
Abstract
Early mortality remains a major challenge for the treatment of hemophagocytic lymphohistiocytosis (HLH), which warrants the need for prompt risk stratification in the early phase of the disease. We retrospectively analyzed clinical features of a cohort of pediatric patients managed at a tertiary hospital in southern China from 2005 to 2015. A total of 116 patients (median age 27.5 months) with predominantly secondary HLH were included. In a multivariate Cox regression model, neutrophils <0.5 × 109/L (risk ratio (RR) = 5.01; 95 % confidence interval (CI) 1.55–16.20; P = 0.007), total bilirubin over twofold upper limit of normal value (RR = 2.86; 95 % CI 0.83–9.88; P = 0.097), and albumin ≤20 g/L (RR = 5.79; 95 % CI 1.70–19.73; P = 0.005) at diagnosis were independent risk factors for 30-day mortality. The 30-day overall survival rate (OS) of patients with three risk factors was significantly lower than that of patients with zero to two risk factors (0 vs 90.7 %; P<0.001). Patients with three risk factors were 64-fold more likely to have early adverse outcome as compared to patients with zero to two risk factors (RR = 64.45; 95 % CI 18.35–226.33; P<0.001). Platelet count normalization in 2 weeks was an independent predictor for resolution after initial therapy with an odds ratio (OR) of 18.4 (95 % CI 2.7–122.9; P = 0.003). Our results indicate that severe neutropenia and liver function damage are prognostic factors for early death in HLH and platelet count normalization in 2 weeks is a critical predictor for resolution after initial therapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hemophagocytic lymphohistiocytosis (HLH) is a rare but life-threatening clinical syndrome primarily affecting infants and children, characterized by common clinical signs and symptoms of extreme inflammation [1]. Typical clinical findings of HLH are indicative of significant immune activation, including prolonged fever, hepatosplenomegaly, elevated levels of serum ferritin, and soluble CD25. Other presentations including cytopenias, hepatitis, decreased fibrinogen, increased triglyceride, hemophagocytosis, and central nervous system (CNS) involvement reflect organ damage as the consequence of immune dysregulation [2].
There are two forms of HLH: primary (genetic) and secondary (acquired). Familial hemophagocytic lymphohistiocytosis (FHL) is an autosomal recessive disease with an estimated incidence of 1.8 per 100,000 live births [3]. Known causative genes including PRF1, UNC13D, STX11, and STXBP2, all encode proteins required for lymphocyte cytotoxicity [4–7]. Other genetic immunodeficiencies can also develop HLH as part of the disease process, such as X-linked lymphoproliferative syndrome (XLP) due to SH2D1A or XIAP gene defects [8]. Patients in the primary category are usually infants or younger children with a clear risk of recurrence, typically requiring hematopoietic cell transplantation (HCT) for cure. In contrast, patients with secondary HLH (sHLH), which is often triggered by overwhelming immune stimuli such as severe infections, malignancies, or rheumatologic disorders, are generally older children or adults. Nevertheless, both forms have variable disease severity and may progress to a fatal outcome [9]. Thus, patients with the severe full picture of HLH require prompt administration of systemic immunochemotherapy per HLH-94 or HLH-2004 protocols to achieve immediate disease control [10, 11]. Approximately 80 % of patients respond to HLH-94 regimen but only half of patients can achieve resolution [12], indicating a significant number of patients not responding adequately or completely may need more aggressive treatment or salvage therapy in order to survive to HCT. In patients with sHLH who achieved resolution at week 8, the routine use of a per-protocol continuation therapy in these cases is not appropriate. So resolution at week 8 is the decision-point on whether to use continuation therapy after induction except for familial, genetically verified HLH. Thus, predictors in the early 2 weeks into therapy with evident relation to resolution could be especially useful for alerting clinicians to consider timely treatment improvement. In addition, early incorporation of cyclosporine into the etoposide-containing regimen appears beneficial to improve neutrophil recovery in HLH [13–15], so the impact of cyclosporine on early outcome has also been assessed in this study.
