therapy, and all patients responded to reintroduction of ibrutinib.
As more novel therapeutic options for CLL become available, the changes this may bring to the incidence, prognosis, and best treatment approach to RT will need to be revisited more thoroughly.
The ideal treatment approach for patients with DLBCL type RT thus generally depends on whether or not there is clonal relationship to the preceding CLL, patient fitness for enrollment in clinical trials, and patient’s candidacy for consideration of autologous or allogeneic SCT [21].
CAR-T cell therapy is a new promising option for patients with relapsed or refractory DLBCL.
With regard to DLBCL arising as a transformation event, the three landmark trials for CAR-T cell therapy in DLBCL differed in their enrollment criteria. The TRANSCEND trial [22] allowed transformed DLBCL from indolent histology, while JULIET [23] and the ZUMA-1 trial [24] allowed enrollment of patients with transformation from a follicular lymphoma (tFL). The full cohort for TRANSCEND trial included one patient with RT.
It is thus not possible to draw any conclusions about whether the response rates noted with the different CAR-T products in DLBCL still apply for those with DLBCL arising from an RT event, and further data is needed in this space. It is notable that a single-center experience reporting the largest cohort of patients with RT (n = 9) treated with commercial axicabtagene ciloleucel (axi-cel) showed promising results, with 5 out of 8 patients who underwent formal response assessment achieving a CR and 3 patients achieving a PR. [25]
A few promising regimens that have been studied in RT are:
-
The combination of nivolumab plus ibrutinib (phase II study), which showed a response rate of 43% and a median overall survival of 13.8 months. [26]
-
VR-EPOCH (phase II study) which showed a response rate of 59% and a median OS of 16.3 months. [27]
-
U2 (umbralisib and ublituximab) plus pembrolizumab (phase I/II) showing a response rate of 38% in RT (n = 8). [28]
Some of the agents and regimens currently under investigation for treatment of RT are zanubrutnib (NCT04271956), combination of venetoclax, atezolizumab, and obinutuzumab (NCT04082897), obinutuzumab, high-dose methylprednisolone (HDMP) and lenalidomide (NCT03113695), and atezolizumab, gemcitabine, oxaliplatin, and rituximab (NCT03321643).
Conclusion
In summary, our study highlights the challenging nature of RT and the overall poor outcomes for this rare phenomenon. Given the evolving landscape in treatment of CLL and DLBCL, more gaps in our understanding of RT are likely to surface. Whether the favorable outcomes for DLBCL with the newer approaches such as CAR-T cell therapy would apply as effectively for RT-DLBCL is yet to be proven. Continued dynamic investigation of RT biology and pathogenesis is thus needed and multi-center collaborations are strongly encouraged.
References
Parikh SA et al (2013) Diffuse large B-cell lymphoma (Richter syndrome) in patients with chronic lymphocytic leukaemia (CLL): a cohort study of newly diagnosed patients. Br J Haematol 162(6):774–782
Richter MN (1928) Generalized reticular cell sarcoma of lymph nodes associated with lymphatic leukemia. Am J Pathol 4(4): 285–292.7
Tsimberidou AM et al (2006) Clinical outcomes and prognostic factors in patients with Richter’s syndrome treated with chemotherapy or chemoimmunotherapy with or without stem-cell transplantation. J Clin Oncol 24(15):2343–2351
Ben-Dali Y et al (2020) Richter’s transformation in patients with chronic lymphocytic leukaemia: a nationwide epidemiological study. Leuk Lymphoma 61(6):1435–1444
Mao Z et al (2007) IgVH mutational status and clonality analysis of Richter’s transformation: diffuse large B-cell lymphoma and Hodgkin lymphoma in association with B-cell chronic lymphocytic leukemia (B-CLL) represent 2 different pathways of disease evolution. Am J Surg Pathol 31(10):1605–1614
Rossi D et al (2011) The genetics of Richter syndrome reveals disease heterogeneity and predicts survival after transformation. Blood 117(12):3391–3401
Jain P et al (2017) Long-term outcomes for patients with chronic lymphocytic leukemia who discontinue ibrutinib. Cancer 123(12):2268–2273
Davids MS, et al (2017) Richter’s syndrome (RS) in patients with chronic lymphocytic leukemia (CLL) on novel agent therapy. J Clin Oncol 35(15_suppl):7505–7505
Anderson MA et al (2017) Clinicopathological features and outcomes of progression of CLL on the BCL2 inhibitor venetoclax. Blood 129(25):3362–3370
Miller CR et al (2017) Near-tetraploidy is associated with Richter transformation in chronic lymphocytic leukemia patients receiving ibrutinib. Blood Adv 1(19):1584–1588
Al-Sawaf O et al (2021). Richter transformation in chronic lymphocytic leukemia (CLL)-a pooled analysis of German CLL Study Group (GCLLSG) front line treatment trials. Leukemia, 35(1), 169–176.