According to the data from HLH-94 trial, the majority of deaths occurred during initial therapy, especially in the first month, indicating different phases of the disease may have distinct odds of death [16]. Thus, we hypothesize that risk stratification in early stage of HLH may be critical to tailor treatment and reduce mortality. In this report, we examined the usefulness of regular clinical parameters for predicting early outcome in pediatric patients with HLH. Epstein-Barr virus-associated HLH (EBV-HLH) appears to have a high incidence in East Asian countries [17, 18]. Patients included in the present study could be a very typical cohort for investigating EBV-HLH because they are from Guangxi province, where has long been known as an endemic region with very high incidence of EBV infection in China [19].
Patients and methods
Patients and data collection
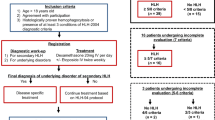
During the period from July 2005 to September 2015, patients (<14 years old) with discharge diagnosis of HLH in a tertiary medical center of Guangxi Province in China were retrospectively analyzed. Since two diagnostic criteria (soluble CD25 and NK cell activity tests) in HLH-2004 protocol were not available in our institution, diagnosis was done when five out of the other six criteria fulfilled [11]. Patients with clear evidence of rheumatologic diseases and malignancies were excluded. Clinical findings and laboratory data before and after therapy were obtained using medical chart review. EBV infections were confirmed serologically from evidence of initial or previous exposure to EBV (elevated viral capsid antigen IgM or IgG) or reactivation status (elevated early antigen IgG or IgA) and/or increased EBV genomic DNA copy number by polymerase chain reaction (PCR) [20]. Cytomegalovirus (CMV) infection was also routinely examined serologically (CMV-IgM and IgG) and by PCR. The evaluation of disease states was done based on HLH-2004 protocol. Clinical response was evaluated at 2 weeks during initial therapy as out-lined: no fever, spleen size reduction, platelets≧100 × 109/L, normal fibrinogen, and ferritin level decreasing by 25 %. Ferritin decrease was calculated as ferritin measure after 2 weeks of therapy minus the measure at diagnosis, divided by the measure at diagnosis. Non-active disease (resolution) was evaluated around 8 weeks after the start of therapy as follows: no fever, no splenomegaly, no cytopenia (hemoglobin ≧90 g/L, platelets ≧100 × 109/L, neutrophils >0.5 × 109/L), no hypertriglyceridemia (triglyceride <3 mmol/L), no hyperferritinemia (ferritin <500 μg/L), and normal cerebrospinal fluid (CSF). Active disease was diagnosed when patients did not have resolution. Overall survival (OS) was defined as the evaluation of the time from HLH diagnosis to the date of death from any cause or last follow-up. Follow-up was done by searching medical records or making phone calls. Early death was the primary outcome, and it was defined as death occurred within 30 days after diagnosis. An event truncation for the outcome of 30 days after onset of therapy was used. Genetic testing was carried out by amplifying all exons of PRF1, UNC13D, STX11, and XIAP genes using PCR and then directly sequencing on ABI 3730XL sequencer (Applied Biosystems, NY, USA). Variations identified in the above genes were compared to the information in NCBI dbSNP, OMIM, and UCSC databases to determine their function significance. Approval to perform this study was given by the First Affiliated Hospital of Guangxi Medical University Institutional Review Board. Informed consent was obtained from all patients for being included in the study.
Statistical methods
Statistical analyses were performed by SPSS (Statistical Package for Social Sciences) software, version 16.0 (SPSS, Chicago, IL). Continuous biologic variables were dichotomized applying usual clinical thresholds. First, univariate Cox regression analyses were used to screen possible predictors for early death. Univariate relations between resolution and prognostic variables were described as cross-tabulations and assessed by using Pearson chi-square test or Fisher’s exact test. Variables with a P value <0.15 in the univariate analysis were incorporated into a multivariate Cox’s proportional hazards regression model or logistic regression model to further assess their independent contribution to outcomes. Missing data were left as missing. A forward conditional selection procedure with significant level for entry into the model set at 0.10 was conducted. Prognostic groups (high or low risk) were defined by categorizing the number of prognostic factors in the final multivariate Cox regression model. Comparison of survival rates between groups was performed by using Kaplan-Meier method and log-rank test. Two tailed P value <0.05 was considered statistically significant.
Results
Patient characteristics
This study included a total of 116 Chinese children (77 males, 39 females) with HLH for analysis. The median age at diagnosis was 27.5 months (range 3 months to 14 years). 9.5 % (11/116) patients were infants (<1 year old). Clinical and laboratory characteristics of patients at diagnosis were shown in Table 1. Strikingly, 92.3 % (24/26) patients with adequate testing (both serologically and by PCR) were EBV infection positive. Almost all patients were tested for CMV infection, and the positive rate was 8.3 % (9/109). No consanguinity was reported, and no patient had a family history of HLH. No known HLH-related mutation was identified in PRF1, UNC13D, STX11, SH2D1A, and XIAP genes of 60 patients in this cohort. Five monoallelic single nucleotide variations (SNV) with uncertain clinical significance in PRF1 exons including R4C (rs12161733) in four unrelated patients, V50L (rs776299562), A424T, L478Q, and R489W in each patient were detected. Three patients harbored each monoallelic SNV in UNC13D exons: R414L (rs768171054), R682G, and R921H. There was no mutation identified in STX11, SH2D1A, and XIAP genes.
Patients were treated basically per HLH-94 or HLH-2004 protocol but off the clinical trials. In total, all patients were treated with dexamethasone and etoposide, and 59/116 (50.9 %) patients had cyclosporine A (CSA) per OS from the start of therapy in 8 weeks of initial therapy. No patient received intrathecal therapy during the 8 weeks. Neutropenic patients were protectively isolated. Antibiotic prophylaxis was not routinely applied. Empirical antibiotic therapy, including broad-spectrum piperacillin-sulbactam or cefoperozone-sulbactam, and intravenous antifungal therapy were prescribed initially for neutropenic patients with clinical signs and/or laboratory indicators of infection and were adjusted according to treatment response and identification of pathogens. A total of 31 HLH-related fatalities occurred in this cohort: 16/31 (51.6 %) deaths occurred within 30 days after onset of therapy, one patient (3.2 %) died during 4–8 weeks, 9/31 (29.0 %) deaths occurred during 2–6 months, and 5/31 (16.1 %) deaths occurred after 6 months. There were 9/116 (7.8 %) patients had reactivation after remission, six of them reactivated in 6 months from the start of therapy and the other three had late recurrence after 6 months. Six patients with reactivation were genetically tested and results only showed two of them harbored the monoallelic R4C variation in PRF1. No patient proceeded to HCT. Patients were followed up at least 3 months.
Prognostic factors for early death
Six patients were lost to follow-up by 30 days after diagnosis, and the dropout rate is 4.9 %. The 30-day OS of this cohort was 86.0 %. The correlations between clinical characteristics at diagnosis and 30-day mortality by univariate analysis were shown in Table 2. Results showed that neutrophils <0.5 × 109/L (RR = 3.82; 95 % CI 1.33–11.00; P = 0.01), albumin level ≦20 g/L (RR = 4.96; 95 % CI 1.80–13.68; P = 0.002), total bilirubin >twofold upper limit of normal value (2ULN) (RR = 3.86; 95 % CI 1.34–11.12; P = 0.01), and lactate dehydrogenase (LDH) >1000 IU/L (RR = 3.71; 95 % CI 1.05–13.16; P = 0.04) were significantly associated with increased early death in HLH. However, variables including age <1 year, gender, hemoglobin <60 g/L, platelets <20 × 109/L, fibrinogen ≦1.5 g/L, triglyceride ≧3 mmol/L, ALT >200 IU/L, ferritin >2000 or>10,000 μg/L, and hemophagocytosis in bone marrow, as well as abnormal CSF at diagnosis were not significantly correlated with early adverse outcome. Administration of CSA from the start of therapy was not significantly associated with a decreased risk of early death (RR = 0.93; 95 % CI 0.35–2.48; P = 0.89). Subsequently, age, gender, neutrophils, hemoglobin, albumin, total bilirubin, LDH, and SF (> or ≦10,000 μg/L) were included into a multivariate Cox regression analysis, and neutrophils <0.5 × 109/L, total bilirubin >2ULN, and albumin ≦20 g/L were retained in the final model as independent risk factors for early death (Table 3). The 30-day OS was significantly different between patients with neutrophils ≧ or <0.5 × 109/L (92.6 % vs 75.4 %; P = 0.007; Fig. 1a), total bilirubin > or ≦2ULN (75.6 % vs 92.7 %; P = 0.007; Fig. 1b), and albumin > or ≦20 g/L (89.8 % vs 62.5 %; P = 0.001; Fig. 1c). Patients with all of the above three risk factors at diagnosis in our cohort had 100 % mortality within 30 days, and the median survival time was 2.0 days (95 % CI 0–5.6). There was no significant difference among 30-day OS of patients with zero, one, and two risk factors at diagnosis (95.2 86.8, and 90.5 %; P = 0.40). The 30-day OS of patients with three risk factors was significantly lower than that of patients with zero to two risk factors (0 vs 90.7 %; P<0.001). Thus, patients with all three risk factors at diagnosis were categorized into high-risk group, while patients with zero to two risk factors were at relatively low risk (Fig. 1d). And patients with three risk factors were 64-fold more likely to have early adverse outcome as compared to patients with zero to two risk factors (RR = 64.45; 95 % CI 18.35–226.33; P<0.001).