Griffiths R et al (2010) Racial differences in treatment and survival in older patients with diffuse large B-cell lymphoma (DLBCL). BMC Cancer 10:625–625
Shenoy PJ et al (2011) Racial differences in the presentation and outcomes of diffuse large B-cell lymphoma in the United States. Cancer 117(11):2530–2540
Abrisqueta P et al (2020). Clinical outcome and prognostic factors of patients with Richter syndrome: real-world study of the Spanish Chronic Lymphocytic Leukemia Study Group (GELLC). British journal of haematology, 190(6), 854–863.
Tsimberidou AM et al (2006) Hodgkin transformation of chronic lymphocytic leukemia: the M. D Anderson Cancer Center experience. Cancer 107(6):1294–1302
Xiao W et al (2016) Hodgkin lymphoma variant of Richter transformation: morphology, Epstein-Barr virus status, clonality, and survival analysis-with comparison to Hodgkin-like lesion. Hum Pathol 55:108–116
Kharfan-Dabaja MA et al (2016) Clinical practice recommendations for use of allogeneic hematopoietic cell transplantation in chronic lymphocytic leukemia on behalf of the Guidelines Committee of the American Society for Blood and Marrow Transplantation. Biol Blood Marrow Transplant 22(12):2117–2125
Cwynarski K et al (2012) Autologous and allogeneic stem-cell transplantation for transformed chronic lymphocytic leukemia (Richter’s syndrome): a retrospective analysis from the chronic lymphocytic leukemia subcommittee of the chronic leukemia working party and lymphoma working party of the European Group for Blood and Marrow Transplantation. J Clin Oncol 30(18):2211–2217
Maddocks KJ et al (2015) Etiology of ibrutinib therapy discontinuation and outcomes in patients with chronic lymphocytic leukemia. JAMA Oncol 1(1):80–87
Hampel PJ et al (2020) Incidental Richter transformation in chronic lymphocytic leukemia patients during temporary interruption of ibrutinib. Blood Adv 4(18):4508–4511
Rossi D, Spina V, Gaidano G (2018) Biology and treatment of Richter syndrome. Blood 131(25):2761–2772
Abramson JS et al (2020) Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet 396(10254):839–852
Schuster SJ et al (2019) Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med 380(1):45–56
Neelapu SS et al (2017) Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med 377(26):2531–2544
Kittai AS et al (2020) Clinical activity of axicabtagene ciloleucel in adult patients with Richter syndrome. Blood Adv 4(19):4648–4652
Jain N et al (2018) A phase II trial of nivolumab combined with ibrutinib for patients with Richter transformation. Blood 132(Supplement 1):296–296
Davids MS, et al (2020) A multicenter phase II study of venetoclax plus dose-adjusted R-EPOCH (VR-EPOCH) for Richter’s syndrome. J Clin Oncol 38(15_suppl):8004–8004
Mato AR et al (2019) Phase I/II study of umbralisib (TGR-1202) in combination with ublituximab (TG-1101) and pembrolizumab in patients with Rel/Ref Cll and Richter’s transformation. Hematol Oncol 37(S2):119–120
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical approval
This article does not contain any studies with human participants performed by any of the authors.
Informed consent
This study utilized a widely available database. Individual patient consent was thus not obtained for the purpose of this study. All data were collected and used following the SEER policies and procedures.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Elnair, R., Ellithi, M., Kallam, A. et al. Outcomes of Richter’s transformation of chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL): an analysis of the SEER database. Ann Hematol 100, 2513–2519 (2021). https://doi.org/10.1007/s00277-021-04603-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00277-021-04603-y