Kaplan-Meier curve of patients with risk factors by 30 days after diagnosis. Overall survival (OS) of patients with a neutrophils ≥ or <0.5 × 109/L, b total bilirubin > or 2 upper limit of normal value (ULN), c albumin > or ≤20 g/L, and d zero to two risk factors (low-risk group) or three risk factors (high-risk group)
Prognostic factors for resolution
Patients with sufficient data to be evaluated for disease states around 8 weeks of treatment were dichotomized according to the resolution or active disease. Patients died before 8 weeks were excluded from this analysis. In this study cohort, 72.0 % (59/82) patients achieved resolution after 8 weeks of therapy. The relation between disease states after 8 weeks and changes of clinical findings over the first 2 weeks of therapy was examined to identify predictors for resolution (Table 4). Remarkably, patients with normal body temperature at 2 weeks into therapy had obviously higher rate of resolution after 8 weeks than those patients with persistent fever (P = 0.02). And patients with platelet count ≧100 × 109/L at 2 weeks also had significantly higher rate of resolution than those patients with platelet count <100 × 109/L (χ2 = 10.2, P = 0.001). In patients with serial ferritin data, 97.1 % (33/34) patients had ferritin level decrease by 25 % at 2 weeks, and 64.7 % (22/34) patients had ferritin level decrease by 80 %. Patients with ferritin decrease over 80 % at 2 weeks also had significantly higher rate of resolution than those patients with ferritin decrease less than 80 % (P = 0.03). Clinical response at 2 weeks during initial therapy was evaluated for patients with sufficient parameters. The clinical response rate at 2 weeks was 51.0 % (26/51). A total of 37 patients with evaluation of both 2-week clinical response and 8-week disease states were analyzed, and results showed that there was no statistically significant association between 2-week clinical response and 8-week resolution (P = 0.08). Subsequently, fever, platelet count, and ferritin decrease after 2 weeks of therapy were incorporated into a multivariate logistic regression analysis, which resulted in a parsimonious model with the main effect of platelet count over 100 × 109/L with an odds ratio (OR) of 18.4 (95 % CI 2.7–122.9; P = 0.003), indicating that patients was 18 times more likely to attain resolution after initial therapy when platelet count was above 100 × 109/L at 2 weeks into therapy as compared to patients with lower platelet count.
Discussion
Early initiation of an appropriate treatment is critical for improving the chance of survival in patients with HLH. However, it is often difficult to decide the treatment intensity without reliable prognostic indicators in the very early phase of the disease. This study reports original findings of predictors for 30-day mortality in Chinese children with predominantly sHLH. In this cohort, over half of all deaths occurred in the first month of therapy, which we termed “early death.” Since the majority of early deaths actually happened in 2 weeks after diagnosis, we believe that the most helpful predictor should be assessed upon diagnosis and guide clinicians to categorize patients into high- or low-risk subgroups. As previously reported, hyperbilirubinemia or jaundice were significantly associated with adverse outcome in HLH [9, 21, 22]. Our results confirmed high bilirubin more than twofold upper limit of normal value (usually with jaundice) as a risk factor for 30-day mortality. We also found that severe neutropenia or agranulocytopenia (neutrophils <0.5 × 109/L), as well as severe hypoalbuminemia (albumin ≦20 g/L) are independent risk factors. No patient with all three risk factors at diagnosis has survived till 30 days in our cohort, as patients with three risk factors altogether could be with extremely overwhelming disease and ongoing severe organ damage. Our finding of the prognostic value of severe neutropenia agrees with the analytic study by another Chinese group, in which leukopenia (white blood cell count <3 × 109/L) upon hospitalization was found to be correlated with inferior survival in pediatric EBV-HLH [23]. Patients with leukopenia or agranulocytopenia are prone to severe opportunistic infection, which is known as a common cause of death in HLH [24]. Ten out of the 16 early deaths in our cohort had severe infection complications including pneumonia and sepsis, which played a major role in early mortality despite aggressive antimicrobial therapy. Prompt short-term CSA infusion followed by CSA per OS has been described as an attractive treatment to improve neutrophil recovery in patients with HLH [13]; however, our result does not support that early administration of CSA per OS can significantly improve early outcomes. Edema has been described as a factor associated with deaths in the first 8 weeks of initial therapy [16], while severely decreased albumin is demonstrated to be a strong prognostic marker in our study, reflecting the similar biological effect as edema but in a more objective manner. Abnormal CSF at diagnosis is associated with poor long-term survival and increased risk of sequelae [25, 26], but it appears not a risk factor for early outcome according to our result. The reason may be that CNS relapse generally occurs as a major cause of late recurrence. Trottestam et al. found that elevated ferritin level (>2000 μg/L) at diagnosis is an independent risk factor for pre-transplant death [9], but our results did not show significant relevance of this parameter to 30-day mortality.
To date, little is known about early indicators of resolution during the HLH treatment. Ferritin has been well recognized as a useful inflammatory marker correlating with current disease activity. Lin et al. has shown that patients with less than a 50 % drop in ferritin level as opposed to a 96 % or greater drop had a 17-fold increased chance of dying [27]. There is also a study by Otrock et al. observed a correlation between percent ferritin decrease less than 75 % and poor survival in adult patients [28]. Similarly, our results showed that patients with ferritin decrease over 80 % by 2 weeks had significantly higher rate of resolution as compared to those with less ferritin decrease in univariate analysis. But this effect was not significant in a multivariate logistic regression model. Thrombocytopenia has been described as a consistent prognostic factor for mortality in adult HLH by several studies [29–32]. Trottestam et al. has reported thrombocytopenia (platelet count <40 × 109/L) 2 weeks into therapy was associated with increased pre-transplant death in pediatric HLH [9]. Notably, an original finding of the present study is that platelet recovery in 2 weeks can largely predict resolution after 8 weeks of initial therapy, which is an important supplement and extension of the prognostic value of platelet count in HLH. Thrombocytopenia may result from multiple mechanisms, such as a dysmegakaryopoiesis due to excess of cytokines release, consumption through hemophagocytosis, hypersplenism, and disseminated intravascular coagulopathy [32, 33]. The conventional supportive care for thrombocytopenia is platelet transfusion, while a prospective study by Wang et al. has recently shown efficacy of recombinant human thrombopoietin (rhTPO) in the treatment of thrombocytopenia in adult HLH [34]. It could be interesting to investigate the benefit of rhTPO to patient outcomes.
Currently, clinical response at 2 and/or 4 weeks according to HLH-2004 protocol is the main index for clinicians to assess the efficacy of therapy in the early phase of treatment and decide on whether to improve treatment or not. Unexpectedly, the association between 2-week clinical response and the achievement of 8-week resolution was not significantly obvious according to our results. We consider that the current criteria for evaluating clinical response might not be specific enough to reflect real disease responsiveness. Particularly, ferritin level decrease (by 25 %) is one of the criteria, but in our cohort, 97 % of the patients reached this criterion at 2 weeks. And almost all patients fulfilled the criteria of normal fibrinogen levels and spleen size reduction by 2 weeks, irrespective of active disease after initial therapy. As these results come from analysis of small sample, these findings should be interpreted with caution.
Imashuku et al. has reported a male/female ratio of 0.64 in Japanese children with EBV-HLH [14]; on the contrary, more boys had this disease in our cohort with a male/female ratio of 2.1, which is consistent with reports from other Chinese groups [23, 35]. The male preponderance may reflect the pediatric population in China. For many decades, it has been concerned that a part of EBV-HLH in boys may actually be XLP, which is linked to mutations of the signaling lymphocyte activation molecule (SLAM)-associated protein (SAP) or X-linked inhibitor of apoptosis protein (XIAP), leading to dysregulated T cell activation in response to EBV infection [8, 14]. We sequenced exons of SH2D1A and XIAP genes of 60 children in this cohort, but no deleterious mutation was identified.
In conclusion, neutrophils <0.5 × 109/L, total bilirubin >2ULN, and albumin ≦20 g/L at diagnosis represent a set of risk factors for 30-day mortality in HLH. The number of risk factors at diagnosis identifies patients with high risk for early death. These results may provide clinical guidance in categorizing patients upon diagnosis, in order to intensify treatments including maximizing supportive care promptly for the subset of patients with a substantial risk of early fatal outcome. Platelet count normalization may aid clinicians in judging real responsiveness of the disease to the etoposide-based regimen as early as 2 weeks into therapy and highlighting poor responders for the consideration of treatment improvement. Large prospective studies with uniform evaluation and treatment are applauded to further verify these findings.
References
Risma K, Jordan MB (2012) Hemophagocytic lymphohistiocytosis: updates and evolving concepts. Curr Opin Pediatr 24(1):9–15. doi:10.1097/MOP.0b013e32834ec9c1
Jordan MB, Allen CE, Weitzman S, Filipovich AH, McClain KL (2011) How I treat hemophagocytic lymphohistiocytosis. Blood 118(15):4041–4052. doi:10.1182/blood-2011-03-278127
Meeths M, Horne A, Sabel M, Bryceson YT, Henter JI (2014) Incidence and clinical presentation of primary hemophagocytic lymphohistiocytosis in Sweden. Pediatr Blood Cancer. doi:10.1002/pbc.25308
Stepp SE, Dufourcq-Lagelouse R, Le Deist F, Bhawan S, Certain S, Mathew PA, Henter JI, Bennett M, Fischer A, de Saint Basile G, Kumar V (1999) Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science 286(5446):1957–1959
Feldmann J, Callebaut I, Raposo G, Certain S, Bacq D, Dumont C, Lambert N, Ouachee-Chardin M, Chedeville G, Tamary H, Minard-Colin V, Vilmer E, Blanche S, Le Deist F, Fischer A, de Saint Basile G (2003) Munc13-4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3). Cell 115(4):461–473
zur Stadt U, Schmidt S, Kasper B, Beutel K, Diler AS, Henter JI, Kabisch H, Schneppenheim R, Nurnberg P, Janka G, Hennies HC (2005) Linkage of familial hemophagocytic lymphohistiocytosis (FHL) type-4 to chromosome 6q24 and identification of mutations in syntaxin 11. Hum Mol Genet 14(6):827–834
Cote M, Menager MM, Burgess A, Mahlaoui N, Picard C, Schaffner C, Al-Manjomi F, Al-Harbi M, Alangari A, Le Deist F, Gennery AR, Prince N, Cariou A, Nitschke P, Blank U, El-Ghazali G, Menasche G, Latour S, Fischer A, de Saint Basile G (2009) Munc18-2 deficiency causes familial hemophagocytic lymphohistiocytosis type 5 and impairs cytotoxic granule exocytosis in patient NK cells. J Clin Invest 119(12):3765–3773. doi:10.1172/JCI40732
Yang X, Miyawaki T, Kanegane H (2012) SAP and XIAP deficiency in hemophagocytic lymphohistiocytosis. Pediatr Int 54(4):447–454. doi:10.1111/j.1442-200X.2012.03683.x
Trottestam H, Berglof E, Horne A, Onelov E, Beutel K, Lehmberg K, Sieni E, Silfverberg T, Arico M, Janka G, Henter JI (2012) Risk factors for early death in children with haemophagocytic lymphohistiocytosis. Acta Paediatr 101(3):313–318. doi:10.1111/j.1651-2227.2011.02501.x
Henter JI, Arico M, Egeler RM, Elinder G, Favara BE, Filipovich AH, Gadner H, Imashuku S, Janka-Schaub G, Komp D, Ladisch S, Webb D (1997) HLH-94: a treatment protocol for hemophagocytic lymphohistiocytosis. HLH study Group of the Histiocyte Society. Med Pediatr Oncol 28(5):342–347. doi:10.1002/(SICI)1096-911X(199705)28:5<342::AID-MPO3>3.0.CO;2-H
Henter JI, Horne A, Arico M, Egeler RM, Filipovich AH, Imashuku S, Ladisch S, McClain K, Webb D, Winiarski J, Janka G (2007) HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 48(2):124–131. doi:10.1002/pbc.21039
Henter JI, Samuelsson-Horne A, Arico M, Egeler RM, Elinder G, Filipovich AH, Gadner H, Imashuku S, Komp D, Ladisch S, Webb D, Janka G (2002) Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood 100(7):2367–2373. doi:10.1182/blood-2002-01-0172
Imashuku S, Hibi S, Kuriyama K, Tabata Y, Hashida T, Iwai A, Kato M, Yamashita N, Oda MM, Kinugawa N, Sawada M, Konno M (2000) Management of severe neutropenia with cyclosporin during initial treatment of Epstein-Barr virus-related hemophagocytic lymphohistiocytosis. Leuk Lymphoma 36(3–4):339–346. doi:10.3109/10428190009148855
Imashuku S (2002) Clinical features and treatment strategies of Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis. Crit Rev Oncol Hematol 44(3):259–272
Imashuku S, Kuriyama K, Teramura T, Ishii E, Kinugawa N, Kato M, Sako M, Hibi S (2001) Requirement for etoposide in the treatment of Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis. J Clin Oncol 19(10):2665–2673
Trottestam H, Horne A, Arico M, Egeler RM, Filipovich AH, Gadner H, Imashuku S, Ladisch S, Webb D, Janka G, Henter JI (2011) Chemoimmunotherapy for hemophagocytic lymphohistiocytosis: long-term results of the HLH-94 treatment protocol. Blood 118(17):4577–4584. doi:10.1182/blood-2011-06-356261
Imashuku S, Kuriyama K, Sakai R, Nakao Y, Masuda S, Yasuda N, Kawano F, Yakushijin K, Miyagawa A, Nakao T, Teramura T, Tabata Y, Morimoto A, Hibi S (2003) Treatment of Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis (EBV-HLH) in young adults: a report from the HLH study center. Med Pediatr Oncol 41(2):103–109. doi:10.1002/mpo.10314
Koh KN, Im HJ, Chung NG, Cho B, Kang HJ, Shin HY, Lyu CJ, Yoo KH, Koo HH, Kim HJ, Baek HJ, Kook H, Yoon HS, Lim YT, Kim HS, Ryu KH, Seo JJ (2015) Clinical features, genetics, and outcome of pediatric patients with hemophagocytic lymphohistiocytosis in Korea: report of a nationwide survey from Korea Histiocytosis Working Party. Eur J Haematol 94(1):51–59. doi:10.1111/ejh.12399
De-The G (1981) The Chinese epidemiological approach of nasopharyngeal carcinoma research and control. Yale J Biol Med 54(1):33–39
Imashuku S, Teramura T, Kuriyama K, Kitazawa J, Ito E, Morimoto A, Hibi S (2002) Risk of etoposide-related acute myeloid leukemia in the treatment of Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis. Int J Hematol 75(2):174–177
Kaito K, Kobayashi M, Katayama T, Otsubo H, Ogasawara Y, Sekita T, Saeki A, Sakamoto M, Nishiwaki K, Masuoka H, Shimada T, Yoshida M, Hosoya T (1997) Prognostic factors of hemophagocytic syndrome in adults: analysis of 34 cases. Eur J Haematol 59(4):247–253
Yu JT, Wang CY, Yang Y, Wang RC, Chang KH, Hwang WL, Teng CL (2013) Lymphoma-associated hemophagocytic lymphohistiocytosis: experience in adults from a single institution. Ann Hematol 92(11):1529–1536. doi:10.1007/s00277-013-1784-3
Xue H, Chen C, Li W, Lin C, Fang J, Li Y, Xu H (2015) Analysis of prognostic risk factors in children with Epstein-Barr virus-associated hemophagocytic syndrome. Minerva Pediatr 67(3):251–261
Park HS, Kim DY, Lee JH, Kim SD, Park YH, Lee JS, Kim BY, Jeon M, Kang YA, Lee YS, Seol M, Lee YJ, Lim YS, Jang S, Park CJ, Chi HS, Lee KH (2012) Clinical features of adult patients with secondary hemophagocytic lymphohistiocytosis from causes other than lymphoma: an analysis of treatment outcome and prognostic factors. Ann Hematol 91(6):897–904. doi:10.1007/s00277-011-1380-3
Horne A, Trottestam H, Arico M, Egeler RM, Filipovich AH, Gadner H, Imashuku S, Ladisch S, Webb D, Janka G, Henter JI (2008) Frequency and spectrum of central nervous system involvement in 193 children with haemophagocytic lymphohistiocytosis. Br J Haematol 140(3):327–335
Kim MM, Yum MS, Choi HW, Ko TS, Im HJ, Seo JJ, Koh KN (2012) Central nervous system (CNS) involvement is a critical prognostic factor for hemophagocytic lymphohistiocytosis. Korean J Hematol 47(4):273–280. doi:10.5045/kjh.2012.47.4.273
Lin TF, Ferlic-Stark LL, Allen CE, Kozinetz CA, McClain KL (2011) Rate of decline of ferritin in patients with hemophagocytic lymphohistiocytosis as a prognostic variable for mortality. Pediatr Blood Cancer 56(1):154–155. doi:10.1002/pbc.22774
Otrock ZK, Eby CS (2015) Clinical characteristics, prognostic factors, and outcomes of adult patients with hemophagocytic lymphohistiocytosis. Am J Hematol 90(3):220–224. doi:10.1002/ajh.23911
Dhote R, Simon J, Papo T, Detournay B, Sailler L, Andre MH, Dupond JL, Larroche C, Piette AM, Mechenstock D, Ziza JM, Arlaud J, Labussiere AS, Desvaux A, Baty V, Blanche P, Schaeffer A, Piette JC, Guillevin L, Boissonnas A, Christoforov B (2003) Reactive hemophagocytic syndrome in adult systemic disease: report of twenty-six cases and literature review. Arthritis Rheum 49(5):633–639. doi:10.1002/art.11368
Li F, Yang Y, Jin F, Dehoedt C, Rao J, Zhou Y, Li P, Yang G, Wang M, Zhang R (2015) Clinical characteristics and prognostic factors of adult hemophagocytic syndrome patients: a retrospective study of increasing awareness of a disease from a single-center in China. Orphanet J Rare Dis 10:20. doi:10.1186/s13023-015-0224-y
Li J, Wang Q, Zheng W, Ma J, Zhang W, Wang W, Tian X (2014) Hemophagocytic lymphohistiocytosis: clinical analysis of 103 adult patients. Medicine (Baltimore) 93(2):100–105. doi:10.1097/MD.0000000000000022
Arca M, Fardet L, Galicier L, Riviere S, Marzac C, Aumont C, Lambotte O, Coppo P (2015) Prognostic factors of early death in a cohort of 162 adult haemophagocytic syndrome: impact of triggering disease and early treatment with etoposide. Br J Haematol 168(1):63–68. doi:10.1111/bjh.13102
Zoller EE, Lykens JE, Terrell CE, Aliberti J, Filipovich AH, Henson PM, Jordan MB (2011) Hemophagocytosis causes a consumptive anemia of inflammation. J Exp Med 208(6):1203–1214. doi:10.1084/jem.20102538
Wang Y, Wang Z, Wu L, Zhang J, Wang J, Yan L (2013) Recombinant human thrombopoietin is an effective treatment for thrombocytopenia in hemophagocytic lymphohistiocytosis. Ann Hematol 92(12):1695–1699. doi:10.1007/s00277-013-1819-9
Zhizhuo H, Junmei X, Yuelin S, Qiang Q, Chunyan L, Zhengde X, Kunling S (2012) Screening the PRF1, UNC13D, STX11, SH2D1A, XIAP, and ITK gene mutations in Chinese children with Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 58(3):410–414. doi:10.1002/pbc.23216
Acknowledgments
This work was supported by grants provided by the National Natural Science Foundation of China (NO. 30860308; NO. 81160070). We thank all of the patients who gave consent to participate in this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Approval to perform this study was given by the First Affiliated Hospital of Guangxi Medical University Institutional Review Board. Informed consent was obtained from all patients for being included in the study.
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Bin, Q., Gao, JH. & Luo, JM. Prognostic factors of early outcome in pediatric hemophagocytic lymphohistiocytosis: an analysis of 116 cases. Ann Hematol 95, 1411–1418 (2016). https://doi.org/10.1007/s00277-016-2727-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00277-016-2727-6